Fig. 2.1
Properties of stem cells. Stem cells have the ability to self-renew and differentiate to a mature cell through an intermediate progenitor. The ability to give rise to cells from different lineages is termed multipotency.
It is hypothesized that variations in self-renewal and proliferative abilities generate a cellular hierarchy within brain tumors, with BTSCs at the apex of this hierarchy [6]. In addition to these obligate properties, BTSCs should be able to initiate tumors and phenocopy the original tumor when injected into animal models [7, 16]. The presence of BTSCs in brain tumors raises another important question: Do brain tumors arise from the oncogenic transformation of normal neural stem cells (NSCs) residing within the brain? Or can differentiated brain cells undergo mutations that lead to their dedifferentiation and tumorigenesis?
This chapter focuses on two aspects of stem cell biology in brain tumors. The first part will cover the role of NSCs and progenitor cells as candidates for the cell of origin in brain tumors. The second part will discuss molecular characteristics of BTSCs and their therapeutic implications. We believe that understanding cancer stem cell biology and its therapeutic implications will be crucial for developing fundamentally new, and hopefully more effective, treatments.
Brain Tumor Initiation
Mechanisms of brain tumor initiation are unclear; mouse models have revealed that a number of mutations are capable of initiating tumors and that the cell of origin may differ amongst different genetic subclasses of brain tumors or even within a given tumor type. Also, the question of how tumor cells acquire a BTSC phenotype during or after tumor initiation remains to be answered. Two dominant hypotheses have emerged to account for the presence of BTSCs within gliomas: (1) Brain tumors arise from the transformation of endogenous NSCs or progenitor cells that acquire aberrant self-renewal and differentiation properties; and (2) differentiated brain cells undergo oncogenic transformation, dedifferentiate and acquire stem cell characteristics. This section will provide background on neurogenesis and normal NSC biology, which will lay the foundation for understanding gliomagenesis and the molecular characteristics of BTSCs.
Neurogenesis in the Adult Brain
The cell of origin in brain tumors, including gliomas, is still a matter of debate [17]. However, there has been intense speculation that brain tumors may arise from neurogenic niches in the brain [18–27]. Since the discovery of adult neurogenesis, new insights have emerged about the mechanisms by which the brain maintains a small population of NSCs that can replenish both neurons and glia [28, 29]. Given the intrinsic ability of both BTSCs and NSCs to self-renew and differentiate, it is important to consider how cancer stem cells’ ability to regulate their own function in the tumor deviates (or remains the same) from that of normal NSCs. NSCs in the adult brain actively generate both neurons and glia that contribute to the brain’s cellular and functional homeostasis, as well as plasticity, remodeling, and response to injury [30–32]. In contrast to post-mitotic neurons, it is the cell types undergoing mitoses that are thought to harbor the greatest potential to give rise to brain tumors. Such mitotically active cells are found in neurogenic niches. Cells in the neurogenic niche perturbed by mutagenic events can theoretically serve as starting points for brain tumor formation and may harbor intrinsically higher oncogenic potential than other dormant neural cell types. Understanding neurogenesis and gliogenesis will allow us to explore concepts relevant to the cell-of-origin question in brain tumors and the regulation of BTSCs during tumorigenesis.
NSCs have been identified in at least two regions of the adult brain: the subventricular zone (SVZ) of the lateral ventricles, and the subgranular zone (SGZ) of the hippocampus [28, 33–35]. NSCs may also exist within the subcortical white matter [36]. During fetal life, radial glia, which are derived from neuroepithelium, are responsible for neurogenesis. Radial glia participate in neural organization; immature neurons that arise from radial glia move along their transcortical extensions for migratory guidance to their respective areas in the cortex, contributing to its stratified organization during the late embryonic stages [37]. With the transition to postnatal life, radial glia differentiate into many different cell types, including neurons, astrocytes, oligodendrocytes, ependymal cells, and the SVZ NSCs, which contribute to adult neurogenesis in the mammalian brain in postnatal life [28, 32, 34, 38, 39]. The adult SVZ NSCs, or type B cells, line the subependymal zone of the lateral ventricles. B cells give rise to intermediate progenitors termed transit-amplifying cells (or type C cells), which proliferate and have the ability to form immature neurons termed neuroblasts (type A cells). Neuroblasts migrate through the rostral migratory stream (RMS) to the olfactory bulb in rodents [40] and, additionally, through the medial prefrontal migratory stream in humans [41], eventually becoming mature post-mitotic neurons. Type B cells additionally give rise to oligodendrocyte precursor cells (OPCs) and astroglia depending on growth or inhibitory signals ([31, 42], see reviews [29, 43]).
