1. Neoplasm or not neoplasm: This hypercellular lesion is composed of pleomorphic cells with atypical nuclei, which indicate neoplasm. If the lesion were not neoplastic, other etiologic categories, including infectious, inflammatory, toxic-metabolic, traumatic, vascular, developmental, and degenerative, would be considered.
2. Primary or metastatic: These cells are fibrillar. They are plump and large but not spindled or vacuolated. The neoplastic cell nuclei are moderately pleomorphic; the cells have small processes emanating from them. The lesion lacks rosettes. There is a vessel cuffed by many small, reactive-appearing mononuclear cells. No epithelial elements are recognized. The findings are those of a primary CNS neoplasm rather than a metastatic lesion (Tables 10.10 and 10.11).
3. Glial, neuronal, and other primary considerations: Possible primary CNS neoplasm diagnoses in Table 10.10 include giant cell astrocytoma, gemistocytic astrocytoma, ganglion cell tumor, or histiocytosis (Fig. 10.1). Although nuclei of the lesional cells have a slight resemblance to neuronal nuclei, their chromatin is more condensed. Their cytoplasm is pink without purple Nissl substance of neurons. Bielschowsky stain shows no silver staining of neoplastic cells (Fig. 10.1); neoplastic cells were also negative for synaptophysin, a neuronal marker (not shown). Aggregate features do not support a ganglion cell tumor. The neoplastic cells are positive for the intermediate filament glial fibrillary acidic protein (GFAP), a marker that accentuates their fibrillarity and confirms their astrocytic nature (Fig. 10.1). The related text in this chapter describes differences between gemistocytic astrocytoma and giant cell astrocytoma. Gemistocytic astrocytomas resemble reactive astrocytes and have abundant, plump, hyaline cytoplasm and peripheralized nuclei. Macrophages (histiocytes) are GFAP-negative and lack the fibrillary structure highlighted by GFAP, arguing against the diagnosis histiocytosis. Neoplastic cells are negative for the T-lymphocyte marker CD45RO (Fig. 10.1D) and the B-lymphocyte marker CD20 (Fig. 10.1E), whereas polyclonal perivascular lymphocytes are positive. Therefore, the aggregate histologic, histochemical, and immunohistochemistry (IHC) findings support the diagnosis of gemistocytic astrocytoma.
4. Grade of astrocytoma: Bielschowsky stain reveals passing axons of brain tissue infiltrated by this neoplasm (Fig. 10.1). Infiltration of brain tissue is seen in astrocytomas of grade II to IV. Grade III astrocytomas have mitotic activity, but there is no evidence of mitotic activity. Features of grade IV, microvascular proliferation and necrosis, are absent. This astrocytoma is grade II. Most gemistocytic astrocytomas are grade II, although they tend to progress to a higher grade. Grading is further discussed in the “Gemistocytic Astrocytoma,” “Diffuse Astrocytoma,” and “Anaplastic Astrocytoma” sections. Other algorithmic approaches to the brain biopsy are available (9,10).
CLINICAL AND RADIOGRAPHIC PERSPECTIVE OF LESIONS
Biopsies should be examined with knowledge of the clinical history. If a specific diagnosis or differential is not suspected preoperatively, at the least, a major neurologic symptom (e.g., weakness or visual loss) or category of neurologic disease (e.g., refractory epilepsy) is usually available to focus the search for a diagnosis (Table 10.2). The pathologist should always know, at a minimum, the age and sex of the patient, the precise location of the targeted lesion, and imaging characteristics. Knowledge about past medical history (e.g., previous CNS or primary neoplasms, connective tissue disease, immunosuppressive disease) is critical to interpretation. Likewise, knowledge about preoperative therapy (e.g., corticosteroids, chemotherapy, radiation therapy, radiosurgery) is also critical to interpretation of findings (e.g., necrosis, vascular fibrosis).
Certain pathologic entities predominate in pediatric, adult, or geriatric age categories as described in the text. A communication system between the operating rooms and the pathology diagnostic rooms is especially important to share relevant clinical and pathologic observations on individual cases examined by frozen section (Table 10.1).

Radiology provides extremely valuable gross pathologic information—most importantly, computed axial tomography (CT) scan and magnetic resonance imaging (MRI); use of contrast and special imaging techniques (e.g., diffusion- and perfusion-weighted imaging) adds additional data in characterizing normal and abnormal structures. It is now possible to map sensitive regions such as corticospinal tracts and perform MRI during the surgical interval to aid localization and removal of a lesion. Emerging imaging techniques including magnetic resonance (MR) spectroscopy (evaluation of chemical components in a lesion) offer additional information about specific lesions including the likelihood that a lesion is neoplastic (6,11).
Major categories of lesions of the brain, spinal cord, and meninges, such as solitary or multiple masses, cysts, vascular malformations, or abscesses, are likely to be recognized by imaging or upon viewing a gross specimen. The neurosurgical lesions summarized in Tables 10.6 through 10.19 are usually focal, whereas the lesions in the biopsies directed toward a neurologic disease tend to be more diffuse (Table 10.2). One particularly problematic diagnosis is vasculitis, which can be focal, multifocal, or diffuse. If you are lucky enough to be consulted on any multifocal case, advise the surgeon to target a new or subacute radiographic lesion.
Multiple lesions can be produced by neoplasms or by inflammatory, vascular, and infectious diseases. If inflammation or infection is suspected prior to or seen at biopsy, cultures for appropriate microorganisms should be sent in sterile containers directly from the operating room to microbiology laboratory. The “M rule” for common multiple CNS neoplasms includes metastases, malignant lymphoma, melanoma, and (late stages only) medulloblastoma.
In some settings (e.g., in a patient with neurologic deficits), a biopsy is performed in a desperate attempt to find a diagnosis. In such a setting, to optimize the probability of a pathologic diagnosis, the biopsy should be directed at a region of recent radiologic abnormality such as contrast enhancement and optimally should include (a) dura, (b) arachnoid, (c) gray matter, and (d) white matter. In the setting of radiologic lesions, biopsy of a normal radiologic region or a so-called unguided or undirected (“blind”) biopsy is, in our experience, a useless waste of everyone’s time. It is a ritual that helps the clinician say that everything was tried on a moribund patient.
The tomographic density of hemorrhage is sufficiently unique at certain stages of organization to identify preoperatively hemorrhage as a major component of a lesion. In general, calcifications and relationships with the skull are resolved well by CT. Gray and white matter, edema, and melanin are resolved well by MRI. Vascular abnormalities are frequently appreciated clinically and angiographically. CT or MRI angiography of flow voids is sometimes sufficient and is less invasive than classic angiography.
INTRAOPERATIVE CONSULTATION
Certain entities that may be either suspected clinically or suggested on cytologic preparation and frozen section can affect the immediate surgical procedure (see “Primary Open Biopsy” section and Table 10.1). Most significantly, attempts at total resection are often made for these neoplasms: meningiomas, schwannomas, solitary metastases, cysts, ependymomas, hemangioblastomas, cerebellar pilocytic astrocytomas, and craniopharyngiomas. Therefore, assessment of such specimens requires careful clinicopathologic correlation at the time of the primary operation, and intraoperative consultation guides the procedure.
Any undefined lesion should be biopsied and inspected by both cytologic preparation and frozen sections. Cytologic preparations add fine nuclear detail and the presence or absence of (a) glial-type processes; (b) discohesiveness in pituitary adenomas, oligodendrogliomas, medulloblastomas, and lymphomas; and (c) “epithelioid” features, with cellular cohesion (suggesting junctions) in carcinomas. The value of such preparations has been demonstrated (12,13) and, in some centers, intraoperative diagnosis is based only on evaluation of cytologic preparations. Although smear and crush preparations may be done, touch preparations minimize effort and artifact. Touch a glass slide to the wet tissue and then immediately fix it in ethanol before it dries. Stain with H&E or with a cytologic stain.
Difficult access to, and fragility of, central nervous tissue places a high premium on obtaining tissue suitable for diagnosis during the first surgical procedure and avoiding secondary biopsy after a primary nondiagnostic biopsy (4). The goal of the pathologist in the intraoperative setting is not to diagnose definitively and grade every case but, rather, (a) to ensure that lesional tissue has been obtained for subsequent diagnosis and grading, (b) to ensure the lesional tissue has been appropriately sampled (e.g., that high-grade features are noted on a suspected glioblastoma based on imaging features), (c) to provide sufficient preliminary diagnostic information to optimize surgery, and (d) to perform appropriate special tissue processing (Table 10.1). Optimizing surgery depends on the individual case and on whether open biopsy or stereotactic needle biopsy is being done (see later sections).
