Chapter 12
Cancer Biology
Neal S. Rote and David M. Virshup∗

Cancer Characteristics and Terminology
The term cancer derives from the Greek word for crab, karkinoma, which the physician Hippocrates used to describe the appendage-like projections extending from tumors. The word tumor originally referred to any swelling that was caused by inflammation, but is now generally reserved for describing a new growth, or neoplasm. Not all tumors or neoplasms, however, are cancer. The term cancer refers to a malignant tumor and is not used to refer to benign growths, such as lipomas or hypertrophy of an organ. Yet it is important to recognize that benign neoplasms also can be life threatening if they enlarge in critical locations. For example, a benign meningioma at the base of the skull may cause symptoms by compressing adjacent normal brain tissue. The definitions of benign vs. malignant are presented in the following text and in Table 12-1.
TABLE 12-1
CHARACTERISTICS OF BENIGN VS. MALIGNANT TUMORS
| BENIGN TUMORS | MALIGNANT TUMORS |
| Grow slowly | Grow rapidly |
| Have a well-defined capsule | Are not encapsulated |
| Are not invasive | Invade local structures and tissues |
| Are well differentiated; look like the tissue from which they arose | Are poorly differentiated; may not be able to determine tissue of origin |
| Have a low mitotic index; dividing cells are rare | High mitotic index; many dividing cells |
| Do not metastasize | Can spread distantly, often through blood vessels and lymphatics |
Tumor Classification and Nomenclature
Some tumors initially described as benign can progress to cancer and then are referred to as malignant tumors. These tumors are distinguished from benign tumors by their more rapid growth rates and specific microscopic alterations, including loss of differentiation and absence of normal tissue organization. One of the hallmarks of cancer cells, as seen under the microscope, is anaplasia, the loss of cellular differentiation, irregularities of the size and shape of the nucleus, and the loss of normal tissue structure. Malignant tumors may present with different degrees of encapsulation; some lack a capsule, and even if a capsule is apparent, its integrity has been compromised so that tumor cells can grow to invade nearby blood vessels, lymphatics, and surrounding structures. The most important and most deadly characteristic of malignant tumors is their ability to spread far beyond the tissue of origin, a process known as metastasis (Figures 12-1 and 12-2; see Table 12-1).
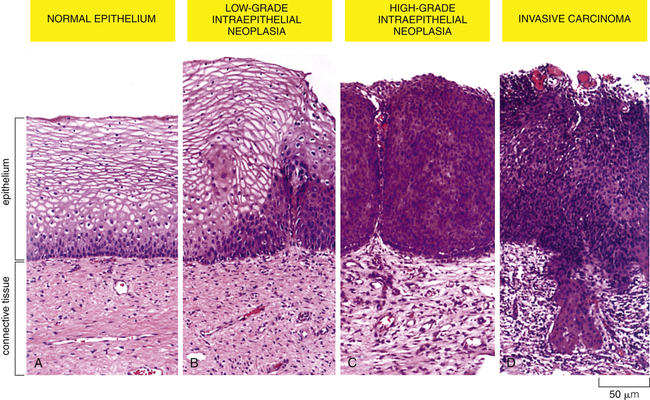
A sequence of cellular and tissue changes progressing from low-grade to high-grade intraepithelial neoplasms (also called carcinoma in situ) and then to invasive cancer is seen often in the development of cancer. In this example of the early stages of cervical neoplastic changes, the presence of anaplastic cells and the loss of normal tissue architecture signify the development of cancer. The high rate of cell division and the presence of local mutagens and inflammatory mediators all contribute to the accumulation of genetic abnormalities that lead to cancer. (From Alberts B et al: Molecular biology of the cell, ed 5, New York, 2002, Garland.)
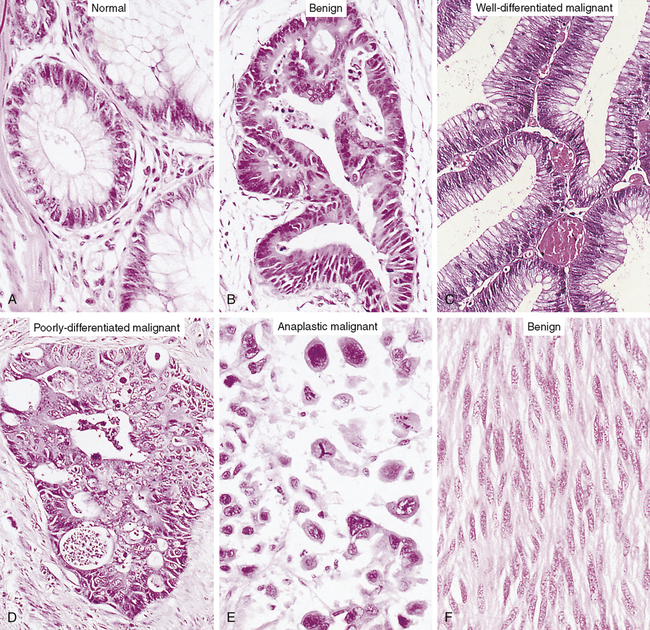
The cells of a benign neoplasm (B) resemble those of the normal colonic epithelium (A), in that they are columnar and have an orderly arrangement. Loss of some degree of differentiation is evident in that the neoplastic cells do not show much mucin vacuolization. Cells of the well-differentiated malignant neoplasm (C) of the colon have a haphazard arrangement and although gland lumina are formed, they are architecturally abnormal and irregular. Nuclei vary in shape and size, especially when compared with those in A. Cells in the poorly differentiated malignant neoplasm (D) have an even more haphazard arrangement, with very poor formation of gland lumina. Nuclei show greater variation in shape and size compared with the well-differentiated malignant neoplasm in C. Cells in anaplastic malignant neoplasms (E) bear no relation to the normal epithelium, with no recognizable gland formation. Tremendous variation is found in the size of cells and their nuclei, with very intense staining (hyperchromatic nuclei). Not knowing the site of origin would make it impossible to classify this tumor by microscopic appearance alone. Well-differentiated tumors often resemble their cell of origin, as shown in the example of a benign tumor of smooth muscles (F). (From Stevens A, Lowe J: Pathology, ed 2, London, 2000, Mosby.)
In general, cancers are named according to the cell type from which they originate. Cancers arising in epithelial tissue are called carcinomas, and if they arise from or form ductal or glandular structures are named adenocarcinomas. Hence, a malignant tumor arising from breast glandular tissue is a mammary adenocarcinoma. Cancers arising from connective tissue usually have the suffix sarcoma. For example, malignant cancers of skeletal muscle are known as rhabdomyosarcomas. Cancers of lymphatic tissue are called lymphomas, whereas cancers of blood-forming cells are called leukemias. However, many cancers, such as Hodgkin disease and Ewing sarcoma, are named for historical reasons that do not follow this naming convention. Table 12-2 presents the nomenclature and classification of selected tumors.