In the hippocampal formation, the dentate gyrus SGZ houses radial astrocytes, which serve as a source of neurogenesis in the region [28, 33]. Radial astrocytes (type 1 cells) form intermediate progenitor cells (type 2 cells), which become immature granule cells (type 3 cells) and subsequently mature granule neurons [44–47]. The SGZ and SVZ house the two identified groups of stem cells responsible for formation of new neuronal and glial cell types in adult mammals. The majority of what we have learned about neurogenesis has emerged from studies conducted using rodent models. Some of these findings have been validated in postmortem human brain samples [30, 35, 41], although such studies are limited in number. The discovery of active neurogenesis in the adult human brain has numerous implications for the pathobiology of brain tumor formation, neurodegenerative disorders, and response to injury.
Gliomagenesis
In the very beginning of brain tumorigenesis, the cellular composition is thought to differ drastically from that of a mature tumor. Given the known heterogeneity of fully developed tumors and the overwhelming possibility that the tumor began from a single cell type, an open question remains: How does this heterogeneity arise? To address this question, we will focus on gliomas, whose developmental biology is perhaps better understood than any other brain tumor.
We know that BTSCs from mature gliomas can phenocopy the tumors they were derived from in animal models. The BTSCs found in gliomas must have arisen from a tumor that lacked cells with stem cell properties or a tumor that arose from an endogenous NSC (Fig. 2.2) The cell of origin is defined as the cell type that has accumulated the correct combination of mutations that induces proliferation and eventually tumor formation (oncogenic transformation). Although not mutually exclusive, the cell of origin differs from the cell type that acquires mutations, which is referred to as the cell of mutation. This is because the cell of mutation, after acquiring the first oncogenic hit, may differentiate or dedifferentiate into another cell type before proliferating, so a distinction must be made for the cases where this happens (see reviews [17, 48]). An important concept to clarify is the difference between the cell of origin and BTSCs. Both can theoretically form the mature tumor, but the cell of origin is responsible for initiating the tumor and may or may not be a stem cell. The cell of origin is of interest in the discussion of BTSCs because evidence from mouse models has pointed to the possibility that NSCs within the brain may serve as the cells of origin. Given that BTSCs have stem cell properties, identifying the cell of origin may provide insight into how BTSCs are derived and how their ability to self-renew or differentiate differs from that of normal stem cells and the cell of origin. It is still unclear if multiple cell types within the brain can give rise to the same type of tumor and whether different types of tumors share the same cell of origin harboring different genetic mutations. To answer some of these questions, glioma models have been developed and used successfully to uncover key aspects of glioma biology.
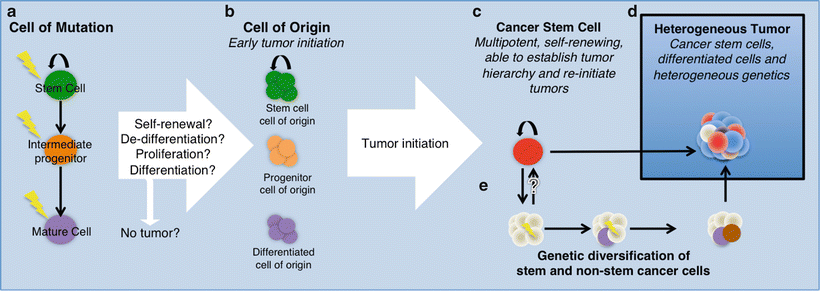
Fig. 2.2
Model of gliomagenesis. (a) Representation of a normal cellular hierarchy where the cell of mutation may give rise to the cell of origin (b). Cancer stem cells (c) are self-renewing and tumor re-initiating cells found in the tumor. Both cancer stem cells and non-stem cells experience genetic changes (e), contributing to the heterogeneous tumor (d). Different colors represent different cell types emphasizing intratumoral heterogeneity.