Given the constraints of intraoperative evaluation of tissue, including sampling limitations and freezing artifact, precise diagnosis is often neither possible nor necessary for a successful intraoperative consultation. The surgical pathologist need only go as far as necessary with his or her diagnosis (e.g., high-grade glioma, metastatic neoplasm, atypical lymphoid infiltrate suspicious for lymphoma, abundant neutrophils suggestive of abscess, granulomatous inflammation) to guide the surgery without attempting to make complex diagnoses that optimally employ paraffin sections. The pathologist’s microscopic impression, even if rendered as a differential diagnosis, guides the surgical procedure.
In the setting of a glial neoplasm, it is not wise to offer a grade during intraoperative frozen section or touch preparation interpretation. Because gliomas are quite heterogeneous, diagnostic features that affect the classification and grade may be revealed only in the “permanent” specimens and not on frozen section. For instance, oligodendroglial elements in a mixed glioma may not be well represented or recognized on the frozen material; the characteristic halo is a paraffin-embedding artifact. A diagnosis of “primary glial neoplasm with abundant mitotic activity, at least World Health Organization grade III,” or “high-grade glioma,” effectively characterizes a biopsy when malignant glial cells are unequivocally identified on cytologic and frozen section preparations and allows for the later identification of oligodendroglial and other elements not evident on the fresh sample.
The intraoperative consultation provides the opportunity to obtain tissue for microbiologic culture, special fixation (e.g., lymphoma), and special processing (e.g., flow cytometry, cytogenetics, molecular evaluation, and electron microscopy) when preliminary assessment deems it necessary or pragmatic (see “Tissue Processing” later in this chapter).
SUMMARY POINTS ABOUT INTRAOPERATIVE CONSULTATION
I. Be prepared:
A. History: Patient age, duration of symptoms, and surgical location are the minimum.
B. Radiography: View images and/or read reports. On reports, objective findings trump interpretation. Are there one or more masses? Infiltrative? Enhancing?
C. Check for prior specimens: Slides and reports.
II. New specimen: Both cytologic and frozen sections preferred.
III. Observations:
A. Cell type: Fibrillar, epithelioid, mixed, small and crowded, or syncytial (e.g., fibrillar*)
B. Major features: Such as mass; marked cellular and nuclear pleomorphism*
C. Differential (Dif): Such as glioblastoma (GbM), giant cell astrocytoma (GCA), pleomorphic xanthoastrocytoma (PXA), sarcoma (Src), or melanoma (Mel)*
IV. Narrow the differential: Check excluding and corroborating features (e.g., for earlier Dif*).
A. Location: Entirely extramedullary (e.g., outside of CNS [excludes GbM, GCA]), skull invasion (favors Src, Mel), subependymal (favors GCA)
B. Radiography: Diffuse margin with CNS (favors GbM)
C. Duration of symptoms: For a new mass, less than 3 months favors GbM, Src, Mel
D. Microscopic features:
1. No mitoses (favors GCA, PXA)
2. Granular bodies (favors PXA)
3. Necrosis (favors GbM, Src, Mel)
4. Microvascular proliferation (favors GbM)
5. Intercellular collagen (favors PXA, Src)
*The differential (set in parentheses) of a mass with fibrillar cells of marked cellular and nuclear pleomorphism is a single algorithmic example of how to use the summary points.
CATEGORIES OF SURGICAL SPECIMENS
Optimal management of a specimen requires knowledge of the aim of the procedure. Three major types of neurosurgical procedures routinely yield tissue: (a) primary biopsies, (b) secondary biopsies, and (c) therapeutic resections. A primary biopsy seeks a diagnosis. Secondary biopsies may try again to establish a diagnosis or monitor consequences of therapy. Biopsies can be performed using CT- or MRI-guided stereotactic needle biopsy or using an open technique. Therapeutic resections attempt gross total excision of lesional tissue. Individual cases may combine more than one procedure at a single operation. For example, open biopsy for diagnosis of an intramedullary spinal cord tumor may proceed directly to a therapeutic resection if the intraoperative evaluation suggests ependymoma.
PRIMARY OPEN BIOPSY
The primary biopsy for diagnosis is critical. The craniotomy and open biopsy, where the neurosurgeon “turns a flap” by sawing through, and temporarily opening, a portion of the calvarium, affords a macroscopic view of the lesion. In some centers, localization of the lesion is aided by intraoperative imaging, including MRI. The pathologist’s database should include the preoperative radiologic data as well as macroscopic findings in situ, either by direct observation or by conversation with the neurosurgeon. Descriptive observations of the margin, color, consistency, precise point of attachment, and immediate surroundings of a lesion are frequently helpful. Because an open biopsy provides surgical latitude from biopsy to total resection, it is important to recognize resectable tumors whenever possible during surgery. Resectable tumors include meningiomas, carcinomas, adenomas, schwannomas, pilocytic astrocytomas, and many ependymomas.
STEREOTACTIC NEEDLE BIOPSY
Radiographically guided stereotactic needle biopsies of vital and sensitive locations through a small hole in the skull are increasing in frequency (14). Needle biopsy specimens have requirements for tissue handling similar to open biopsy specimens, with the following exceptions.
Radiographs substitute for direct visual inspection of the lesion in situ. This and the small size of the specimen confound even the most careful attempts at proper sampling of the lesion. Movement of a centimeter or less may result in totally necrotic or gliotic nondiagnostic tissue. This so-called black box effect requires that sampling of needle biopsies for diagnosis be monitored with frozen sections. However, the intraoperative interpretation does not affect the immediate surgical procedure with regard to therapy because excision of the lesion is not intended.
Better results come from examining both whole cell cytology and frozen section histology than from either alone, but special care is needed to avoid harming tiny stereotactic biopsies by touching and drying. One approach is to freeze the entire tissue and touch only the gauze pad it came on to a slide for cytology. When the pathologist indicates that a diagnostic region has been sampled, the neurosurgeon should take a second sample for permanent section from the same region, placing it directly into fixative. Those who excessively ponder the macroscopic appearance of this tiny tissue fragment under powerful lights risk a dried, nondiagnostic sample.
The most important task for the pathologist during stereotactic surgery is to advise the surgeon of the likelihood that he or she is sampling the lesion itself, rather than normal, gliotic, or otherwise nondiagnostic tissue (Fig. 10.2). Frequently, initial biopsies yield minimally abnormal tissue and lesional tissue is identified on subsequent biopsies, or an initial biopsy may yield low-grade features when high-grade features are suspected radiologically. The features to look for are addressed in the “Tumor and Tumor Margin” section later in this chapter.
The most frequent lesions to be approached by stereotactic biopsy are suspected gliomas in critically sensitive regions of the brain, including speech and motor cortex, thalamus, basal ganglia, and brainstem. Lesions in less sensitive regions should be approached by open biopsy to provide a better tissue sample than needle biopsy. Lesions firmer than gliomas, particularly common in the pineal region, tend to resist the needle. The relative difficulty of capture of the stereotactic specimen is an important clue to its diagnosis because gliomas and lymphomas tend to be easily obtained, whereas others are difficult. The presence of a large vessel in a needle biopsy specimen is a matter of concern that should be reported to the neurosurgeon.
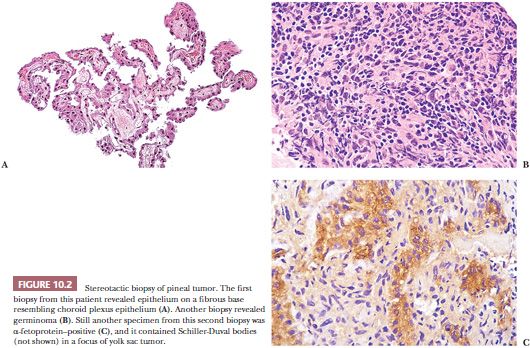
Stereotactic biopsies of cystic lesions yield a lower percentage of positive diagnoses than do those of solid tumors because of the difficulty of obtaining diagnostic tissue from a mural nodule or the wall of a lesion. Stereotactic biopsies of tumors with architectural and cytologic appearances similar to normal tissue (e.g., choroid plexus and choroid plexus papilloma, pineal and pineocytoma) can be reported as the appropriate type of tissue, with a comment about the need for neuroanatomical and clinical correlation to rule out the possibility of normal structure. Stereotactic biopsies of pineal tumors present problems analogous to the problems the fabled elephant presented to the six blind men (Fig. 10.2).
SECONDARY BIOPSY
Secondary biopsies are repeat biopsies of the lesion or its immediate vicinity. The need for a secondary biopsy following a nondiagnostic primary biopsy can usually be avoided by adequate sampling of the tissue at the primary biopsy. Most helpful in this regard is monitoring the primary biopsy procedure with frozen sections (Table 10.1). The proper time to render the judgment that a specimen is nondiagnostic is during the biopsy procedure, allowing the surgeon the opportunity to obtain another specimen.