TABLE 12-2
NOMENCLATURE AND CLASSIFICATION OF BENIGN AND MALIGNANT TUMORS∗
| CELL OR TISSUE OF ORIGIN | BENIGN TUMOR | MALIGNANT TUMOR |
| Tumors of Epithelial Origin | ||
| Squamous cells | Squamous cell papilloma | Squamous cell carcinoma |
| Basal cells | — | Basal cell carcinoma |
| Glandular or ductal epithelium | Adenoma | Adenocarcinoma |
| Cystadenoma | Cystadenocarcinoma | |
| Transitional cells | Transitional cell papilloma | Transitional cell carcinoma |
| Bile duct | Bile duct adenoma | Bile duct carcinoma (cholangiocarcinoma) |
| Liver cells | Hepatocellular adenoma | Hepatocellular carcinoma |
| Melanocytes | Nevus | Malignant melanoma |
| Renal epithelium | Renal tubular adenoma | Renal cell carcinoma |
| Skin adnexal glands | ||
| Sweat glands | Sweat gland adenoma | Sweat gland carcinoma |
| Sebaceous glands | Sebaceous gland adenoma | Sebaceous gland carcinoma |
| Germ cells (testis and ovary) | — | Seminoma (dysgerminoma) |
| Embryonal carcinoma, yolk sac carcinoma | ||
| Tumors of Mesenchymal Origin | ||
| Hematopoietic/lymphoid tissue | ||
| Leukocytes | Leukemias | |
| Granular leukocytes and precursors | Granulocytic leukemia | |
| Myelocytic leukemias | ||
| Myelogenous leukemias | ||
| Plasma cells | Multiple myeloma | |
| Lymphoid | ||
| Nongranular leukocytes and prelymphocytes | Lymphomas | |
| Proliferating lymphocytes and monocytes | Lymphocytic leukemia | |
| Proliferating immature precursor monocytes | Lymphoblastic leukemia | |
| Solid tumors of lymph tissue (thymus, spleen, lymph nodes) | Lymphoma or lymphosarcoma | |
| Neural and retinal tissue | ||
| Nerve sheath | Neurilemoma, neurofibroma | Malignant peripheral nerve sheath tumor |
| Nerve cells | Ganglioneuroma | Neuroblastoma |
| Retinal cells (cones) | — | Retinoblastoma |
| Connective tissue | ||
| Fibrous tissue | Fibromatosis (desmoid) | Fibrosarcoma |
| Fat | Lipoma | Liposarcoma |
| Bone | Osteoma | Osteogenic sarcoma |
| Cartilage | Chondroma | Chrondrosarcoma |
| Muscle | ||
| Smooth muscle | Leiomyoma | Leiomyosarcoma |
| Striated muscle | Rhabdomyoma | Rhabdomyosarcoma |
| Endothelial and related tissues | ||
| Blood vessels | Hemangioma | Angiosarcoma |
| Kaposi sarcoma | ||
| Lymph vessels | Lymphangioma | Lymphangiosarcoma |
| Synovium | — | Synovial sarcoma |
| Mesothelium | — | Malignant mesothelioma |
| Meninges | Meningioma | Malignant meningioma |
| Tumors of Uncertain Origin | — | Ewing tumor |
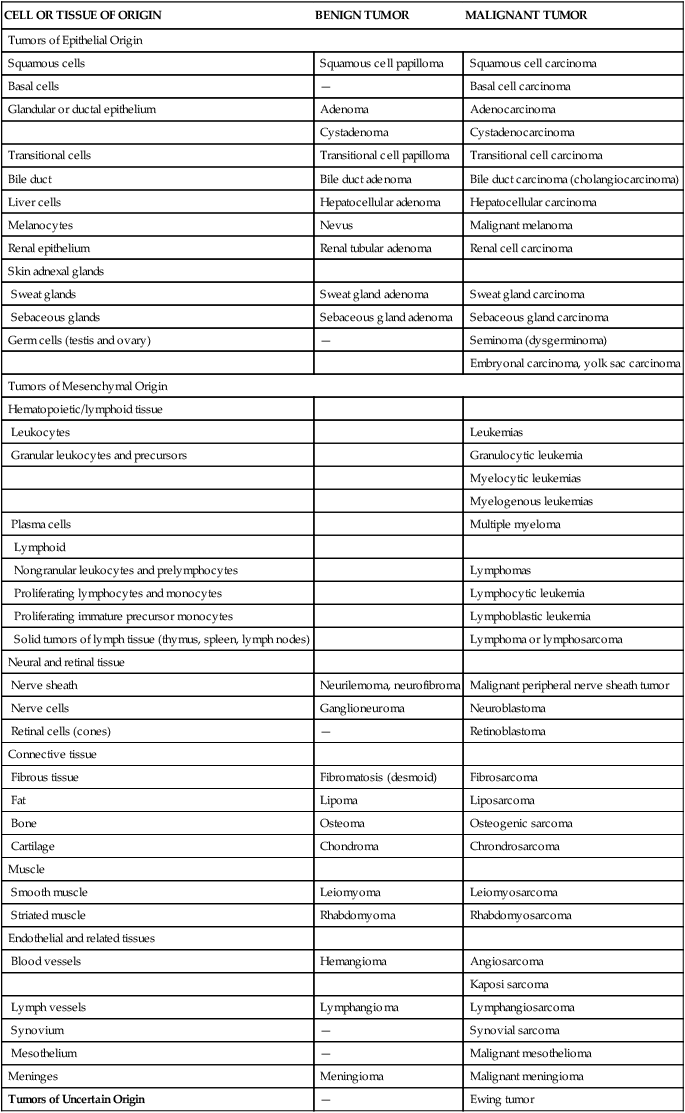
∗Note: This list is intended to provide only an introduction to tumor nomenclature.
Modified from Murphy GP et al: American Cancer Society’s textbook of clinical oncology, ed 2, New York, 1995, American Cancer Society.
Classification of Tumors—Classical Histology and Modern Genetics
Carcinoma in situ (often abbreviated CIS) refers to preinvasive epithelial malignant tumors of glandular or squamous cell origin. Cancers develop incrementally, as they accumulate specific genetic lesions. Careful surveillance for cancer often detects abnormal growths in epithelial tissues that have atypical cells and an increased proliferation rate compared with normal surrounding tissues. These early stage cancers are localized to the epithelium and have not penetrated the local basement membrane or invaded the surrounding stroma (see Figure 12-1). Based on these characteristics, they are not malignant but are often called carcinoma in situ (CIS). CIS is recognized in a number of sites, including the cervix, skin, oral cavity, esophagus, and bronchus. In glandular epithelium, in situ lesions occur in the stomach, endometrium, breast, and large bowel. In the breast, ductal carcinoma in situ (DCIS) fills the mammary ducts but has not progressed to local tissue invasion. DCIS lesions are readily treatable, although the optimal therapeutic approach is controversial. CIS lesions can have one of the following three fates: they can remain stable for a long time, they can progress to invasive and metastatic cancers, or they can regress and disappear. CIS can vary from low-grade to high-grade dysplasia, with the high-grade lesions having the highest likelihood of becoming invasive cancers. The time that such preinvasive lesions remain in situ before becoming invasive is unknown.1 Some carcinomas of the cervix appear as preinvasive lesions in situ for several years before they progress to invasive carcinoma and metastatic tumors (see Figure 12-1). Knowing how to best treat low-grade CIS lesions is challenging because the proportion that progress to cancer vs. the proportion that will never cause clinical problems is usually not known. Although most persons prefer removal of any CIS as opposed to “watchful waiting,” this topic continues to be a source of great debate.
