Chapter 12
Cancer chemotherapy
12.1 Overview
The purpose of cancer chemotherapy is analogous to that of antimicrobial chemotherapy, namely, to selectively kill the pathogenic cells. Of the two, cancer chemotherapy tends to be more toxic, yet less reliably effective; or in other words, it fails to achieve the level of selective toxicity that is possible with antimicrobial chemotherapy. This is not surprising, since the cells that we aim to destroy originate in our own bodies, and therefore contain few or no drug targets that are truly distinct from those found in healthy cells. Considering this lack of biochemical distinctiveness, it is rather remarkable that cancer chemotherapy works even as well as it does.
Like pathogenic microbes, cancer cells may develop resistance to anticancer drugs under therapy. To mitigate this problem, anticancer drugs are almost always used in combination.
12.1.1 Forms of cancer therapy
- Surgery
- Radiation
- Chemotherapy
Criteria for therapy selection
- Benign or malignant tumor
- Tissue of origin, histological variant of tumor
- Stage of tumor—early and localized vs advanced and disseminated
Notes: Broadly speaking, surgery is applied locally, radiation regionally, and chemotherapy systemically.
Benign tumors don’t infiltrate the surrounding tissue and don’t give rise to metastases; therefore, they can usually be cured through surgery alone. In most solid malignant tumors, surgery is used to remove the main tumor mass as far as possible. Subsequently, radiation or chemotherapy are applied alone or in combination in an attempt to extirpate any regionally or systemically disseminated tumor cells and thus prevent resurgence of the cancer. However, leukemias and lymphomas, as well as some specific solid tumors, are treated primarily with chemotherapy.
As we had seen in Chapter 7, the cells in some of our organs depend on growth stimulation by hormones. Tumor cells originating in these organs may retain this hormone dependency; important examples are breast and prostate cancers, which often remain dependent on growth stimulation by sexual steroid hormones. In such cases, withdrawal of the hormone using receptor antagonists or inhibitors of hormone synthesis can be an effective part of chemotherapy. Where such specific hormone dependency does not exist, broadly cytotoxic drugs that interfere with mitosis, DNA synthesis, or other fundamental aspects of cell biology are often the only option. In any case, such cytotoxic drugs are part of most combination regimens.1
It is important to understand that cytotoxic drugs don’t simply overwhelm and crush the cancer cells; instead, they trigger regulatory pathways within the cell that initiate programmed cell death, or apoptosis. Accordingly, mutations that inactivate apoptotic pathways are often responsible for resistance of tumor cells to chemotherapy.
12.2 Cellular pathways that control proliferation and apoptosis
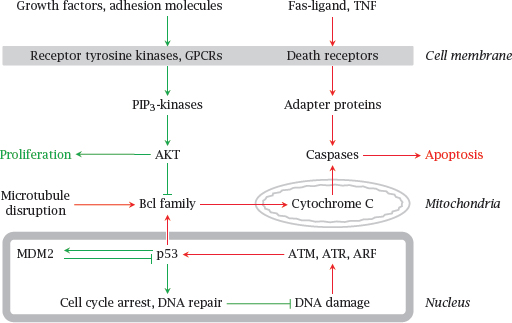
Notes: The lifetimes and rates of proliferation of the cells in our bodies are highly variable. Most nerve cells in the brain don’t ever divide but just stay put throughout life,2 while those in the bone marrow proliferate rapidly; lymphocytes often end their days through apoptosis, which is necessary to avoid damage to the body through autoimmune reactivity. The pathways that determine whether a cell is to live, proliferate, or commit apoptosis are subject to both intra- and extracellular influences.
Growth factors and adhesion molecules stimulate the PIP3 kinase pathway and thereby stimulate proliferation and inhibit apoptosis. Conversely, mediator proteins such as Fas-Ligand and tumor necrosis factor (TNF) act on so-called “death receptor” proteins in the cell membrane. This results in the activation of caspases, a family of cysteine proteases that cleave their numerous substrates at aspartate residues, and thereby destroy the cell.