Models of glioma aim to recapitulate human glioma pathology most commonly through two model systems: genetically engineered rodent models with mutations found in human gliomas, or xenografts of primary human glioma lines derived from patients. Other model systems also exist, including in vitro cultures of glioblastoma multiforme (GBM) cell lines, allografts of rodent glioma lines and virally mediated oncogenesis. These approaches have been used to provide reproducible platforms to study many aspects of tumor biology, including BTSCs, the cell of origin, and therapeutic implications [19, 21, 22, 49–54].
Primary lines derived from patient tumors carry a distinct advantage due to their genetic make-up, which most faithfully represents the disease [49]. The drawbacks of primary lines include: (1) the necessity to inject the mouse brain to create a xenograft, thus potentially altering the microenvironment; (2) the possibility that, in derivation of each culture, a subpopulation of the tumor is selected for; and (3) the fact that these tumors are grown in an immunodeficient background. Murine models have the disadvantage of a rodent genetic background and are limited in recapitulating the order and number of mutations that occur in a sporadic human glioma. Modeling efforts will likely continue to sample the phenotype produced by different combinations of mutations, location of the tumor, and the developmental point of induction of these mutations. In both cases, the tumor is grown in the mouse brain microenvironment, which raises an additional degree of separation from human glioma biology for both the mouse and xenograft model systems.
Experimental evidence from murine models of glioma suggests the cell of origin to be adult NSCs or proliferating progenitor/precursor cells, but not mature glia. However, this is highly controversial and remains an open question for the many different subtypes of glioma (see reviews [53, 55]). Following the discovery of adult neurogenesis, there was a paradigm shift in thinking about the origins of glioma, as the discovery of NSCs and their progenitor/precursor cell progeny became new candidates (Fig. 2.2) In a landmark effort to link the differentiation stage to tumor initiation, Holland et al. found that NSCs or neural progenitor cells expressing Nestin in the mouse brain were preferentially forming tumors with GBM characteristics after activation of the K-Ras/Akt pathway [25]. The same oncogenic insult did not produce tumors under the control of a GFAP promoter, suggesting that not all cell types within the same lineage could serve as the cell of origin [25]. Parada and colleagues have developed multiple tumor suppressor knockout models of gliomagenesis via inducible loss of p53, PTEN, and NF1, which are some of the most commonly mutated tumor suppressors in GBM [56, 57]. Analysis of the high-grade gliomas generated from these models revealed that Nestin-expressing cell types found in the SVZ are likely to contain the cell of origin. More recent studies with PDGF-driven tumor formation and p53/NF1 knockout have shown that oligodendrocyte precursor cells (OPCs) are capable of giving rise to GBM tumors in mice [20, 58].
Mouse models using Ink4a-ARF loss, K-ras activation, or PDGF signaling give rise to gliomas in areas and cell types found outside of the neurogenic niches, suggesting that mature glial cells can produce malignancy when given the correct combination of oncogenic mutations [54, 59–62]. Interestingly, it has also been reported that mature neurons, in addition to GFAP-positive astrocytes, are capable of acting as the cell of mutation in a p53/NF1 model of glioma by undergoing dedifferentiation [24]. Some consideration should be given to the fact that some of these murine models represent functional genetic alterations that may or may not be the initiating events in glioma formation despite their oncogenic transforming abilities in this context. It remains an open question as to what the initial events in the different subtypes of glioma are, and how restricted the cell of origin truly is for any given tumor type, considering the unique combination of microenvironment, genetic changes, and organism.