Because our knowledge about the molecular alterations in cancer can influence the choices of therapy, it becomes increasingly important for clinicians to accurately molecularly classify each cancer. The classification, and hence the treatment decisions, of cancers was originally based on gross and light microscopic appearance and is now commonly accompanied by immunohistochemical analysis of protein expression. Increasingly, this is supplemented by a more extensive molecular analysis of the tumors. Sometimes a single gene is examined (for example, to determine if there is a characteristic chromosomal translocation diagnostic of chronic myelogenous leukemia [CML]), and sometimes a panel of genes and proteins are examined (e.g., in breast cancer) to determine if the tumor expresses estrogen receptor, progesterone receptor, and the epidermal growth factor (EGF) receptor HER2, or if there are mutations in specific genes that include response to therapy. In a research setting, and increasingly in clinical settings, expression and mutation analysis of a large number of genes can be measured using polymerase chain reaction (PCR), microarray, or advanced DNA sequencing technology. These analyses can be used to classify tumors more precisely and may predict the most effective therapy. This detailed analysis of each tumor is a form of personalized medicine that offers therapy based on a very detailed knowledge of each individual’s characteristics and their specific cancer.2 This enhanced molecular characterization subdivides cancers into therapeutically and prognostically relevant smaller groups. As an example, breast cancers can now be subclassified into more than four types (luminal A, luminal B, basal-like, and others) based on their expression of specific markers, such as estrogen receptor, HER2/Neu, and other specific genes and proteins. Each subtype has a different response to therapy and a different prognosis (Figure 12-3).
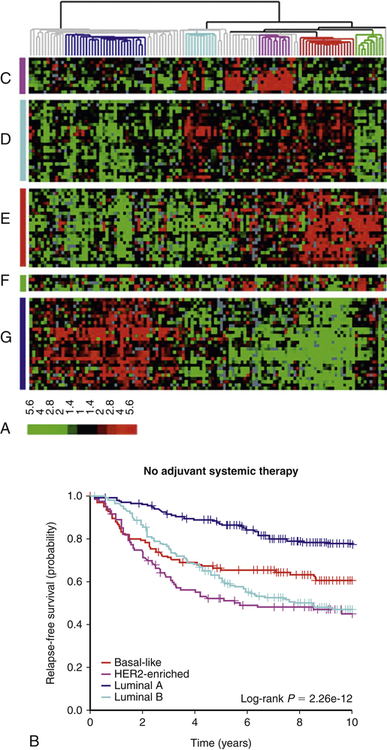
A, Cancers can be classified based on gene expression patterns. In this breast cancer study, gene expression was measured in tumors from 115 patients. Each row is a different gene, and each column is a different patient sample. Red denotes high gene expression; green signifies low gene expression. Using this molecular subtyping method, breast cancer can be subdivided into at least four molecular subtypes—luminal A, luminal B, HER2, and basal. The group on the far right is normal breast tissue. B, Molecular classification predicts overall survival. In this group of women, breast cancer molecular subtypes were determined by gene expression profiles in a group of women with similar stage tumors, as in (A). The molecular subtype is a good predictor of response to chemotherapy and survival. This molecular subtyping method can help select the best therapy for women. (A from Sorlie T et al: Proc Natl Acad Sci U S A 100[14]:8418–8423, 2003; B from Parker JS et al: J Clin Oncol 27[8]:1160–1167, 2009.)
Tumor Markers
During surveillance or diagnosis of cancer as well as following therapy, specific biochemical markers of tumors have proven to be helpful. These tumor markers are substances produced by both benign and malignant cells that either are present in or on tumor cells or are found in blood, spinal fluid, or urine (Table 12-3). Some tumor markers have been known for many decades. For diseases associated with a tumor marker, there is indeed a “blood test for cancer.” Tumor markers include hormones, enzymes, genes, antigens, and antibodies. Liver and germ cell tumors secrete a protein known as alpha fetoprotein (AFP) into the blood, and prostate tumors secrete prostate specific antigen (PSA) into the blood. If the tumor marker itself has biologic activity, then it can cause symptoms, a phenomenon known as a paraneoplastic syndrome. For example, the adrenal medulla normally secretes the catecholamine epinephrine (adrenaline). Benign tumors of the adrenal medulla (pheochromocytoma) can produce catecholamines (e.g., adrenaline) in vast excess, leading to rapid pulse rate, high blood pressure, diaphoresis (i.e., sweating), and tremors. Detection of elevated blood or urine levels of catecholamines helps to confirm the diagnosis, and treatment of the disease relieves the symptoms. Tumor markers can be used in three ways: (1) to screen and identify individuals at high risk for cancer; (2) to help diagnose the specific type of tumor in individuals with clinical manifestations relating to their tumor, as in adrenal tumors or enlarged liver or prostate; and (3) to follow the clinical course of a tumor. For example, a falling PSA level after radiation or surgical therapy for prostate cancer indicates successful treatment, and a later rise in the PSA level may indicate a recurrence.
TABLE 12-3
| MARKER NAME | NATURE | TYPE OF CANCER |
| Alpha fetoprotein (AFP) | 70-kDa protein | Hepatic, germ cell |
| Carcinoembryonic antigen (CEA) | 200-kDa glycoprotein | GI, pancreas, lung, breast, etc. |
| β-Human chorionic gonadotropin (β-hCG) | Glycopeptide hormone β-chain | Germ cell |
| Prostate-specific antigen (PSA) | 33-kDa glycoprotein | Prostate |
| Catecholamines | Epinephrine and precursors | Pheochromocytoma (adrenal medulla) |
| Homovanillic acid/vanillylmandelic acid (HVA/VMA) | Catecholamine metabolites | Neuroblastoma |
| Urinary Bence Jones protein | Ig light chain | Multiple myeloma |
| Adrenocorticotropic hormone (ACTH) | Peptide hormone | Pituitary adenomas |
The Biology of Cancer Cells
Transformation and Differentiation
Cancer cells behave differently than normal cells in several important ways. Transformation refers to the process by which a normal cell becomes a cancer cell. Autonomy refers to the cancer cell’s independence from normal cellular controls and is part of the transformational process. These differences are most readily seen in specialized laboratory assays, especially those examining the growth patterns of normal and cancerous cells in laboratory incubators. Transformed cells lack many of the normal “social controls” seen in nontransformed cells. They often have markedly decreased requirements for external growth factors. Normal cells cease to divide when they fill a petri (or tissue culture) dish. Transformed cells, unlike normal cells, lack contact inhibition and continue to crowd, eventually piling up on each other (Figure 12-4). Normal cells usually will not grow unless they are attached to a firm surface (like a petri dish). However, cancer cells are often anchorage independent; that is, they continue to divide even when suspended in a soft agar gel. Normal cells have a limited life span in the laboratory; they may divide in a petri dish 10 or 50 times, but then they cease growing. Cancer cells usually are immortal in that they seem to have an unlimited life span and will continue to divide for years under appropriate laboratory conditions. One of the most commonly used laboratory cell lines, HeLa cells, was derived from a cervical cancer specimen obtained in 1951 that continues to grow and divide in laboratories around the world.3 Cancerous cells can be assayed in mice as well; when normal human cells are injected into a special type of mouse (genetically engineered to lack an immune system to prevent rejection of human cells) they will not grow. However, cancerous cells from humans can continue to grow and even metastasize in these mice.
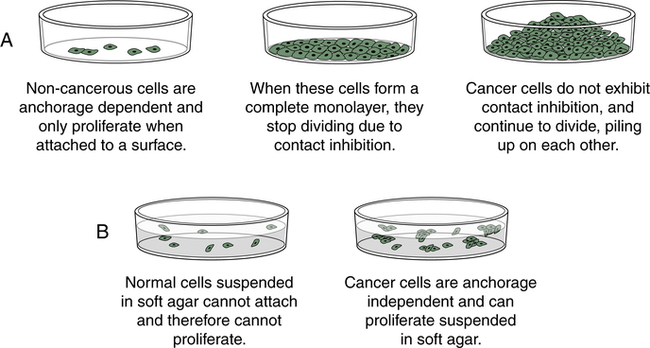
Cancer cells, unlike most normal cells (A), usually continue to grow and pile on top of one another after they have formed a confluent monolayer in culture (loss of contact inhibition) and (B) can grow without being attached to a surface, called anchorage independence.