BIOPSY AFTER THERAPY
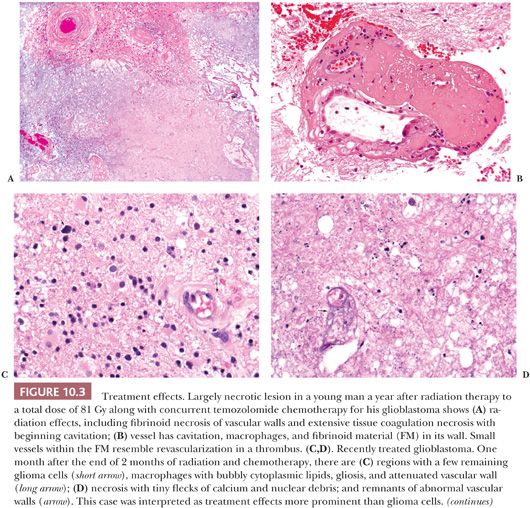
Accurate diagnosis is enhanced when the patient’s clinical status and the type of neurosurgical procedure are understood. Moreover, ancillary microscopic findings may be helpful. One of many examples is the significance of necrosis in a cerebral astrocytoma. Spontaneous coagulation necrosis in a diffuse cerebral astrocytoma is a feature of a glioblastoma multiforme. On the other hand, if necrosis is found in an astrocytoma biopsied after cytotoxic or radiotherapy, it may simply represent a good therapeutic response (Fig. 10.3A,B). Geographic necrosis is less reliable than necrosis with pseudopalisades found in cellular tumors. “Pseudopalisades” are rings of increased cellular density around the necrosis. Geographic necrosis alone has been seen in white matter infiltrated by glioma as part of the therapeutic response to radiotherapy.
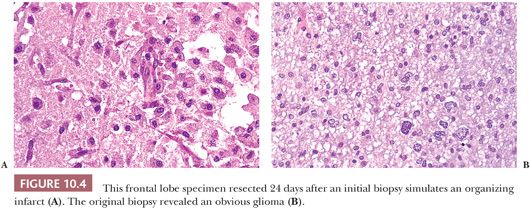
Exceptionally problematic are biopsies after radiotherapy, chemotherapy, or vascular embolization of tumors. Even the previous surgical procedure can radically alter the appearance of a neoplasm (Fig. 10.4). Previous treatment information should be obtained prior to rendering an opinion. Biopsies for diagnosis before the initiation of such therapy should be encouraged.
Glioma Regrowth or Progression versus Treatment Effect
Some biopsies, often prompted by brain swelling, intentionally monitor the effects of therapy. Determining status of a glioma during or soon after therapy is often critical. Procedures including choline to N-acetyl aspartate (NAA) ratios on MR spectroscopy have improved radiographic estimation of glioma versus necrosis in a glioma treated with radiation and/or chemotherapy. However, glioma regrowth and treatment effects (TEs) are more complex than glioma versus necrosis. When radiographic procedures do not work, biopsy is needed to distinguish TE from tumor regrowth (Fig. 10.3C,D).
Tumor progression and TE can both involve necrosis, vascular changes, and cellular pleomorphism. The necrosis of TE often shows fibrinoid change (Fig. 10.3E,F). Either tumor progression or TE can show vascular proliferation, but only TE shows fibrinoid necrosis or extravasation affecting vascular walls. Radiation necrosis is not pseudopalisading, and during its maturation, it shows tiny flecks of calcification. Both the tumor and white matter are particularly sensitive to radiation, grey matter being more resistant. Mitoses and tumor cell density are decreased with TE.
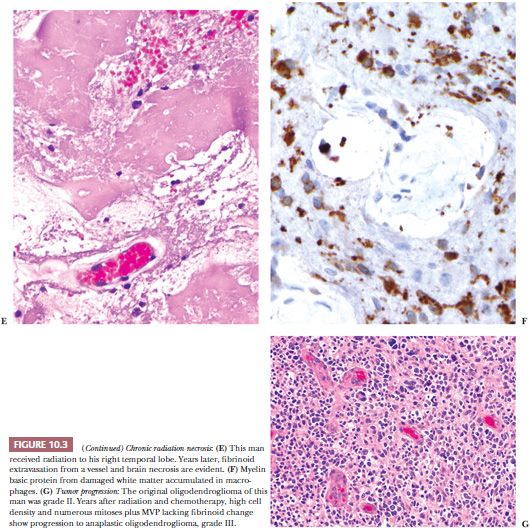
A few scattered glioma cells are usually present in biopsies done to monitor TE versus glioma regrowth and/or progression. Scattered glioma cells do not dissect the differential. Instead, look for significant glioma cell density and mitoses as signs of regrowth. Tumor cell density and mitoses also are usually increased with tumor progression (Fig. 10.3G). The character of necrosis in tumor progression is often oddly shaped (geographic) and may include adjacent increased cell density (pseudopalisades). Pseudopalisades are more reliable indicators of progression, in my experience. Microvascular proliferation in tumor progression lacks fibrinoid changes.
Months or years following tissue diagnosis and radiotherapy of a glioma, a radiologic abnormality or declining clinical status may prompt another operation to distinguish between recurrence of tumor and radiation necrosis (Fig. 10.3E–G).
THERAPEUTIC RESECTION
If the initial operation for diagnosis was incomplete, a second operation may be done to achieve a therapeutic effect. Other operations are primary resections of brain, often the temporal lobe, to treat medically refractory seizure foci. Categories of therapeutic resection include gross total (100% removal of visible mass), radical subtotal (95% to 99% removal), subtotal (75% to 95% removal), and partial (10% to 75% removal) resection.
If the lesion was removed with a Cavitron ultrasonic aspirator (CUSA), tissue obtained from the suction collection device retains diagnostic features and immunoreactivity. However, tissue artifact is common and easy to confuse with necrosis in aspirated specimens. Blue-tinged necrosis and linear streaks of nuclear material suggest artifact.
The pathologist should compare prior diagnostic findings to the remaining lesion. Familiarity with the histologic appearance of a lesion on previous biopsy is critical for accurate interpretation (Fig. 10.4). If the resection is performed months or years following the primary biopsy, the pathologist may be expected to render an opinion about the effect of therapy. Evidence of treatment response, such as necrosis and vascular fibrosis, should be noted (Fig. 10.3).
Alternatively, the second procedure is performed with therapeutic intent, such as to place a wafer impregnated with a chemotherapeutic agent. In such cases, specific histologic features (e.g., definitive features of glioblastoma) may be necessary to qualify for specific treatment protocols for placement of the wafer.
MACROSCOPIC SPECIMEN INTERPRETATION AND PROCESSING
All specimens should be examined for arachnoid, gray matter, and white matter. To optimize evaluation, sectioning of specimens with identifiable architecture should be performed perpendicular to the meninges, from arachnoid through gray matter to white matter.
Many lesions are macroscopically evident in biopsy specimens. Yellow or firm gray or white matter typically corresponds to gliosis. Solid tissue that is not discernible as either gray matter or white matter typically represents an abnormal mass. Some tumors are often semiliquid and gelatinous in nature (oligodendrogliomas, lymphomas, pituitary adenomas). See Tables 10.11 through 10.13 for diagnosis by distinguishing features. Multiplicity of masses is usually impossible to establish from the neurosurgical specimen alone; it requires radiographic correlation.
Flat tissue no thicker than 3 mm that is neither dura nor arachnoid may represent the wall of a cyst (see Tables 10.18 and 10.19). This can be compared and verified with the in situ or radiographic impressions of the lesion. Intracranial and intraspinal cysts are often fragmented in the course of their removal.
A vascular conglomerate larger than the usual aggregates of arachnoidal vessels may represent a vascular malformation (Table 10.6). Vascular malformations involve meninges and/or parenchyma and are often associated with recent or chronic hemorrhage.
Necrosis of the CNS parenchyma or of tumors is initially very soft and friable. It cavitates with age more rapidly in CNS parenchyma than in tumors. It can be clear, white, or yellow depending on the state of microcavitation and macrophage reaction. The arachnoid is typically preserved despite underlying parenchymal necrosis.
Although clinical diagnosis is variable, radiographic and macroscopic examination often do not distinguish inflammatory from other lesions. This must be done microscopically, as described in Tables 10.2 through 10.19 and the accompanying text and figures.
Some lesions have many of the features described earlier. A mass may contain regions of necrosis or hemorrhage. Arteriovenous malformations and cavernous angiomas are commonly linked with hemorrhage. Macroscopic cysts may occur within a mass, especially within cystic astrocytomas, oligodendrogliomas, craniopharyngiomas, and teratomas. Therefore, all tissue regions should be adequately sampled, particularly any interface between lesion and recognizable brain.