It should be highlighted that in many of the aforementioned murine models, there is a propensity of early events in murine gliomagenesis to occur near the SVZ when the NSC population is targeted [56, 57, 63]. There is clinical evidence in humans that initiating events in glioma formation occur in or near the neurogenic zones of the brain. Human GBM has a propensity to occur most frequently in the periventricular area and less so in the surrounding cortex, albeit this evidence is controversial [63–65]. GBM also occurs infratentorially, but with much lower frequency [66]. The tendency for GBM to occur in the periventricular area within the cerebrum suggests that a cell-of-origin also resides within the same region; however, this correlation has not been directly linked to human neurogenesis. It is possible that tumors found far from neurogenic regions may be initiated by migrating cells that originated in the neurogenic niche. This is an interesting but unexplored hypothesis.
Grade II/III gliomas mutated for isocitrate dehydrogenase (IDH) tend to arise in different anatomic locations as compared to their grade IV GBM counterparts. Although IDH1 normally functions to convert isocitrate to α-ketoglutarate, the mutation leads to the production of oncometabolite 2-hydroxyglutarate (2HG) which stereo-chemically resembles α-ketoglutarate and is hypothesized to cause tumorigenic epigenetic changes [67–70]. In IDH-mutated gliomas, the cytosolic variant IDH1 is most frequently mutated, whereas mutations in IDH2 can be found less commonly [71]. Strikingly, the IDH1-mutated gliomas are most commonly found in the frontal lobe in an area that overlaps with the rostral and medial migratory streams used by neuroblasts to replenish interneurons in the olfactory bulb and frontal cortex, respectively [30, 63]. Nevertheless, IDH1-mutated tumors can also be found in other regions of the brain, albeit at a lower frequency. The fact that low- and high-grade tumors tend to arise in different anatomic locations raises the possibility of differing cells of origin. Alternatively, it may signify that IDH1 mutations promote gliomagenesis only in restricted cell types or lineages. The lack of mouse models that produce IDH1-mutated tumors has inhibited the dissection of the cell of origin in this class of tumors [72–74].
Despite the possibility that the cell of origin may originate from a stem cell or a more restricted progenitor/precursor cell, there is evidence that the tumor either gains (via dedifferentiation) or maintains a portion of its population as cells with stem cell properties. The continuum between the cell of origin and BTSCs is not well understood (Fig. 2.2) Murine models have allowed the detection and study of BTSCs in the context of gliomas and medulloblastomas primarily. Much of our understanding of BTSC biology has derived from the study of primary human GBM.
Identification of Brain Tumor Stem Cells
As mentioned earlier, the concept of cancer stem cells was initially developed in studies involving leukemias, where the cellular hierarchy is well established [14]. In such tumors, an abundance of lineage-specific cell surface markers made the isolation of distinct cell types within this hierarchy feasible. Some of the same surface markers were later used to isolate cancer stem cells in solid tumors. Before going into the details of surface markers and molecular characteristics of BTSCs, we will describe the main approaches used to identify them.
BTSCs, which are a subpopulation of cells within the tumor, are defined by their ability to initiate tumors in animal models that recapitulate patient tumor phenotype and heterogeneity [7, 8, 49]. As mentioned earlier, two critical properties of BTSCs are self–renewal and multipotency (Fig. 2.3) Self-renewal is tested with the following two standard assays. First, clonogenicity is assessed by in vitro tumor sphere formation ability over serial passaging [49, 75]. Briefly, cells that have been isolated according to their surface markers are seeded in suspension in low density or as single cells and the formation of spheres is analyzed. Serial sphere formation over time shows that cells have clonogenic potential, consistent with the ability of BTSCs to self-renew. A second critical assay is xenograft tumor formation, where these isolated cells form tumors when injected into immunodeficient or isogenic mice [15]. Such xenograft tumors are expected to recapitulate the original disease phenotype. Re-isolation of BTSCs from xenograft tumors and secondary tumor formation from those cells shows in vivo self-renewal. Differentiation potential, or multipotency, is another required functional property of BTSCs. In the case of gliomas, for example, BTSCs have been shown to give rise to glia, neurons, endothelium and pericytes [12, 13, 76–79]. These findings underscore the stem-like properties of BTSCs and provide a mechanism for BTSC-driven tumor heterogeneity.