Cancer cells often show defects in the normal process of differentiation; that is, the process of acquiring a specialized function and organization, such as evolving into a muscle cell (see Chapter 43) or a nerve cell (see Chapter 15). Anaplasia is the absence of differentiation (see Figure 12-2) and means literally “without form.” In clinical specimens, anaplasia is recognized by a loss of organization and a marked increase in nuclear size with evidence of ongoing proliferation. In contrast to normal cells, which are uniform in size and shape, anaplastic cells are of variable size and shape, or pleomorphic. For example, a benign muscle tumor (benign myoma) will retain the ability to make muscle, whereas in a malignant muscle tumor (rhabdomyosarcoma), new muscle formation is seen only rarely, and even then appears highly disorganized. Thus the muscle cancer cells appear undifferentiated. The most malignant tumors tend to have the most anaplasia and be the least differentiated.
Cancer Metabolism
Cancer cells live in a distinct milieu from normal cells and have different nutritional requirements from nonproliferating cells. The successful cancer cell divides rapidly, with the consequent requirement for the building blocks of new cells. Cancers often must grow in a hypoxic and acidic environment. Cancers also are parasites, able to selectively extract nutrients from the bloodstream without any evolutionary pressure for balanced metabolism. Nonmalignant cells in the presence of adequate oxygen normally generate adenosine triphosphate (ATP) by mitochondrial oxidative phosphorylation (OXPHOS), generating 36 ATP molecules from each glucose molecule that is broken down to water and carbon dioxide. Only in the absence of sufficient oxygen do normal cells perform anaerobic glycolysis, generating only two ATP molecules per molecule of glucose, with lactic acid as a byproduct. However, even in the presence of oxygen, cancer cells perform glycolysis, not OXPHOS4 (Figure 12-5). Although this aerobic glycolysis was originally postulated as some form of cancer-specific mitochondrial dysfunction, it is now apparent that this is instead a highly regulated and beneficial adaptation for cancer cells. This shift from OXPHOS to glycolysis allows lactate and its metabolites to be used for the more efficient production of lipids and other molecular building blocks needed for rapid cell growth. Furthermore, many cancer genes promote this switch to aerobic glycolysis. Alterations in a number of cancer genes, including receptor tyrosine kinases, AKT, PTEN, TP53, and MYC, inhibit OXPHOS and promote the activity of glycolytic and related metabolic pathways that support the rapid growth of cancers.4,5
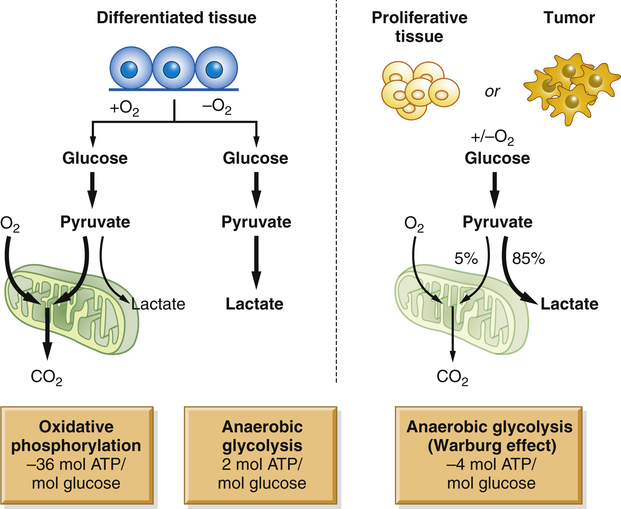
Normal tissues use oxidative phosphorylation (OXPHOS) to turn glucose into CO2 and energy (in the form of ATP). Cancers take a different approach; even in the presence of oxygen, they do not use OXPHOS. Instead, they consume large quantities of glucose to make cellular building blocks, supporting rapid proliferation. ATP, Adenosine triphosphate. (From Van der Heiden MG, Cantley LC, Thompson CB: Science 324:1029–1033, 2009.)
Clinically the high glucose utilization of a cancer can be exploited for its detection. 18F-Fluorodeoxyglucose (FDG) is incorporated into cells in the same way as glucose, with two key differences. Because it is missing a key hydroxyl group it cannot be broken down by glycolysis and, thus, FDG accumulates in cells. Because it is tagged with 18F, it can be imaged by positron emission tomography (a PET scan). Small metastatic tumor masses that are consuming huge amounts of glucose can readily be detected with this imaging method (Figure 12-6).
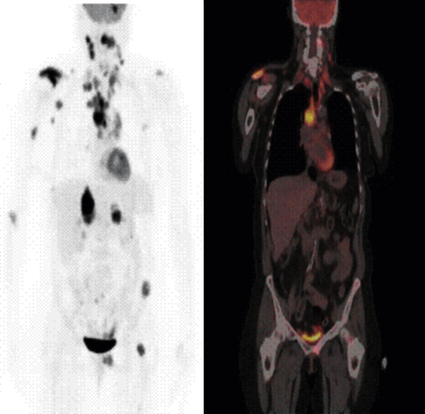
This 54-year-old woman had a non–small cell lung cancer (NSCLC) resected from the left upper lobe. Five years later, these studies were obtained. The positron emission tomography (PET) scan using 18-fluorodeoxyglucose (FDG) shows metastatic lesions in the brain, right shoulder, and mediastinal and cervical lymph nodes, as well as in the liver, left pelvis, and proximal femur. PET whole-body image (left): Representative coronal image from the whole-body FDG-PET/CT–fused image of the same patient (right). The fused image consists of the computed tomography (CT) image with the metabolic information superimposed in color. The pattern of spread is most likely from the primary tumor to the large mediastinal lymph nodes, followed by lymphatic spread to cervical nodes. Blood-borne dissemination produced the bone, brain, and liver metastases. Normally, only the heart, brain, and bladder show a strong signal in PET scans. (Images courtesy John Hoffman, MD, Huntsman Cancer Institute, Salt Lake City, UT.)
Cancer Stem Cells
Many tissues, most notably the skin, intestines, and blood-forming cells, continuously renew themselves. The human gut sheds and replaces hundreds of grams of cells each day. This ongoing proliferation of these tissues with a high turnover rate depends on their regeneration from a small fraction of cells known as adult stem cells. Adult stem cells have two essential characteristics: first, they self-renew (that is, some fraction of the cell divisions creates new stem cells); and second, they are multipotent, or have the ability to differentiate into multiple different cell types. In the bone marrow, it is estimated that only 0.05% (1 in 20,000) of the blood-forming cells are stem cells, yet this small pool of stem cells can be stimulated to divide and to repopulate all the mature bone marrow–derived cells in approximately 2 weeks after bone marrow transplantation. As few as 10 stem cells are sufficient to entirely repopulate the entire bone marrow of a mouse in bone marrow transplantation experiments. Similarly, the absorptive and goblet cells lining the intestine have a life span of less than 1 week, after which they undergo cell death, or apoptosis, and slough into the intestinal lumen. They too must be replenished by ongoing proliferation of intestinal stem cells. A key feature of stem cells is that they can divide asymmetrically (i.e., during division, the cytoplasmic contents are unevenly distributed between the daughter cells); they can give rise to another stem cell and one daughter cell that ultimately terminally differentiates into diverse cell types, depending on the needs of the tissue (Figure 12-7). Multipotent bone marrow stem cells can self-renew and differentiate into all types of bone marrow–derived cells, such as red cells, lymphocytes, and neutrophils. Certain adult stem cells have a broader range of potential fates. For example, mesenchymal stem cells are able to differentiate into multiple types of cells, such as blood vessels, neurons, and muscle cells. Theoretically, such a stem cell might be able to give rise to all cell types in the body, and as such, could be useful in regeneration of diseased tissues.