In the intrinsic pathway of apoptosis, DNA damage leads to the activation of protein kinases such as ATM and ATR, which in turn activate the transcriptional regulator p53. Subsequently, increased expression of certain regulatory proteins in the Bcl family leads to the permeabilization of mitochondria by Bax, another Bcl family member. Cytochrome C released from the mitochondria then again activates caspases.
Mutations in many of the proteins shown here have been implicated in the induction or progression of cancer. One such protein, p53, is disabled by mutation in as many as 50% of all malignant tumors, which highlights its central role in tumor suppression.
12.2.1 Dysregulation of growth in tumor cells
Normal body cells
- grow or persist only when stimulated by growth factors, which is mediated by growth factor receptors
- undergo apoptosis when deprived of growth factor stimulation
Tumor cells contain mutations that
- create surrogate growth stimuli: Constitutively active receptors, autocrine secretion of growth factors
- inhibit activation of apoptosis downstream of growth factor deprivation
- make tumor cells more susceptible to some apoptotic stimuli than normal cells
Notes: It was noted above that p53 is very frequently inactivated in malignant tumors. This protein has an ambivalent role: while it promotes apoptosis when maximally activated, at lower degrees of activation it can actually promote cell survival. It does this by initiating a pause in cell division and activating DNA repair systems. Failure to induce DNA repair may be one of the reasons why tumor cells actually tend to be more susceptible to certain apoptotic stimuli than normal cells. On the other hand, failure of DNA repair also increases the mutation rate, which in turn accelerates the progression of the tumor to more malignant and invasive behavior, and the development of resistance to chemotherapy.
The readiness to plunge into apoptosis varies not only between normal cells on one hand and tumor cells on the other, but also between normal cells from different tissues. For example, lymphocytes and their precursors are driven into apoptosis extremely easily (cf. slide 10.3.2). Tumors derived from lymphocytes—lymphomas and lymphatic leukemias—often retain this trait, and therefore are much more readily amenable to chemotherapy than most other tumors. On the other hand, tumors derived from normal cells with little inclination to commit apoptosis may inherit this trait also, and therefore tend to be quite impervious to chemotherapy. Examples are cancers originating in the brain and kidneys.
12.2.2 Cellular models of dysregulated apoptosis
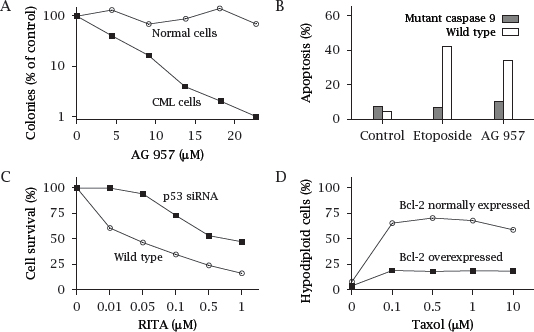
Notes: This slide shows a few experiments that illustrate some of the points made above on the dysregulation of apoptosis in tumors.
The plots in this slide were redrawn from data found in various sources [90, 91, 92].
12.3 The cell cycle and its checkpoints
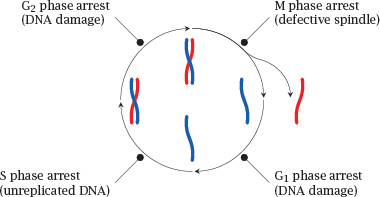
Notes: Cell proliferation goes through successive coordinated phases, which together constitute the cell cycle. Daughter cells arise by mitosis, which within the cell cycle is referred to as the M phase. After the G1 phase,4 a cell prepares for the next round of division by entering the S phase, in which the DNA is replicated, that is, a second double-stranded DNA copy is synthesized for each chromosome. Following another short intermission, the G2 phase, mitosis repeats.
In each of these phases, corresponding checkpoint proteins are activated that will arrest the cell cycle if they detect specific forms of genetic damage. Depending on the extent of damage, the arrest may be followed by DNA repair and resumption of the cell cycle, or apoptosis may be triggered.