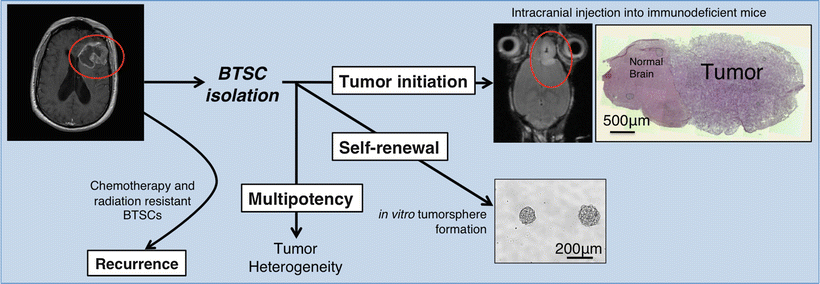
Fig. 2.3
Hallmarks of BTSC biology. Upon surgical resection and primary culture generation, BTSCs are isolated via expression of molecular markers. Isolated BTSCs are studied for their tumor initiation, self–renewal, and multipotency. Therapy-resistant BTSCs cause disease recurrence.
By using these important assays, initial evidence for BTSCs came from pediatric brain tumors. Isolating cells by fluorescence-activating cell sorting (FACS) using cell surface markers originally found in human fetal brain, cells with tumorigenic properties were identified [80]. Shortly thereafter, Dirks and colleagues successfully isolated tumor-initiating cells in human GBM, the most malignant form of glioma, where they showed that CD133, a surface marker also expressed in embryonic NSCs and other adult stem cells, identifies cells with the ability to generate tumors when injected into immunodeficient animals [7, 8, 81]. After these seminal papers, the field of brain tumor stem cells expanded exponentially. However, we still need a better understanding of these cells in terms of their molecular signatures and niches, as well as their relevance to tumor growth and recurrence.
Molecular Characteristics of Brain Tumor Stem Cells
Molecular Markers
Functional similarities between BTSCs and NSCs directed researchers to analyze the expression of markers that were shown to be important for NSC biology and neural development. One of the best-defined molecular markers for brain tumors, including pediatric tumors, ependymoma, and especially GBM, is the cell surface marker CD133. CD133 is a pentaspan, glycosylated transmembrane protein. Apart from being expressed in fetal brain NSCs during embryonic development, its expression is highly associated with other tissue-specific stem cells and cancer stem cells of blood and solid malignancies [14, 81–85]. CD133-knockout murine models show photoreceptor degeneration, but the signaling functions of CD133 remain unknown [86]. In the context of GBM and medulloblastoma, it was shown that when injected into animals in limiting dilutions, CD133+ cells generate tumors more efficiently than their CD133− counterparts, suggesting that they have BTSC properties [87]. Furthermore, downregulation of CD133 via short hairpin RNA (shRNA) suppresses self-renewal of BTSCs in GBM [88].
Although CD133 is one of the best-studied markers in brain tumors, it is now well established that some CD133− cells do possess tumorigenic potential, suggesting that CD133 is not a universal marker and that CD133− BTSC populations exist as well [11, 89–91]. Furthermore, the fact that not all GBM tumors have CD133+ cells supports the hypothesis that CD133− BTSCs exist in these tumors [92–94]. Another important marker associated with BTSCs is the intermediate filament protein Nestin, a well-established NSC marker. Nestin + cells were shown to have tumor-initiating ability in animal models and to generate tumor recurrence after chemotherapy [10, 61, 95]. BTSCs are also enriched for other NSC and stem cell markers, such as Nanog, Musashi-1, Bmi-1, Sox2, and Oct4 [96–98]. In addition, some BTSCs were shown to express other surface markers and transmembrane proteins, such as CXCR4, integrin α6, SSEA-1/CD15, L1CAM, and A2B5 [11, 99–101]. Finally, the side population (SP), defined as cells with the ability to extrude Hoechst dye via ABC-type transporters on the cell surface on flow cytometric analysis, has been shown to contain stem-like cells in a variety of brain tumors [102, 103].