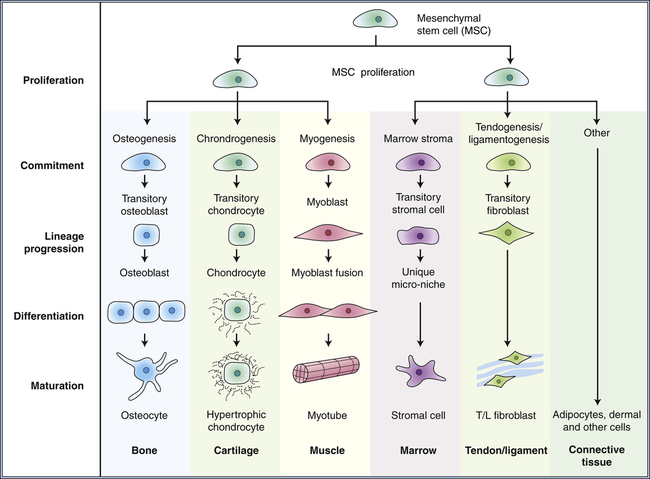
Differentiation occurs progressively as the offspring of stem cells commit to various lineages. Mesenchymal stem cells can renew themselves and give rise to multiple tissue types. (Modified from Caplan A, Bruder S: Trends Mol Med 7[6]:259–264, 2001.)
Many cancers, like normal tissues, are heterogeneous, with differences in cell shape, size, behavior, and protein expression. There are two models to explain this heterogeneity within tumors. In the clonal evolution model, all the cancer cells divide regularly, and heterogeneity arises from proliferation, mutation, epigenetic changes, differences in local environment, and natural differentiation of cells. In this model, most of these cells are still robust cancer cells, capable of forming complex tumors in experimental animals. In contrast, in the cancer stem cell model, heterogeneity arises because there is a rare cancer stem cell whose offspring do not have stem cell properties, but undergo a limited number of divisions while generating heterogeneity through epigenetic and environmental alterations. However, similar to the adult tissue stem cells, only the cancer stem cell, if transplanted, is postulated to be capable of forming complex and heterogeneous tumors. The typical cancer stem cell experiment separates cancer cells into different pools depending on some measurable characteristic such as cell surface proteins, and then assays how many cells must be injected into an experimental mouse to form a cancer. In some cases, these tumor-initiating cells are very rare, with only 1 in 10,000 human colon cancer cells able to re-form a complex and heterogeneous colon cancer in mice (Figure 12-8).6 However, these tumor-initiating cells are not always rare; in human melanomas, one in four cells can initiate a complex tumor in the appropriate mouse model.7 This is an experimentally difficult area of research, because human cancers are examined in mice where there are multiple barriers to transplantation, including immune responses, as well as species differences in the various growth factors, cytokines, and microenvironment that can all influence the results. Therefore most progress is likely to be made by studying cancer stem cells arising in mouse tumors rather than human tumors.
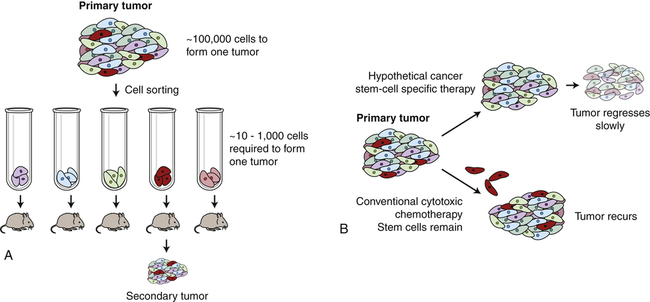
A, Only rare cells within a cancer can initiate cancer regrowth. In laboratory experiments it takes as many as 100,000 breast cancer cells injected into a mouse mammary fat pad to form a new cancer. If the breast cancer cells are sorted, one rare subtype (shown here in red) is much more proficient at forming new cancers. B, Conventional chemotherapy can destroy the bulk of a cancer. However, if the cancer stem cells (red cells) are not destroyed, the cancer may regrow. If therapies can be devised that kill the cancer stem cells, then durable long-term responses may be achieved.
The Genetic Basis of Cancer
Cancer-Causing Mutations in Genes
Before the advent of modern molecular biology, many different causes of cancer were postulated, based on epidemiologic studies as well as investigations of specific carcinogens and viruses. We now understand that changes in the genes of the cancer cell cause the cell to become cancerous. As our knowledge of cancer biology continues to increase so too does our understanding of the many ways that heritable changes in cells can contribute to cancer. These changes include deoxyribonucleic acid (DNA) mutations, but also include changes in DNA and histone chemical modification (epigenetic changes), and, most recently recognized, changes in non–coding micro-ribonucleic acid (miRNA) expression (also see Chapters 4, 6, and 13). Although the word mutation is used extensively here, it can also refer to heritable changes in gene and non–coding RNA expression (epigenetics) that do not involve changes in DNA sequence.
Cancer is predominantly a disease of aging. Perhaps the most revealing epidemiologic data are presented in Figure 12-9. The incidence of most cancer, that is, the fraction of individuals in each age group who develop cancer, increases dramatically with age. The best explanation for these epidemiologic data is that each individual acquires a number of genetic “hits” or mutations over time. When sufficient mutations have occurred, cancer develops. These epidemiologic data are consistent with the observation of mutations in early and advanced cancers and with data obtained from the study of experimental cancers created in the laboratory—the accumulation of four to seven specific hits over time is required to cause a full-blown cancer.8
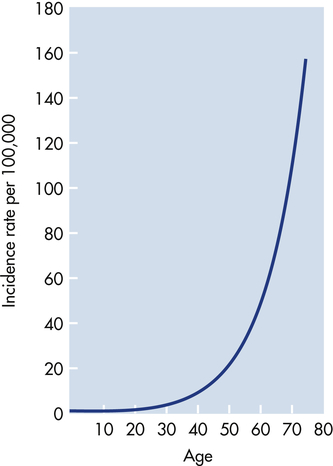
The graph depicts the number of cases of colon cancer diagnosed per 100,000 women in England and Wales in 1 year. The incidence of cancer increases dramatically with advancing age. These data suggest that accumulation of genetic and epigenetic alterations over time increases the risk of developing cancer. The slope of the curve suggests that five to seven mutations must occur before full-blown cancer develops. (Modified from Alberts B et al: Molecular biology of the cell, ed 4, New York, 2002, Garland.)
Clonal Selection
As a cell accumulates specific mutations, it can acquire, step-by-step, the characteristics of a cancer cell, for example, anchorage-independent growth, lack of contact inhibition, and immortality. That mutant cell may then have a selective advantage over its neighbors; its progeny can accumulate faster than its nonmutant neighbors. This is referred to as clonal proliferation or clonal expansion (Figure 12-10). As a clone with a mutation proliferates, it may become an early stage tumor, for example, a carcinoma in situ or a benign colonic polyp. Additional heritable changes can occur in these early lesions that permit progression to more advanced tumors. The process of tumor development is a form of darwinian evolution; cells with a genetic change that confers a survival advantage out-compete their neighbors. The progressive accumulation of distinct advantageous (from the point of view of the cancer cell, not the individual!) mutations leads from normal cells to fully malignant cancers.