MARGINS
Assessment of margins in the resection of many CNS lesions, particularly diffuse gliomas, is an unrealistic goal. Due to limitations of access, CNS lesions are frequently resected piecemeal and with the use of combined ultrasound and suction. Although a gross total resection is attempted in some cases, guided by imaging studies, most adult primary gliomas manifest a diffusely infiltrative growth pattern that defies even radical resections attempted in the past and are considered nonresectable. Some exceptions exist, with, for example, a pilocytic astrocytoma from the cerebellum of a child or a dysembryoplastic neuroepithelial tumor from the cerebrum. See further considerations later in the “Gliomas” section. In rare cases, an en bloc excision of an entire neoplasm (e.g., a meningioma) is obtained. It is best to mark the margin of tissue around such an intact specimen with ink, unless the surgeon requests otherwise.
TISSUE PROCESSING
General Considerations
1. Squash/smear/touch preparations
2. Unfrozen routine permanent paraffin sections
3. Frozen sections
4. Electron microscopy (EM) (retain a small portion in a fixative for EM, if necessary)
5. Frozen tissue, for possible future molecular diagnostic studies (freeze fresh tissue as soon as possible and store)
6. Other (microbiology, flow cytometry, cytogenetics, molecular diagnostics)
Routine Fixation
In most cases, with the critical exception of suspected hematopoietic neoplasms, fixation of biopsy and resection specimens in formalin and routine processing are sufficient to meet diagnostic needs.
Electron Microscopic Fixation
Glutaraldehyde fixation of a small portion of a lesion can be done any time after formalin fixation, making it easy to save tiny pieces of wet tissue in formalin in case EM is needed. It may be needed for (a) poorly differentiated or unusual tumors, (b) differential that includes a lesion best confirmed by EM (like ependymoma), (c) toxic-metabolic disease, and (d) infection. Saving a bit of tissue for possible future EM evaluation until completion of a challenging case is smart. Keeping it in formalin in its original jar is easy.
Hematopoietic Specimens
When a hematopoietic lesion is identified intraoperatively or suspected clinically, the specimen should be treated like a lymph node biopsy. Nuclear morphology is a key element of hematopoietic diagnoses. As such, a portion of the specimen should be fixed in a mercuric chloride-formalin fixative (e.g., B5) or, alternatively, zinc-containing fixative. The cassette must be marked to indicate that removal of the metallic fixative is required after the appropriate fixation interval. If enough tissue is available, a suspected hematopoietic lesion can also be placed in cell culture medium and sent for flow cytometric evaluation for phenotypic characterization. Touch preparations are often useful, especially intraoperatively. A portion can also be flash frozen to accommodate molecular evaluation and labile antigen evaluation by IHC; retaining a frozen section specimen in the frozen state can accomplish this goal simply and effectively.
Microbiologic Workup Recommendations
Cultures usually are more sensitive than other methods (including molecular methods) to detect organisms that grow in vitro. If an infectious etiology is suspected intraoperatively, it is critical that the surgeon be notified of the possibility immediately so that optimal samples for bacterial (aerobic and anaerobic), mycobacterial, fungal, and viral cultures can be obtained by the surgeon from the sterile operating field and sent directly to the microbiology laboratory. Saving a small, fixed portion of the lesion for possible EM evaluation and freezing another small piece are wise.
Molecular Diagnosis
Incorporation of genetic data into the assessment of a CNS neoplasm is now well established for glial neoplasms and is emerging for other tumors and lesions including infections. Formalin-fixed, paraffin-embedded tissue sections can be used for most of the techniques, including IHC, fluorescence in situ hybridization (FISH), in situ hybridization (ISH), and polymerase chain reaction (PCR) techniques. Other techniques require unfixed tissue for evaluation. When abundant lesional tissue is present, the pathologist is wise to snap-freeze a portion and store it at −80°C. Retaining the frozen specimen in a frozen state can accomplish this, assuming adequate tissue is available for permanent sections.
Toxic-Metabolic Processes
When a biopsy from a patient with suspected toxic-metabolic disorder is performed (e.g., for a suspected lysosomal storage disease, mitochondrial cytopathy, or cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy [CADASIL]), representative gray and white matter should be fixed in formalin for routine processing, retained in formalin or glutaraldehyde for possible electron microscopic evaluation, and frozen for possible biochemical studies if enough tissue is available to meet histologic needs.
HIGH-RISK INFECTIOUS DISEASE CASE CONSIDERATIONS
Universal precautions prevent transmission of most infectious diseases from central nervous specimens to pathologists and assistants. However, biopsies obtained from (a) patients with known HIV infection/AIDS and (b) patients with a clinical differential diagnosis that includes Creutzfeldt-Jakob disease (CJD) require special consideration and handling (4,15).
Minimization of risk to laboratory personnel includes (a) advance warning by neurosurgeons when operations are performed on high-risk patients, (b) cytologic preparations instead of frozen sections on specimens from these patients and only if needed to confirm lesional tissue, (c) declining frozen sections from patients with a clinical differential diagnosis that includes CJD, and (d) proper handling of specimens once received in the laboratory.
Human Immunodeficiency Virus
Brain biopsies obtained from patients with known HIV infection or acquired immunodeficiency syndrome (AIDS) are usually tested for infectious processes (e.g., toxoplasmosis) and neoplasm (e.g., lymphoma or other neoplasms). HIV is inactivated by formalin, so, after fixation, there is minimal risk of handling specimens from such patients. Cytologic preparations often provide assurance of lesional tissue. However, if a frozen section is necessary from such a specimen, decontamination of the cryostat should be carried out by using and replacing a disposable blade, removing all freezing medium debris, and wiping surfaces that came into contact with the specimen with a solution of 10% bleach in water. Use of a separate cryostat for such specimens, when available, can minimize downtime in a busy frozen section laboratory.
Prion Diseases, Including Creutzfeldt-Jakob Disease
Biopsy for possible CJD requires a compelling clinical reason, such as a treatable disease being in the clinical differential. A neurologist with expertise in prion diseases should certify all biopsies for dementia: (a) Is there a possibility of prion disease or not? (b) Could the patient have another treatable cause of the dementia? Without an affirmative answer to both questions, biopsy is not appropriate because of the concern for contamination of surgical and pathology personnel.
If biopsy is undertaken, frozen sections should not be done. Specimens must include (a) tissue for formalin fixation and (b) snap-frozen tissue for Western blot and gene sequencing evaluation. It is also desirable to collect blood for germline comparison. Prior to biopsy, the neurosurgeon should confer with the neurologist to sample an appropriate portion of brain, depending on symptoms. In addition to gray matter, white matter and meninges should be sampled.
The National Prion Disease Pathology Surveillance Center (NPDPSC) at Case Western Reserve University is funded and equipped for molecular diagnosis of prion diseases. For Western blot, the NPDPSC requests at least 10 mg of tissue (pea-sized or 0.5 cm diameter) for biopsies. Put the tissue into a small plastic vial and screw on the cap. Put the vial into a biohazard bag and close the bag. Store initially in a −70°C freezer until it can be sent by overnight mail to the NPDPSC. Further instructions and contact information can be obtained by contacting the NPDPSC directly.
In the rare biopsied case in which CJD is in the differential, decontamination of tissue for histologic processing, sectioning, and examination can be obtained as follows. Specimens should be sectioned to fit into cassettes (see special notes in the following section); if desired, a small portion of cortex can be placed into glutaraldehyde for possible electron microscopic evaluation. The cassettes are (a) placed into 10% buffered formalin for fixation for at least 24 hours; (b) transferred to undiluted, 95% to 100% formic acid for 1 hour; and (c) then returned to fresh 10% formalin for another 48 hours or more before routine processing. Although of very low risk, embedded tissue should be handled as if infectious—sectioned by an experienced technologist using disposable blades, with incineration of all section waste and wiping of microtome and cutting station with bleach or 2N NaOH. Histologic and many IHC evaluations are minimally impeded by the formic acid processing. Disposable towels, gloves, and equipment should be used in initial processing and subsequently incinerated. Contaminated surfaces can be cleaned using 5% bleach (corrosive for steel instruments) or 2N NaOH (incompatible with aluminum) with prolonged exposure (15 minutes up to 2 hours) for equipment (4,15). More information is available online at http://www.pathology.med.umich.edu/Safety%20Manual/CJD%20Final.pdf.
Special Notes
1. Cassettes should be labeled with pencil as formic acid will dissolve away even the ink specially formulated for histologic processing.
2. Normal plastic cassettes are resistant to formic acid, but the thin plastic netting of cassettes designed for small biopsies will dissolve in formic acid, so a small biopsy should be wrapped in lens paper to keep it in the cassette. Metal tops should not be used.