Besides from traditional coding genes, the importance of noncoding RNAs in the regulation of gene expression has been increasingly recognized in recent years, making them important biomarkers in cancer biology. In particular, microRNAs are responsible for the posttranscriptional fine-tuning of gene expression by binding the 3′ UTR of messenger RNAs (mRNAs) and causing their translational arrest or degradation. MicroRNAs have been implicated in the regulation of stem cell self-renewal and differentiation, as well as the control of cell cycle and apoptosis [104].
MicroRNAs that are upregulated in gliomas are mostly associated with antiapoptotic, pro-proliferative, pro-invasion, and antidifferentiation pathways [105, 106]. On the other hand, some microRNAs, which are important for neural differentiation, were shown to be downregulated in gliomas, functioning as tumor suppressors [107]. An example of a microRNA critical to brain tumor biology is miR-124. Normally, miR-124 is known to promote neural differentiation in the brain. In gliomas, however, it was shown to be downregulated and its overexpression promotes differentiation [108, 109].
Signaling Pathways
Dissecting major signaling pathways that are important for BTSC biology have been, and will continue to be, a challenge due to the complex interplay and cross talk between different signaling pathways. Besides molecular markers shared between NSCs and BTSCs, signaling pathways are also conserved between these two populations. This conservation has highlighted several signaling pathways in BTSC biology, some of which are described below.
Pathways Supporting Self-Renewal
Similar to NSC culturing, BTSCs are propagated as suspension culture, in serum-free media, under the influence of two mitogens, epidermal growth factor (EGF), and fibroblast growth factor (FGF), which are believed to induce self-renewal of BTSCs in vitro [49]. However, in the in vivo scenario, signals supporting self-renewal are dependent on the complex interplay of multiple pathways.
The Notch signaling pathway was originally identified by genetic screens in Drosophila as a master regulator of neural development [111–113]. Further investigation showed that Notch signaling is essential for maintaining NSCs in an undifferentiated state and represents a key component of fate decisions in neural and glial lineages [114, 115]. Apart from its critical role in neural development, Notch signaling has been highly associated with tumorigenesis, regulating both the self-renewal and differentiation of BTSCs in GBM [12, 77, 116–118]. Notch signaling is critical for the self-renewal of CD133+ GBM BTSCs, as evidenced by the fact that blockade of Notch signaling with γ-secretase inhibitors leads to depletion of CD133+ GBM BTSCs and decreases tumorigenicity [119, 120]. Notch signaling is activated in the vascular niche where GBM BTSCs reside [121]. More specifically, in this niche the endothelium provides Notch ligands to maintain the undifferentiated state of BTSCs [122].
The PI3K/Akt/mTOR pathway is critical for gliomagenesis and glioma BTSC self-renewal [2–5, 123, 124]. Commonly find mutations in gliomas are found in the components of PI3K/Akt pathway, such as loss of function of PTEN or gain of function of EGFR [3]. Furthermore, CD133 knockdown leads to inhibition of Akt activation and impaired self-renewal and tumorigenicity of glioma BTSCs, further confirming its crucial role in BTSC biology [123, 124].
Hedgehog-gli signaling has been implicated in medulloblastoma formation [125, 126]. It is important for BTSC clonogenicity, survival, tumorigenicity, and proliferation by operating through the key cell cycle regulators Cyclin D and Cyclin E [87, 127].