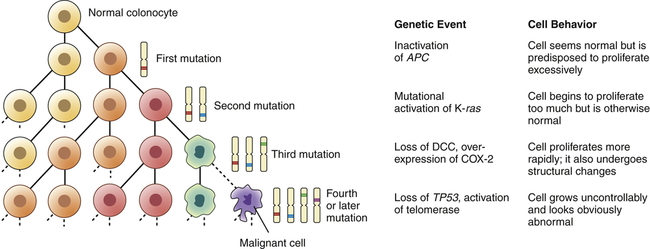
During clonal proliferation, progressively altered populations of cells arise over time. As genetic and epigenetic changes occur, different subclones (indicated by different color cells) coexist for a time. Clones that grow the fastest out-compete other clones, producing increasingly more malignant, and abnormal-appearing, growths. The sequential accumulation of mutations has been well studied in the progression from a normal colon cell to a benign intestinal polyp to a malignant colon cancer. One of the earliest mutations in colon cancer is loss of the tumor-suppressor gene APC. Additional mutations, often in the oncogene ras, activation of COX-2, and loss of the tumor suppressors DCC and TP53 occur as the lesion progresses from a benign polyp to an invasive carcinoma. APC, Adenomatous polyposis coli; COX-2, cyclooxygenase-2; DCC, deleted in colon cancer. (Modified from Mendelsohn I et al: The molecular basis of cancer, ed 2, Philadelphia, 2001, Saunders; Kumar V, Cotran RS, Robbins SL: Basic pathology, ed 6, Philadelphia, 1997, Saunders.)
One organ in which this correlation of genetic and clinical progression has been especially well studied is the colon.9 The colon is accessible to inspection with a colonoscope, and so neoplastic lesions of varying size can readily be detected and removed. Intestinal polyps are benign neoplasms and the first stage in development of colon cancer. Small polyps tend to have only a few detectable mutations. Large polyps have more mutations, whereas frank colon cancers have even more mutations. This type of genetic information provided the framework for the now widely accepted concept that it is the stepwise accumulation of alterations in specific genes that is required for the development of cancer (see Figure 12-10).
Oncogenes and Tumor-Suppressor Genes: Accelerators and Brakes
Passengers and Drivers
The previous discussion refers to the heritable changes in cells as being key in the development of cancer. New technologies that provide massive amounts of information about chromosome and gene structure allow us to see multiple alterations in cancer cells. These methods include massively parallel high-throughput DNA sequencing (the process by which the sequence of nucleotides along a strand of DNA is determined), high-density single-nucleotide polymorphism (SNP), and chromosome copy number analysis (CNA). These approaches confirm that there are a small number of very common genetic alterations in cancer and a very large number of alterations that individually are rare.10 It is clear that some mutations can contribute to cancer progression (e.g., mutations in p53 or RAS). These mutations are driver mutations; they drive the progression of cancer. Conversely, not all mutations in cancer contribute to the malignant phenotype. Some are just random events, and are referred to as passenger mutations; they are just along for the ride. The increasing amount of cancer genome analysis data poses the following question: how do we determine which mutations are drivers and which are passengers?
What Are the Driver Mutations?
To understand how genetic mutations cause cancer, first it is important to distinguish between oncogenes and tumor-suppressor genes. Table 12-4 compares the two types of cancer genes. Oncogenes are mutant genes that in their normal nonmutant state direct synthesis of proteins that positively regulate (accelerate) proliferation. Conversely, tumor-suppressor genes encode proteins that in their normal state negatively regulate (halt, or “put the brakes on”) proliferation. Hence, they also have been referred to as anti-oncogenes.
TABLE 12-4
COMPARISON OF CANCER GENE TYPES
| GENE TYPE | NORMAL FUNCTION | MUTATION EFFECT |
| Caretaker | DNA and chromosome stability | Chromosome instability and increased rates of mutation |
| Dominant oncogenes∗ | Encode proteins that promote growth (e.g., growth factors) | Overexpression or amplification causes gain of function |
| Tumor suppressors (recessive oncogenes) | Encode proteins that inhibit proliferation and prevent or repair mutations | Requires loss of function of both alleles to increase cancer risk |
In its normal, nonmutant state, an oncogene is referred to as a proto-oncogene. An example of a proto-oncogene would be a growth factor (e.g., epidermal growth factor) or a growth factor receptor (e.g., epidermal growth factor receptor). Other positive regulators of proliferation are in the signal transduction pathway that transmits the signal from the growth factor receptor to the cell nucleus. Normally, Ras is a proto-oncogene (Figure 12-11).
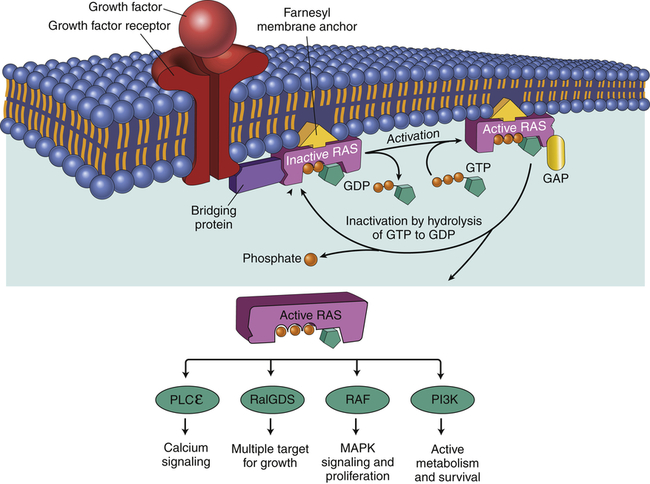
When a normal cell is stimulated through a growth factor binding to its receptor, inactive RAS exchanges GDP for GTP, becoming active. Active (GTP-bound) RAS sends growth signals throughout the cell by interacting with a number of signaling proteins, including RalGDS, RAF, PLCε, and PI3K. RAS is normally inactivated when it hydrolyzes GTP to GDP. Oncogenic Ras has a mutation that blocks hydrolysis of GTP, thereby locking RAS in the active configuration. GDP, Guanosine diphosphate; GAP, guanosine triphosphate-activating protein; GTP, guanosine triphosphate; MAPK, mitogen-activated protein kinase; PI3K, phosphoinositide 3-kinase; PLCε, phospholipase C-epsilon; RAF, serine/threonine-specific protein kinase; RalGDS, Ral guanine nucleotide dissociation stimulator; Ras, RAS protein. (Adapted from Kumar V, Abbas AK, Aster J: Robbins basic pathology, ed 9, Philadelphia, 2013, Saunders.)
Oncogene Addiction
There are a number of common driver mutations in cancer. Cancers that arise because of these mutations often depend on these mutant genes and proteins for their continued growth and survival. If the mutations and abnormal proteins can be returned to their normal states, the cancers often stop growing and even regress. The cancers are addicted to their mutant cancer genes, a concept known as oncogene addiction. This also provides a key example of how targeted cancer therapy can work—for example, if the oncogene is a protein kinase (e.g., BCR-ABL, EGFR, HER2, or BRAF) that can be inhibited by a drug, then the cancer can be deprived of the function of the oncogene. Treating the addiction treats the cancer.11
Gene Changes That Occur in Cancer
Mutation of Normal Genes into Oncogenes
Point Mutations
Several types of genetic events can activate oncogenes (Box 12-1 and Figure 12-12). Perhaps the most common events are small-scale changes in DNA such as point mutations, the alteration of one or a few nucleotide base pairs (see Chapter 4). This type of mutation can have profound effects on the activity of proteins. A point mutation in the ras gene converts it from a regulated proto-oncogene to an unregulated oncogene, an accelerator of cellular proliferation. Activating point mutations in ras are found in many cancers, especially pancreatic and colorectal cancer.12 Specialized tests, such as direct DNA sequencing, can detect such point mutations in clinical samples.