HISTOCHEMICAL AND IMMUNOHISTOCHEMICAL EVALUATION
Special stains (16), IHC stains, and cytologic techniques that aid the diagnosis of specific entities at biopsy are described in Tables 10.2 and 10.10 through 10.20. Routinely use a standard tissue control that contains regions both positive and negative for the IHC marker under evaluation. If possible, choose a block with the lesion of interest that also contains tissue with known positive and negative regions to use as an internal standard tissue control (i.e., the edge of a tumor with gliotic brain and vessels for GFAP IHC).
Polarization microscopy quickly reveals mature collagen fibers and hair shafts (see Fig. 10.133 in the “Dermoid Cyst” section later in this chapter). On the Congo red stain, polarization distinguishes light yellow-green amyloid from whiter collagen (17).
Individual neoplasms may lack a marker generally representative of its category. Others may lose expression of markers as they progress to higher grades of malignancy. Because these factors or technical reasons may cause individual neoplasms to fail to label for a marker, a positive immunostaining result is more meaningful than a negative result.
Based on initial histologic findings, one can construct a differential diagnosis for which a group of appropriate special stains can be assembled from information in the text and tables here. To minimize redundancy, positive rather than negative features are emphasized in the following sections, under each entity. Depending on difficulty, the differential can be solved on H&E or IHC (18).
There are many Web sites for locating IHC expertise, reagents, and suppliers. www.biocompare.com searches for very specific reagents and their suppliers. www.histonet.org provides numerous comments, recipes, and images from histology and histochemistry professionals. Our University of Michigan (UM) IHC laboratory professionals often use the antibody name in Google searches to find new antibodies.
ELECTRON MICROSCOPY
The importance of EM in cranial and spinal diagnostic neuropathology has been diminished by faster IHC. However, EM is valuable in defined settings in which IHC cannot distinguish among specific subtypes of tumor (19). Unlike largely confirmatory IHC, EM shows many structures not well seen on light microscopy that may point to a new direction in interpretation. At times, it is the only path to a definitive answer. For example, among various clear cell tumors, basal bodies, cilia, elongated junctions, and microvillous lumens distinguish clear cell ependymoma from the other tumors, including oligodendroglioma. EM can be a valuable adjunct to diagnosis in specific settings; see the “Tissue Processing” section earlier in this chapter.
EM is needed to confirm certain metabolic diseases. Specimens from sites other than brain are frequently biopsied for evaluation of various metabolic diseases that affect the CNS, such as neuronal ceroid lipofuscinosis, the various lysosomal storage diseases, leukodystrophies, mitochondrial encephalopathies, and vascular diseases including CADASIL (19).
REACTIVE CHANGES
GLIOSIS
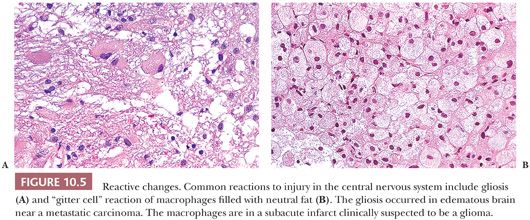
Gliosis is a reaction of the CNS to injury of the brain or spinal cord (see Fig. 10.5A). It identifies abnormal CNS tissue. Gliosis is defined as either (a) increased number of astrocytes or (b) increased number and length of processes or increased cytoplasmic synthesis of GFAP representing gemistocytic astrocytes. Although subtle changes such as astrocyte swelling with fluid occur earlier, gliosis is usually appreciated on H&E-stained slides within 2 weeks after an injury. Because nearly any injury of the CNS can cause gliosis, it is not diagnostic of a specific pathologic entity.
Anti-GFAP IHC differentiates subtle forms of gliosis (Table 10.3). When this differentiation is critical, a control slide from the same region of normal CNS can help. Compared to control, look for a greater number of astrocytes, a greater number and density of cell processes, and the presence of GFAP-positive gemistocytes with reactive nuclei, cells spaced apart from one another.
The problem of distinguishing gliosis from low-grade glioma is challenging and is described later (see “Gliomas”). Gliosis does not expand, although gliotic parenchyma may surround an expanding tumor. “Pure” gliosis, which lacks noticeable CNS parenchyma, is associated with long-standing cysts (Table 10.18). In contrast to microcysts of gliomas (see Fig. 10.32 in the “Gliomas” section), these cysts are macroscopic, confluent, and often have numerous residual macrophages. However, in the absence of evidence of another origin, the possibility of neoplastic origin of such cysts must still be considered. In gliosis, astrocytes respect each other’s space and show more uniform spacing between individual glial cells than in the margin of a glioma (see Fig. 10.5A; also see Figs. 10.23, 10.24, and 10.35 later in this chapter). The nuclear-to-cytoplasmic ratio of gliosis is less than that of a glioma. IDH1 mutation and p53 overexpression are not found in gliosis. This distinguishes gliomas with IDH1 mutation and p53 overexpression from gliosis (20). Gliosis has a less than 5% Ki-67 proliferation index, assessed on hot spots within 100 cells (see “Assays of Proliferative Capacity” section). Excessive fibrillarity and Rosenthal fibers do not distinguish gliosis from glioma (see Figs. 10.35 and 10.40 in the “Gliomas” section).
MACROPHAGES, MICROGLIAL CELLS
Macrophages present as (a) granular or foamy macrophages, (b) microglial cells with rod-shaped nuclei, or (c) epithelioid macrophages with or without multinucleated giant cells. They may be seen, in variable and sometimes striking abundance, in any condition that involves tissue disruption, including infectious, inflammatory, necrotic, traumatic, degenerative, and neoplastic processes (21–23). The origin of these cells is variable among lesions and consists of some combination of (a) “native” parenchymal microglial cells, which can proliferate along with (b) blood-derived macrophages entering the brain parenchyma in response to damage (22). The fate of the latter cells may be to remain at the site of injury or migrate to the perivascular space (23). Because distinction among these origins is not useful diagnostically, the inclusive term macrophages can be employed.
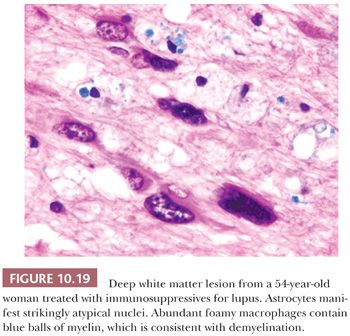
In the brain, a useful marker for macrophages is CD68 (KP-1) IHC (see Fig. 10.25 in the “Demyelination” section later in this chapter), and it frequently identifies considerably more macrophages than are anticipated by H&E evaluation. A special stain for myelin (e.g., Luxol fast blue–hematoxylin and eosin [LFB-H&E]) (see Fig. 10.19 in the “Progressive Multifocal Leukoencephalopathy” section later in this chapter) identifies engulfed myelin debris, and a stain for axons (see Fig. 10.26 in the “Demyelination” section later in this chapter) helps to distinguish between necrotic CNS tissue (axons disintegrate and leave axonal debris in macrophages) and demyelination (foamy macrophages contain myelin but not axonal debris). GFAP IHC is helpful to discern gemistocytic reactive astrocytes from macrophages. Reactive macrophages must be distinguished from the histiocytoses; see the “Histiocytoses” section later in this chapter.
Foamy macrophages are common in infarcts and demyelination; their lysosomes are loaded with cellular debris and may contain yeast forms and other organisms. Macrophages swollen by phagocytosis within the CNS are called endocytic or gitter cells (Figs. 10.4A and 10.5B). Hemosiderin-laden macrophages can be noted around organizing hemorrhages, traumatic lesions, and some vascular malformations. Large, rounded cells with foamy cytoplasm and eccentric nuclei, having the appearance of macrophages, present occasionally as a part of such rare lesions as xanthogranulomas of choroid plexus. They are occasionally evident in various brain tumors, along with lymphocytes. Lipid-laden macrophages can be distinguished from amebae in the rare amebic brain abscess by their larger and more distinct nuclei and less chromatin clumping than amebae and by their CD68 positivity (see the “Parasites” section under “Infectious Diseases” later in this chapter).
Epithelioid macrophages are activated but nonfoamy macrophages are identified in granulomatous inflammation in infectious (e.g., fungus, acid-fast bacillus [AFB]) and inflammatory (e.g., sarcoid) diseases of the CNS.
Giant cell macrophages are usually seen in granulomatous inflammation. Individual multinucleated giant cells in brain tissue are evidence of HIV encephalitis.
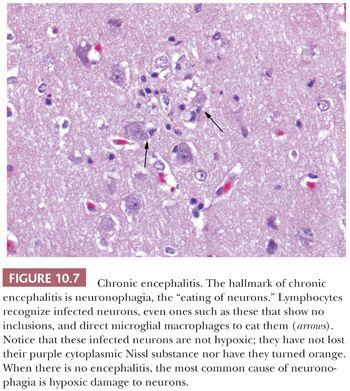
Microglial cells are macrophages that closely resemble antigen-presenting dendritic cells seen elsewhere in the body and play a key role in response to a variety of etiologic insults (22,23). They have a spindle-shaped nucleus and little discernible cytoplasm and must be distinguished from the so-called naked nuclei of infiltrating glioma and from endothelial cell nuclei. They are CD68-positive (see Fig. 10.25). They are notably seen diffusely in brain tissue or as small aggregates, so-called microglial nodules; in viral and rickettsial diseases; and around dying neurons (neuronophagocytosis; see Fig. 10.7).