The Wnt/β-catenin signaling pathway functions by inducing progenitor cell proliferation and differentiation in gliomas [128]. Some reports also show that Wnt signaling is important for GBM BTSC self-renewal [91, 129].
Recent studies have shown that transforming growth factor-β (TGF-β) signaling regulates GBM BTSC biology [130–132]. TGF-β plays a role in regulating GBM BTSC self-renewal via acting through Sry-related HMG box factors (Sox2 and Sox4) [131]. Furthermore, inhibition of TGF-β in GBM tumors decreases perivascular CD44high/Id1high BTSCs by repressing inhibitors of DNA-binding proteins Id1 and Id3 [130].
Another important signaling pathway relevant to BTSC self-renewal is mediated by hypoxia. GBM tumors are histologically heterogeneous and include regions that lack blood vessels and are highly necrotic and hypoxic [133]. Hypoxia has been previously shown to promote self-renewal of embryonic and adult stem cells [102, 134]. Along these lines, in gliomas hypoxia induces BTSC self-renewal via hypoxia-induced factors (HIF-1α and HIF-2α) [135–139]. These same factors also induce angiogenesis and neovascularization via upregulation of vascular endothelial growth factor (VEGF) [140]. Hypoxia is also known to reprogram CD133− BTSCs to become CD133+ [102]. Furthermore, hypoxia induces Notch signaling, whose importance in BTSC self-renewal was mentioned above [134]. Microscopic analysis has shown CD133 immunoreactivity around necrotic areas in GBM, a finding consistent with a hypoxic niche for BTSCs [141].
Pathways Promoting Differentiation
Bone morphogenetic protein (BMP) signaling functions as a strong differentiation signal. BMP4, important for astrocytic differentiation, induces GBM BTSC differentiation. BMP4 treatment was shown to block GBM BTSC self-renewal by suppressing asymmetric division, thereby depleting the stem cell compartment within tumors and leading to differentiation and proliferation block [76]. However, in a subset of glioma BTSCs, BMP-driven differentiation is impaired due to epigenetic silencing of the BMP receptor 1B (BMPR1B). This modification desensitizes glioma BTSCs to normal differentiation cues, thereby leading to their proliferation [142].
In GBM, some reports have suggested that the Notch pathway is critical for tumor-driven endothelial cell trans-differentiation of BTSCs [12, 13]. Similar to Notch signaling, TGF-β is known to regulate glioma progression by modulating the tumor microenvironment, including angiogenesis and immune response. TGF-β was also shown to induce differentiation of glioma BTSCs into vascular pericytes, which leads to further tumor growth by supporting vessel formation [78].
Stem Cell Niche and Tumor Microenvironment
Understanding the stem cell niche for BTSCs is highly important in unraveling the processes responsible for their self-renewal and signals inducing differentiation. Furthermore, the stem cell niche and microenvironment are highly critical in the context of drug design and delivery. Without understanding the different niches and signals provided within them, effective drug design is not possible.
Vascular Niche
NSCs were shown to reside within a vascular niche, adjacent to endothelial cells, which are believed to provide signals required for self-renewal [143]. BTSCs were also shown to reside closer to endothelial cells [144]. In medulloblastoma, CD133+ cells where shown to be in proximity to endothelial cells [121, 122]. Similarly, glioma BTSCs acquire a vascular niche, in which CD34+ endothelial cells present Notch ligands to BTSCs, keeping them in an undifferentiated state via activation of Notch signaling [122]. However, the complex architectural features of GBM suggest that BTSCs may also reside in relatively avascular microenvironments.
Hypoxic Niche
The importance of hypoxia in promoting self-renewal in embryonic stem cells and NSCs suggests that it may also regulate the self-renewal of BTSCs, especially in GBM, which is a highly hypoxic tumor. When considering this possibility, the question that arises is whether there are hypoxic areas within GBM. One such plausible histologic area is represented in pseudopalisading necrosis (PPN), in which densely packed cells surround necrotic regions [145]. The etiology and biological significance of PPNs is not well understood. However, others and we speculate that they may represent areas of active tumor growth and revascularization. A tantalizing hypothesis to explain such tumor growth and angiogenesis is, in turn, the presence of BTSCs within a hypoxic niche. Indeed, some studies have shown enriched CD133 immunoreactivity in PPNs, supporting this hypothesis [141]. Importantly, the putative presence of BTSCs in hypoxic areas devoid of blood vessels raises doubts about the effectiveness of systemic drug delivery methods.