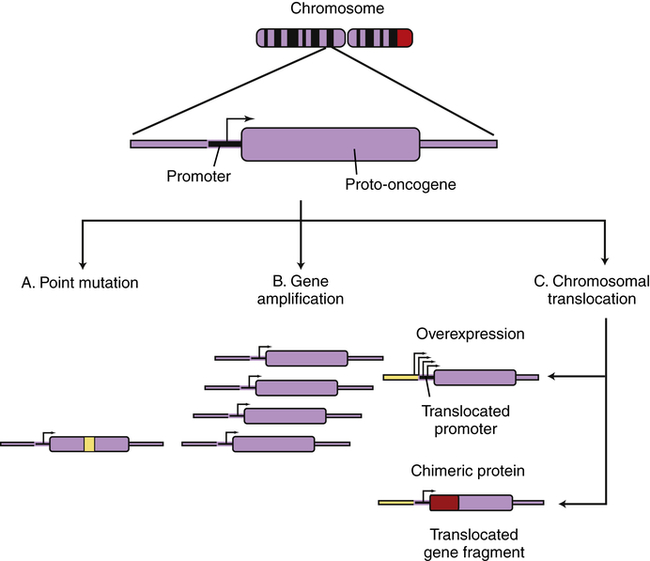
Cellular genes may become cancerous oncogenes as a result of (A) point mutations that alter one or a few nucleotide base pairs, causing the production of a protein that is activated as a result of the altered sequence (e.g., ras); (B) amplification of the cellular gene, resulting in higher levels of protein expression (e.g., N-myc in neuroblastoma); or (C) chromosomal translocations that either lead to the juxtaposition of a strong promoter, causing increased protein expression (c-myc in Burkitt lymphoma), or produce a novel fusion protein that is derived from gene fragments normally present on different chromosomes (Bcr-Abl in chronic myeloid leukemia). (From Haber DA: Molecular genetics of cancer. In ACP medicine, Danbury, CT, 2004, WebMD.)
Chromosome Translocations and Copy Number Variation
Chromosome translocations are large changes in chromosome structure in which a piece of one chromosome is translocated to another chromosome. Translocations can activate oncogenes in one of two distinct mechanisms. First, a translocation can cause excess and inappropriate production of a proliferation factor. One of the best examples is the t(8;14) translocation found in many Burkitt lymphomas;13 t(8;14) designates a chromosome that has a piece of chromosome 8 fused to a piece of chromosome 14 (see Chapter 29). Burkitt lymphoma is an aggressive cancer of B lymphocytes. The myc proto-oncogene found on chromosome 8 is normally activated at low levels in proliferating lymphocytes and is deactivated in mature lymphocytes. The MYC protein is part of the positive signal for cell proliferation. If accidental formation of the t(8;14) translocation occurs, the myc gene is aberrantly placed under the control of a B-cell immunoglobulin gene (Ig) present on chromosome 14. The Ig gene is very active in maturing B lymphocytes. The t(8;14) translocation alters the control of myc; its normal low level is switched to high levels, as directed by an Ig gene promoter. MYC protein, when inappropriately high, drives proliferation and blocks differentiation. Hence, the t(8;14) translocation causes cancer of maturing B cells (see Figure 12-12, C).
Second, chromosome translocations also can lead to production of novel proteins with growth-promoting properties. In a different type of leukemia, chronic myeloid leukemia (CML), a specific chromosome translocation is almost always present. This translocation, t(9;22), was first identified in association with CML in Philadelphia in 1960 and so is often referred to as the Philadelphia chromosome.14 This translocation fuses two chromosomes in the middle of two different genes, bcr on chromosome 9 and abl on chromosome 22. The result is production of a BCR-ABL fusion protein containing the first half of BCR and the second half of ABL. BCR-ABL is a misregulated protein tyrosine kinase that promotes growth of myeloid cells. Imatinib, a drug that specifically targets this tyrosine kinase, represents the first successful chemotherapy targeted against the product of a specific oncogenic mutation. Imatinib and related tyrosine kinase inhibitors (TKIs) are highly effective in the treatment of CML and, because of their specificity, lack the toxic side effects noted with nonspecific anticancer drugs.15 However, imatinib is not effective in cancers that do not have the t(9;22) translocation or related mutations. In modern personalized cancer therapy, knowledge of the specific genetic alteration can dictate the optimal drugs for the individual.
Just as single nucleotides can be gained or lost, larger regions of DNA encompassing entire genes can be gained or lost, a phenomenon known as copy number variation (CNV).16 CNV can be inherited and accounts for a significant fraction of human genetic diversity. CNV also can occur in the course of cancer development, where it can amplify oncogenes or delete tumor-suppressor genes.
Gene Amplification
Another type of genetic abnormality that can activate oncogenes is gene amplification (see Figures 12-12, B, and 12-13). Amplifications are the result of duplication of a small piece of a chromosome over and over again, so that instead of the normal two copies of a gene, tens or even hundreds of copies are present (see Chapter 4). Gene amplification results in increased expression of an oncogene, or in some cases, drug resistance genes. The N-myc oncogene is amplified in 25% of childhood neuroblastoma cases and confers a poor prognosis.17 The epidermal growth factor receptor erbB2 is amplified in 20% of breast cancers.18 Individuals whose cancers have erbB2 amplification respond well to drugs specifically targeted to this oncogene.19
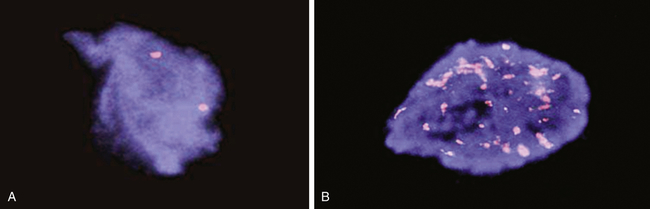
The N-myc gene is detected in human neuroblastoma cells using a technique called FISH (fluorescent in situ hybridization). A, A single pair of N-myc genes are detected in normal cells and in low-grade neuroblastoma. B, Multiple, amplified copies of the N-myc gene are detected in some cases of neuroblastoma. Amplification of the N-myc gene is strongly associated with a poor prognosis in childhood neuroblastoma. (Courtesy Arthur R. Brothman, PhD, FACMG, University of Utah School of Medicine, Salt Lake City, UT.)
Tumor-Suppressor Genes
Tumor-suppressor genes are genes whose major function is to negatively regulate cell growth and prevent mutations. Tumor suppressors may normally slow the cell cycle, inhibit proliferation resulting from growth signals, or stop cell division when cells are damaged. Examples of several tumor suppressors are given in Table 12-5. One of the first discovered tumor-suppressor genes, the retinoblastoma (Rb) gene, normally strongly inhibits the cell division cycle (see Chapter 1). When it is inactivated, the cell division cycle can proceed unchecked. Rb is mutated in childhood retinoblastoma, and in many lung, breast, and bone cancers as well.