GRANULOMATOUS INFLAMMATION
Granulomatous inflammation is defined by both cell types and architecture. Cells include infiltrates of (a) activated, epithelioid macrophages with or without multinucleated giant cells and (b) lymphoid cells. Foamy macrophages may or may not be seen. Architecturally, the infiltrate may form nodules, or granulomas, with or without central necrosis. Identification of granulomatous inflammation raises the differential of infectious or autoimmune inflammatory conditions. Granulomatous inflammation is common in the setting of bacterial (tuberculosis, syphilis, Whipple disease), fungal, and parasitic infections. These entities must be distinguished from rare, noninfectious granulomatous processes that can affect the CNS, including systemic lupus erythematosus (SLE), Wegener granulomatosis, sarcoidosis, rheumatoid meningitis, and Crohn disease. If a histiocytic process is in the differential diagnosis, IHC for S-100 and CD1a can be performed (see the “Histiocytoses” section later in this chapter).
PERIVASCULAR INFLAMMATION
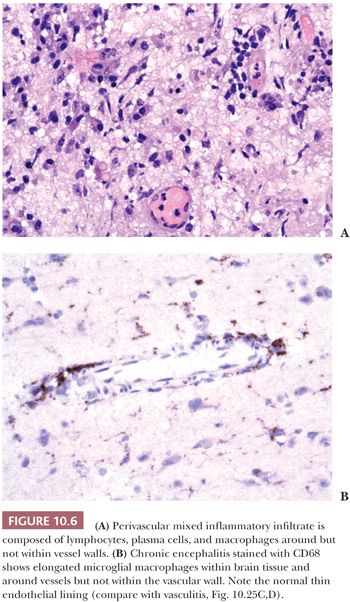
Virtually, any irritation of the CNS can stimulate accumulation of cells around blood vessels (Fig. 10.6), including surgical wounds and implants (24). Macrophages ingest the irritant or injured cellular constituents and move them to the perivascular space (23). In the absence of classic lymph nodes in the brain, this perivascular region is where cells that respond to antigens intermingle. Depending on severity and duration of illness, the perivascular inflammation varies substantially. Old hemorrhage is characterized mainly by perivascular macrophages laden with hemosiderin. Viral or autoimmune encephalitis produces a maximal response, with abundant perivascular macrophages and lymphocytes along with parenchymal infiltrates of microglial cells and microglial nodules.
MENINGEAL FIBROSIS
Meningeal fibrosis develops following traumatic injuries, meningitis, vasculitis involving meningeal vessels, radiation therapy, or as a desmoplastic response to a tumor, especially if the tumor triggers massive vascular proliferation. Multifocal fibrosclerosis with hypertrophic intracranial pachymeningitis is a firm, fibrous lesion with dural thickening, frequently found in conjunction with an intracranial pseudotumor (25). Arachnoiditis ossificans deposits bone within the chronic inflammatory response.
INFECTIOUS DISEASES
In a symptomatic patient with imaging findings suggesting an infectious or inflammatory disease, when evaluation of cerebrospinal fluid (CSF) is unrevealing, biopsy is undertaken to attempt establish a definitive diagnosis and rule out other processes (see Fig. 10.9). Optimal sampling by biopsy that (a) includes dura, arachnoid, and gray and white matter and (b) is performed in the site of a radiologically enhancing lesion markedly improves the chance of reaching a diagnosis.
When biopsy is performed, the principal goal of the surgical pathologist in this setting is to evaluate features of infectious and inflammatory processes and rule out ischemic damage and neoplasm. Check the history; the immunocompromised patient is particularly susceptible to CNS infection. Specific susceptibilities are found in the following discussions.
MICROBIAL CULTURE
If an infectious etiology is suggested at the time of biopsy, the surgeon must be notified to send cultures (see “Tissue Processing”).
SPECIAL STAINS
A combination of histochemical microbial stains is employed in the setting of inflammation to evaluate for organisms and assess for specific cell types. IHC or ISH may help to identify organisms, particularly toxoplasma, viruses (including herpes, cytomegalovirus [CMV], and JC virus), fungi, and Rickettsia (26). Available molecular biologic approaches for identification of microorganisms vary among institutions. Some require frozen section, but PCR usually can be performed on DNA extracted from paraffin-embedded sections of tissue following deparaffinization steps. Most useful is the ability to detect mycobacteria, in the setting of necrotizing granulomatous inflammation, and assess tuberculosis (TB) or not TB. Cultures for TB are more sensitive but they take much longer.
EM has a role in the identification of microorganisms that do not grow in culture and are hard to stain, including viruses, Whipple bacillus, amebae, and spirochetes.
MENINGITIS, CEREBRITIS (FOCAL ENCEPHALITIS), MYELITIS
The terms meningitis, cerebritis, and myelitis literally mean inflammation of the meninges (dura or leptomeninges), cerebral parenchyma, or spinal cord parenchyma, respectively (27). Acute (neutrophils), chronic (lymphocytes and macrophages), and granulomatous (round, fibrous clusters of lymphocytes and epithelioid macrophages) inflammation can be identified. These forms of inflammation can be seen in infectious processes and in autoimmune inflammatory processes. In general, inflammation is not a prominent feature in neoplastic processes, and caution must be exercised in a hypercellular lesion with a large number of macrophages and other inflammatory cells. However, inflammatory infiltrates can be seen in neoplastic processes, especially perivascular lymphocytic infiltrates (particularly common in lymphomas, gangliogliomas, gemistocytic astrocytomas, and oligodendrogliomas) and macrophages mixed with neoplastic cells, but they are a secondary feature.
The term cerebritis has been used to describe a focal cerebral encephalitis of bacterial origin that is not sufficiently encapsulated to be an abcess (27,28). Unfortunately, cerebritis is confusing because it excludes CNS structures posterior to the cerebrum, leaving cumbersome alternatives like cerebellitis, mesencephalitis, or brainstemitis. I prefer the term focal encephalitis to cerebritis. Focal encephalitis (FE) precedes frank abscess formation but requires early biopsy to be seen. FE may be sterile or may contain an infectious agent. Its inflammatory infiltrate is composed of neutrophils, macrophages, lymphocytes, and plasma cells, with or without parenchymal necrosis (Figs. 10.6 and 10.7). FE is found around septic thromboemboli, neoplasms, ruptured vascular malformations, infarcts, or traumatic lesions. Meningitis may precede FE. The aforementioned features also apply to focal myelitis. Septic FE is usually caused by bacterial agents, most often streptococci or staphylococci and less frequently by Gram-negative organisms such as Escherichia coli, Pseudomonas, or Haemophilus influenzae.
ENCEPHALITIS
The term encephalitis literally means brain inflammation. Viruses and Rickettsia usually cause inflammation of brain tissue that is more diffuse than that caused by bacteria and may involve more than the cerebrum. By convention, this brain inflammation is usually called encephalitis (27). Most viral infections are self-limited and cause inflammation that resolves without permanent brain damage. Other infections, emphasized in the section “Viruses” later in this chapter, can result in a serious necrotizing process.
MYELITIS AND TRANSVERSE MYELITIS
Myelitis and so-called transverse myelitis (specifically inflammation of the spinal cord parenchyma) present clinically with symptoms can involve sensory, motor, and autonomic modalities of variable severity. The etiology is often elusive, but etiologies include infectious (herpes simplex virus [HSV], CMV, etc.), inflammatory (multiple sclerosis [MS], connective tissue disease, etc.), vascular (infarct due to thrombosis), developmental (vascular malformation), traumatic, and iatrogenic processes (29,30). Workup is based on imaging and CSF and serum evaluation; biopsy is occasionally performed to search for an etiology.
ABSCESS
An abscess, typically preceded by FE or cerebritis, combines features of inflammation, gliosis, and fibrosis. A mixture of polymorphonuclear leukocytes, lymphocytes, macrophages, and plasma cells on H&E-stained sections confirms inflammation (Fig. 10.8). Polymorphism of inflammatory components can be verified by finding mixed B and T lymphocytes admixed with CD68-positive macrophages.