In the M phase, cell cycle arrest may be induced if defects are found in the mitotic spindle, that is, the cytoskeletal apparatus that distributes the chromosomes evenly between the two daughter cells. Defects in the M phase checkpoint proteins will permit the survival of cells that have gained or lost chromosome fragments, entire chromosomes, or even acquired extra copies of the whole genome. Cells whose chromosome complement deviates from the regular diploid one are called aneuploid. The occurrence of aneuploid cells is a hallmark of cancer and contributes to genetic variability and chemoresistance.
12.3.1 Progressive cell aneuploidy in a recurring tumor
Notes: In the experiment shown, the nuclei of cells obtained from a sarcoma, that is, a malignant tumor derived from non-epithelial tissue, were isolated and incubated with a fluorescent dye such as propidium iodide, which emits fluorescence only after intercalation into DNA. The stained nuclei were then passed through a flow cytometer. As the nuclei pass the instrument’s laser beam in single file, each nucleus causes a fluorescence pulse, the intensity of which is proportional to its DNA content.
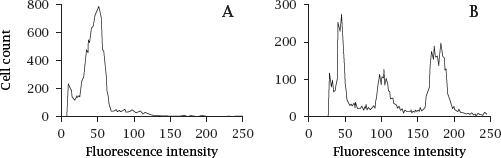
In the original tumor (A), the DNA content per cell shows one major peak, corresponding to the diploid chromosome complement. In the recurring tumor (B), there are multiple peaks, indicating aneuploidy and clonal divergence.
Aneuploidy is very common with malignant tumors. The additional or missing gene copies change the expression levels of individual proteins, including ones that influence tumor proliferation, apoptosis, and susceptibility or resistance to anticancer drugs. From the genetically heterogeneous cell population, particularly resistant cell clones may be selected under drug therapy. Figure prepared from original data in [93].
12.4 Cell type-specific anticancer drugs
- Hormone antagonists: Most significant with breast and prostate cancer
- Hormones and growth factors: Interferon in hairy cell leukemia
- Tissue-specific prodrug activation: Mitotane in adrenal gland tumors
- Tissue-specific accumulation of radioactive iodine: Thyroid cancer
Notes: Among the numerous anticancer drugs, we can broadly distinguish those that act only on specific cell types from those that are broadly cytotoxic. We will now consider some examples of drugs in the first category.
12.4.1 Female sexual hormones and receptor antagonists
Notes: Estrogens like estradiol or progestins like progesteron are required by many breast or uterine cancers, which can therefore be treated with estrogen receptor antagonists like tamoxifen or progestin receptor antagonists like mifepristone.
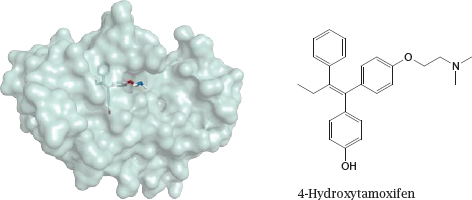
The structure (rendered from 2p7z.pdb) in this slide shows 4-hydroxy-tamoxifen, an active metabolite of tamoxifen, bound to the ligand-binding domain of the estrogen receptor; the formula of the free compound is rendered in a similar orientation. Mifepristone is shown in slide 7.4.5.
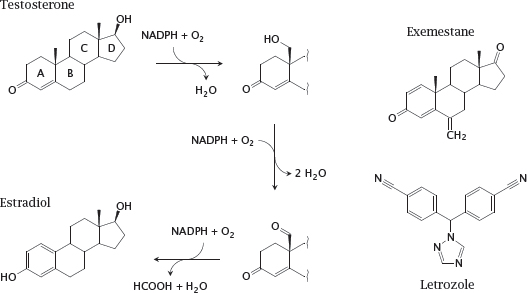
12.4.2 Aromatase and two of its inhibitors
Notes: Aromatase is a cytochrome P450 enzyme that converts androgens to estrogens (see slide 7.4.8). The left part of this slide shows details of the aromatase reaction. The methyl group between rings A and B is converted in two steps to an aldehyde. The keto group in ring A then becomes its enol tautomer, which creates a second double bond in the ring. The third double bond is introduced into ring A concomitantly with oxidative cleavage of the exocyclic aldehyde as formic acid.