TABLE 12-5
SOME FAMILIAL CANCER SYNDROMES CAUSED BY LOSS OF TUMOR-SUPPRESSOR GENE FUNCTION
| SYNDROME | GENE |
| Retinoblastoma | RB1 |
| Li-Fraumeni syndrome | p53 (TP53) |
| Familial melanoma | p16INK4a (CDKN2A) |
| Neurofibromatosis | Neurofibromin (NF1) |
| Familial adenomatous polyps | APC |
| Breast cancer | BRCA1 |
Whereas oncogenes are activated in cancers, tumor suppressors must be inactivated to allow cancer to occur (see Table 12-5 and Figure 12-14). A single genetic event can activate an oncogene because it can act in a dominant manner in the cell. However, we have two copies, or alleles, of each gene, one from each parent. It therefore takes two hits to inactivate the two alleles of a tumor-suppressor gene. The first allele of a tumor suppressor is often inactivated by point mutations. For example, the Rb gene may be inactivated on one chromosome by a point mutation (e.g., the copy inherited from the father). Because the other copy of the retinoblastoma gene (in this example, the one from the mother) is intact, a functional RB protein can still be made and therefore the cell division cycle can be regulated appropriately. If the remaining gene is mutated or silenced, then all RB function is lost and another step toward cancer occurs (see Chapter 4).
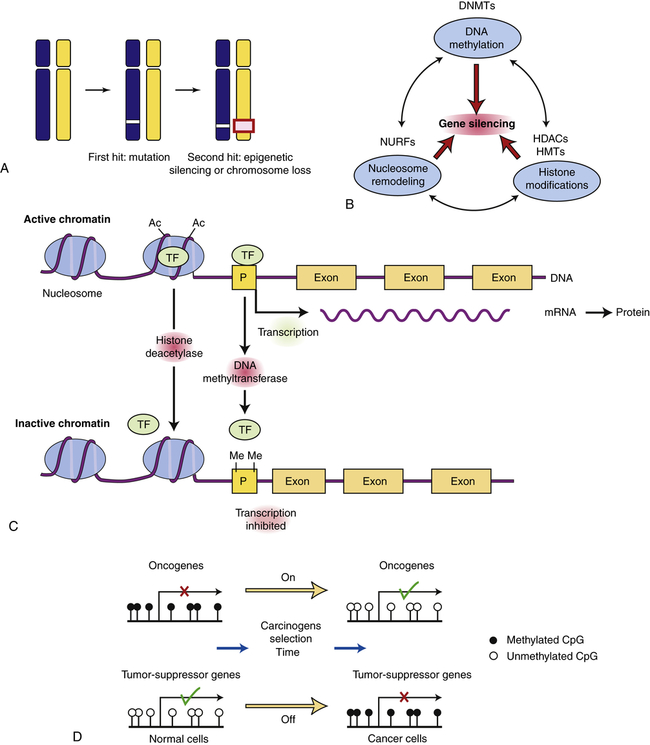
Tumor-suppressor genes can be turned off by a variety of mechanisms. A, In this example, the first hit is a point mutation in a tumor-suppressor gene (white box), followed by either epigenetic silencing or chromosome loss of the second allele (red box). B, Genes can normally be silenced by a variety of interacting processes including DNA methylation, histone modifications, nucleosome remodeling, and microRNAs (not shown). A number of cellular enzymes contribute to these modifications, including DNA methyltransferases (DNMTs), histone deacetylases (HDACs), histone methyltransferases (HMTs), and complex nucleosomal remodeling factors (NURFs). Gene silencing is essential for normal development and differentiation. C, Histone modification and promoter methylation regulate gene expression. Genes are transcribed when chromatin is modified by addition of acetyl (Ac) groups to specific lysine groups in histones. Gene expression can be turned off when specific acetyl groups are removed by HDACs or when the CpG-rich promoter regions of genes are modified by direct DNA methylation (by DNA methyltransferase). In addition, small endogenous RNA molecules (microRNAs or miRNA) can bind to mRNA and reduce gene expression. D, Changes in promoter methylation turn cancer genes off and on. Oncogenes can be turned on by promoter hypomethylation, and tumor-suppressor genes can be turned off by promoter hypermethylation. Each of these changes can produce selective growth and survival advantage for the cancer cell. (B adapted from Jones PA, Baylin SB: Cell 128:683–692, 2007; C from Gluckman PD et al: N Engl J Med 359[1]:66, 2008; D from Shames DS, Minna JF, Gazdar AF: Curr Mol Med 7:85–102, 2007.)
Loss of Heterozygosity
For the function of a tumor suppressor to be lost, both chromosomal copies (alleles) of the gene must be inactivated. This is because they act in a recessive manner at the level of the cell. Although it may seem intuitive that simple inactivating mutations might disrupt both alleles, in fact this is not what usually happens.20 Instead, the first allele (in the preceding example, the paternal copy) is inactivated by simple mutation, but the second allele (in this example, the maternal copy) is lost because entire regions of the maternal chromosome are epigenetically silenced or a piece of the chromosome is simply lost (see Figure 12-14, A). Because humans have two chromosomes, one from each parent, they are always heterozygous for nearby multiple genetic markers; loss of one copy (allele) of a chromosome region in a tumor is referred to as loss of heterozygosity, or LOH. Loss of heterozygosity, like silencing, can unmask inactivating mutations in recessive tumor-suppressor genes. For example, the Rb gene resides on chromosome 13, in a region referred to as q14 (13q14). Most individuals with Rb mutations have a subtle mutation in one allele and have lost the other copy of Rb through loss of the 13q14 chromosome region on the other chromosome.
Turning Off Genes Without Mutation
Epigenetic Silencing
Abnormal gene silencing is emerging as a major cause of cancer. Gene expression can be regulated in a heritable manner (i.e., passed from a parent to a child or from a single cell to its progeny) by an “epigenetic” mechanism called silencing. Inheritance of silencing occurs during cell division and does not require mutations or changes in DNA sequence (also see Chapter 6). More simply, the same DNA sequence can produce dramatically different phenotypes depending on chemical modifications that alter the expression of genes. Epigenetic silencing is caused by reversible chemical modification (methylation [addition of a methyl group] or acetylation [addition of an acetyl group]) of histones and related chromatin components, as well as methylation of cytosine residues in DNA (known as DNA methylation) (see Figure 12-14, Figures 6-2, 6-3, and Chapter 16). Whole regions of chromosomes are normally shut off by silencing, so that the pattern of gene expression is different than that seen in other cells with the same genes. In this way, the progeny of liver cells remain liver cells, and skin cells remain skin cells. Notably, global changes in epigenetic silencing can turn these cells back into stem cells.21
Changes in gene silencing contribute to the development of cancer.22 Many cancers have increased methylation of DNA in the promoter region of tumor-suppressor genes (in CpG islands or C-cytosine and G-guanine, p-phosphodiester bond, rich sequences that are often located near promoter regions; see Chapters 4 and 13). They also have associated changes in the modification of histones in the chromatin, including methylation of lysines 9 and 27 in histone H3. In addition, overexpression of chromosome silencing proteins known as the polycomb complex, and loss of expression of histone deacetylases (enzymes that remove acetyl groups from histone proteins) SIRT1 is seen in human cancers. These changes in chromatin-modifying genes alter the promoter regions of genes leading to their silencing. The boundaries of the normally silenced regions can also spread in cancer cells, thereby inactivating previously active genes. In either case, silencing can shut off critical tumor-suppressor genes in the absence of mutations in the gene. Early in the development of cancer, these changes in gene expression can lead to a selective advantage for affected cells, perhaps leading to their immortalization and clonal expansion. Silencing of tumor suppressors may be a faster way to create cancer cells than mutational or genetic loss of tumor suppressors.23 Conversely, loss of silencing can contribute to inappropriate expression of oncogenes. Chemotherapeutic drugs that can regulate gene silencing, including histone deacetylase (HDAC) inhibitors and 5-azacytidine (which reverses the effects of DNA methyltransferases [DNMTs]), have proven effective in reactivating silenced tumor-suppressor genes in the treatment of selected cancers24 (see Figure 12-14).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