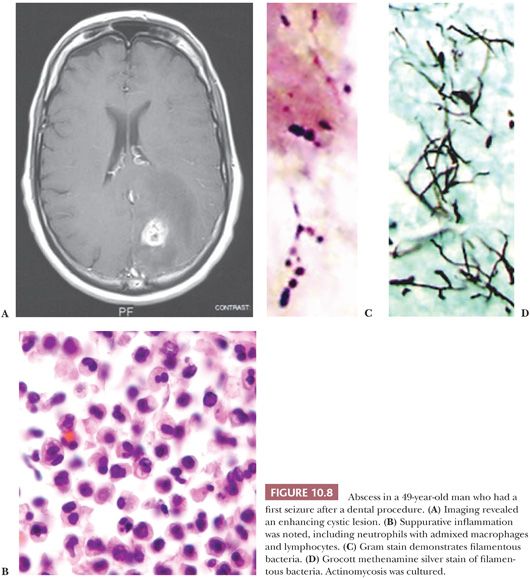
The wall of a brain abscess consists of collagen and reactive gliosis. The collagen layer is formed by fibroblasts that migrate out from the surrounding blood vessels. Thus, the thickness of this layer varies depending on the number of viable vessels and their distance from the abscess as well as the age of the abscess. Because collagen is rare within the CNS, its presence is an important diagnostic feature of an abscess (Table 10.18). Collagen may be difficult to distinguish from fibrillary gliosis on a slide stained with H&E. Points to remember are the extracellular and often wavy appearance of collagen compared with the intracellular fibrillar character of gliosis. Collagen will polarize yellow-white on H&E- or Congo red–stained sections, whereas gliosis will not. If there is any doubt, it is best to stain histochemically for collagen or reticulin or stain immunohistochemically for collagen type IV.
Other entities that produce collagen within the CNS may be confused with the wall of an abscess (Tables 10.10 through 10.12, 10.18, 10.19). Sarcomatous and desmoplastic neoplasms, and various cysts with collagenous walls, generally lack an inflammatory component. Notable exceptions are cysts that have ruptured and extruded material foreign to the CNS, such as colloid or squamous epithelial cells (see Fig. 10.132 in the “Common Metastatic Tumors” section later in this chapter and Table 10.19). If this material is not detectable within the inflammatory reaction to the cyst, clinical and pathologic correlation assists the interpretation. These other lesions that combine collagen and inflammation are sterile in situ and do not contain microorganisms.
Nocardial abscesses of the cerebrum and the spinal cord are rising in frequency in drug addicts. Multiple microabscesses should be considered in the differential diagnosis of encephalopathy in hospitalized patients with chronic disease, immunosuppression, and sepsis (27).
MYCOBACTERIA, SPIROCHETES, AND WHIPPLE DISEASE
Suppurative bacterial infections have been discussed earlier under meningitis, cerebritis (focal encephalitis), myelitis; and shown in Fig. 10.8B. Here, consideration will be given to several special diseases, including TB, syphilis, and Whipple disease (31,32).
TB and atypical mycobacteria can involve any region of the CNS, principally manifesting as meningitis and meningoencephalitis (32). AFB stains are useful, but slower cultures are more sensitive. A role for PCR in identifying and speciating mycobacteria has been established (33). In human immunodeficiency virus (HIV)–infected patients, newly acquired TB can spread readily and progress rapidly to active disease. TB can be the first clinical manifestation of immunodeficiency in these patients.
The frequency of neurosyphilis continues to increase in AIDS patients (31). Microscopic examination reveals focal lymphoplasmacytic inflammatory infiltrates in a predominantly perivascular arrangement. Syphilis contributes to the list of the differential diagnoses of granulomatous inflammations. Silver stains for spirochetes do not work well in brain due to background staining. Special identification procedures are available at the Centers for Disease Control and Prevention (CDC).
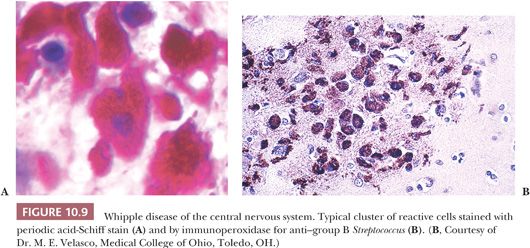
Whipple disease rarely presents as a primary brain disease and even then usually with concomitant gastrointestinal symptoms. The diagnosis can be made by brain biopsy. Histologic features include periodic acid-Schiff (PAS)–positive, diastase-resistant macrophages; microgranulomas; giant multinucleated cells; perivascular lymphocytes; and reactive microglia (Fig. 10.9). Supplemental data come from immunohistochemical evaluation for group B streptococci and EM (34). Biopsy of the central cingulate gyrus, mediobasal temporal region, and insular cortex yields the most diagnostic tissue. EM can confirm the presence of a characteristic bacillus that has been named Tropheryma whippeli.
FUNGI
Fungal organisms that involve the CNS include those presenting as (a) yeast forms (Fig. 10.10), including Cryptococcus neoformans, Coccidioides, histoplasmosis, Candida, and blastomycosis; and (b) hyphal forms (Fig. 10.11), including the Zygomycetes (Mucor and Rhizopus, Rhizomucor), Aspergillus, and Pseudallescheria. Involvement of the CNS can be via a hematogenous route (e.g., from a primary pulmonary source) or via direct extension from an infected sinus.
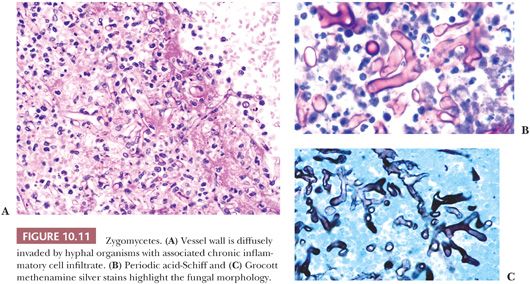
Fungal organisms tend to involve the meninges, in some cases presumably passing from subarachnoid space along perivascular spaces to involve parenchyma. Vascular wall invasion is common in hyphal forms, which may lead to catastrophic rupture and hemorrhage.
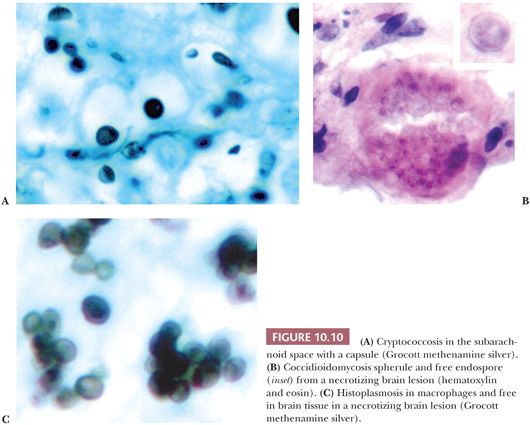
Variable inflammatory infiltrates are elicited by fungi, most often chronic and granulomatous. When fungi are suspected, use of both Grocott methenamine silver (GMS) and PAS is recommended because some forms stain better with one than the other stain (Fig. 10.11). Mucicarmine is of value in evaluating the capsule of Cryptococcus. This relatively nonantigenic capsule delays the inflammatory response to Cryptococcus.
Cryptococcal meningitis is the most common form of fungal meningitis, with a variable inflammatory response, often minimal in acute infections and in the setting of AIDS (Fig. 10.10). Chronic infections can produce granulomatous inflammation. Histoplasmosis, blastomycosis, and coccidioidomycosis can present as CNS parenchymal lesions, after presumed dissemination from lung, with variable suppurative to granulomatous inflammatory reactions; characteristic yeast forms can be identified by H&E, GMS, and PAS (Fig. 10.10). Candida can also involve the CNS as intraparenchymal masses; inflammation is typically acute (28). Zygomycetes organisms tend to be angiocentric and angioinvasive (Fig. 10.11). Zygomycetes may involve brain by direct extension from a nasal sinus infection; diabetic patients appear to be at particular risk for such infections (35).
Assessment includes the identification of hyphal or yeast forms and an attempt at identification of the organism. Ideally, a portion of the specimen is sent for culture and specific identification. The report of hyphal organisms as suspicious for aspergillosis (based on dichotomous branching and septa formation) or Zygomycetes (based on ribbonlike hyphae of varying width, absence of definite septa, and right-angle branching) should be confirmed by culture, IHC, or ISH. Pseudallescheria can appear histologically as hyphae with acute branches and septa, and treatment is different (36). Histologic consultation with a microbiologist prior to reporting is wise.
PARASITES
A variety of parasitic organisms can involve the CNS, including endemic organisms or exotic organisms most frequently seen in immigrants or travelers (37–40). Important parasites include (a) protozoa, including toxoplasma and amebae; (b) the pork tapeworm Taenia solium; and (c) schistosoma, a trematode. The organisms can be seen at various stages of their development and involve the leptomeninges and neuropil of the brain and spinal cord.