Invasion
Invasion of glioma cells through the brain parenchyma represents perhaps their most malignant feature [146]. Single GBM cells have been shown to infiltrate normal brain tissue and travel more than several centimeters from the bulk of the tumor [147–149]. After surgical resection, the recurrent tumor occurs within the borders of the resection cavity, suggesting that these infiltrating cells have the capacity to regenerate tumors.
Although the mechanisms of invasion of BTSCs are not clear, C–X–C chemokine receptor type 4 (CXCR4) and its ligand, stromal-derived factor-1α (SDF-1α), which are highly important for vascularization, were shown to be crucial for the invasive behavior of GBM cells [140, 150]. Enrichment of SDF-1α/CXCR4 expression in glioma BTSCs highlights their invasive properties. This signaling was also shown to mediate recruitment of BTSCs toward endothelium, leading to further invasion and differentiation. Furthermore, this chemoattractant signaling induces endothelial proliferation via attracting tumor cells and inducing VEGF expression in gliomas and other systems, such as the gastrointestinal tract [151].
Therapeutic Importance of Brain Tumor Stem Cells
Besides understanding how tumors are formed and how the cellular hierarchy within the tumors is maintained, BTSCs are of particular interest because of their intrinsic resistance to current chemoradiotherapeutic approaches (Fig. 2.3). In GBM, postsurgical therapy consists of concomitant temozolomide administration and radiotherapy.
Therapy Resistance
Chemotherapy Resistance
The side population of GBM cells is believed to have the ability to actively transport chemotherapeutic agents to the extracellular space, due to the expression of ABC-type transporters on their plasma membrane [152]. Furthermore, analysis of cells that are resistant to lethal doses of chemotherapeutic drugs has revealed that they express stem cell markers, such as CD133, CD117, CD90, CD71, and CD45, and are able to regenerate tumors when injected into immunodeficient mice [153]. Gene expression analysis of CD133+ BTSCs in glioma showed increased expression of antiapoptotic genes. In relation to this finding, CD133 expression was shown to be significantly higher in recurrent tumors, further suggesting that BTSCs have intrinsic mechanisms of chemoresistance [99].
Recently, Parada and colleagues used a mouse model for GBM to demonstrate that upon treatment with temozolomide, an alkylating agent, which represents the standard of care in GBM, a restricted Nestin + BTSC population re-propagated the tumor. Selective ablation of this population arrested tumor growth, suggesting that BTSCs are the reason for GBM recurrence [10].
Radioresistance
Besides their resistance to chemotherapy, BTSCs were shown to be highly resistant to radiation. Similar to chemoresistant populations, radioresistant GBM cells express BTSC markers. Molecular mechanisms involved in glioma BTSC self-renewal, such as Notch and TGFβ signaling, are thought to underlie such radioresistance [116, 154]. CD133+ BTSCs were also shown to have increased DNA repair capacity via selective activation of Chk1 and Chk2 kinases [155].
Most chemotherapeutic drugs target cycling cells. By the same token, response to radiation depends on cell cycle checkpoints. However, BTSCs are mostly dormant and quiescent, which spares them from cell cycle-dependent therapeutic approaches, and further highlights the importance of developing new therapies that directly target BTSCs [156].
Updates on Clinical Trials Targeting BTSCs
Because of their central role in promoting growth and recurrence of primary brain tumors, BTSCs are major candidates for targeted therapeutic approaches. In particular, signaling pathways promoting BTSC self-renewal and inducing therapy resistance represent critical drug targets.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