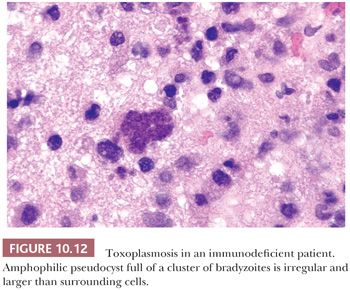
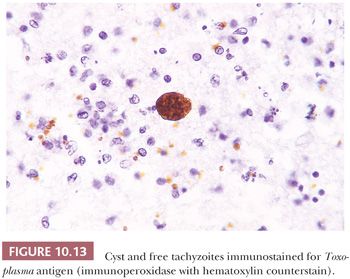
Opportunistic infections are common in AIDS and other immunosuppressed patients. Toxoplasmosis is the most frequent of these infections, and clinically, the differential between toxoplasmosis and lymphoma can be difficult, requiring biopsy if antimicrobial therapy fails (40). A TORCH (toxoplasmosis, other infections, rubella, CMV, and HSV) organism that affects the intrauterine developing brain, toxoplasmosis in an adult likely represents a reactivation of dormant parasites introduced by previous exposure. It presents with a necrotizing encephalomyelitis, typically multifocal by imaging, which is characterized by ill-defined necrotic lesions containing thin-walled cysts filled with bradyzoites (“slow [-growing] animal”), dark-staining dotlike structures representing nuclei, and individual tachyzoites (“fast [-growing] animal”) (Figs. 10.12 and 10.13). IHC or immunofluorescence pinpoints the individual organism not easily found on routine H&E sections. Toxoplasma may coexist with Candida in the same patient as well as with Cryptococcus, CMV, and other organisms. Rarely, toxoplasmosis is complicated by cerebral hemorrhage.
Neurocysticercosis, the result of migration to the CNS of larval forms of the tapeworm Taenia solium, is a common parasitic infection of the CNS found occasionally in people who have been in developing countries. About 50% to 70% of patients with neurocysticercosis present with seizures (37). Some resected specimens are tan, firm nodules containing a cavity with either a cystic region (Fig. 10.14A) or grumous, gray-yellow material. Microscopically, pale eosinophilic membranes separate the cavity from gliotic brain parenchyma with chronic perivascular inflammation. The characteristic invaginated scolex may be seen.
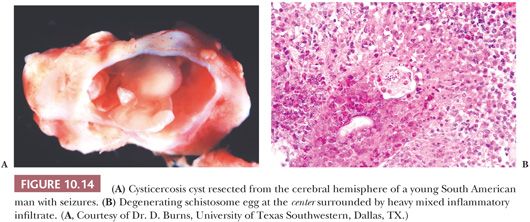
Schistosomiasis in the CNS is the result of migration of eggs of the organisms. The eggs characteristically elicit an intense, granulomatous inflammatory response and can be misinterpreted radiologically as a high-grade neoplasm (Fig. 10.14B).
Amebae including Naegleria and, less commonly, Acanthamoeba, Sappinia diploidea, and Balamuthia (seen mostly in the setting of immunocompromise) can affect the CNS (38,39). The organisms, containing a nucleus (some with a distinct karyosome), have vacuolated cytoplasm and can easily be mistaken for foamy macrophages. They may contain ingested red cells. Macrophages can be distinguished from amebae in the rare amebic brain abscess by their larger and more distinct nuclei and less chromatin clumping than amebae; the organisms are negative for CD68.
Exotic CNS infections often occur in AIDS patients. They include trypanosomiasis and strongyloidiasis.
VIRUSES
A large number of viruses affect the CNS, with or without systemic (especially respiratory system) involvement, with variability among seasons and geographical regions (e.g., presence of specific vectors), and with considerable variation in clinical presentation (41,42). It is highly likely that viral infections have been significantly underreported as the result of difficulty in identifying the etiologic agent.
Most diagnostic tools are clinical and include serologic (serum or CSF, usually with a rise between acute and convalescent specimens), PCR (serum, CSF, or brain), culture on mosquito or mammalian cell line or by intracranial inoculation into suckling mice (blood, CSF, brain tissue), or immunocytochemical (brain and other organ tissues) evaluations. CSF or postmortem brain specimens can be evaluated at reference laboratories or the CDC.
Histologically, most viral encephalitides induce perivascular lymphocytic cuffing. This (an increased number of parenchymal microglial cells) and neuronophagia (“neuron eating”; Fig. 10.7) should raise suspicion for a viral infection. Some viruses produce recognizable inclusions (Table 10.4), many of which can be identified with IHC or ISH. Neuronal inclusion bodies are (a) in nuclei with herpesviruses and measles virus of subacute sclerosing panencephalitis (SSPE) and (b) in cytoplasm in rabies. Oligodendrocyte nuclear inclusions are seen in progressive multifocal leukoencephalopathy (PML) and SSPE. CMV induces inclusions in the nucleus and cytoplasm of endothelial cells, ependymal cells, and glial cells. Many viruses do not manifest inclusions, including HIV, poliovirus, and arboviruses including West Nile virus.

Enteroviruses and Arboviruses
In acute viral meningitis and meningoencephalitis, it is estimated that enterovirus infections (e.g., coxsackievirus, echovirus) cause between 60% and 90% of cases and that arbovirus infections (e.g., Eastern equine, St. Louis, West Nile, etc.) comprise many of the remaining cases (42). Pathologic findings in cases described to date are nonspecific, without characteristic inclusions.
West Nile Virus
This arbovirus, a member of the Flaviviridae family, has recently emerged in the United States and Canada, with mosquito and bird vectors and hosts and involvement of humans (41). Transmission by blood transfusion and organ transplantation has been documented recently. Symptoms are variable, multisystemic, and nonspecific and include fever, myalgia, lymphadenopathy, rash, and headache. Neurologic symptoms include headache, encephalopathy, and diffuse weakness, including acute flaccid paralysis (41,43,44). A proportion of cases are fatal, and fatalities occur principally in the elderly—most with underlying systemic diseases.
The findings are nonspecific. They include a patchy meningitis, encephalitis, and polio-like myelitis with variable but, in some cases, severe involvement of the substantia nigra, brainstem (particularly the medulla), and cerebellum. Perivascular inflammation with evidence of meningoencephalitis and, in some cases, microglial nodules and neuronophagia can be seen (43,44). Anterior horn cells appear to be targeted in some patients, providing a substrate for the weakness (43). Of the involved regions, only the cerebrum and cerebellum are reasonable biopsy candidates. Laboratory diagnosis is based predominantly on serology (45). Additional information on this and other arboviruses is available from the Arbovirus Diagnostic Laboratory in Fort Collins, Colorado.
Herpes Simplex Virus
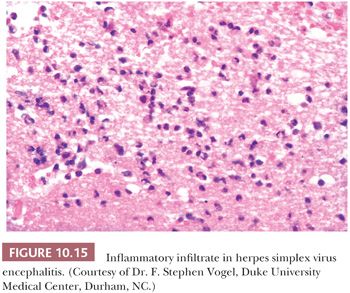
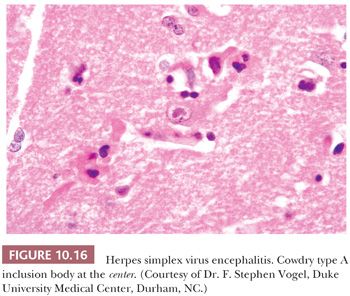
The most common cause of nonepidemic encephalitis is HSV (Tables 10.2 and 10.3). The process is usually localized to the temporal and frontal lobes. When suspected by clinical and imaging findings, the diagnosis of HSV can be made by CSF culture, serology, and PCR evaluation rather than biopsy. Biopsy of the medial portion of a temporal lobe is still carried out in some cases. In the acute and subacute phase, perivascular inflammation, composed predominantly of lymphocytes mixed with macrophages, is accompanied by varying degrees of necrosis and hemorrhage (Fig. 10.15). Inclusion bodies are not easy to find in small brain biopsies (Fig. 10.16). Sensitive and specific methods of identification include ISH and IHC. EM also may demonstrate viral particles but is less sensitive and less specific.
Cytomegalovirus
CMV infection in an adult likely represents reactivation of dormant disease (28,46). Histologic findings vary from cells with typical CMV inclusions with virtually no associated inflammation to severe necrotizing ependymitis and meningoencephalitis. Other lesions may be complicated by CMV infection. IHC, ISH, and PCR are useful techniques for detecting the virus in paraffin-embedded tissue.
Progressive Multifocal Leukoencephalopathy
PML is the result of activation of the DNA papovavirus, predominantly JC virus (rarely SV-40 virus), in the brain. The disease is typically seen in the setting of severe immunodeficiency (e.g., AIDS) and lymphoproliferative disorders and associated treatment; occasional cases occur in the setting of immunosuppression after solid organ transplant and in connective tissue diseases (e.g., lupus) treated with immunosuppression (47).
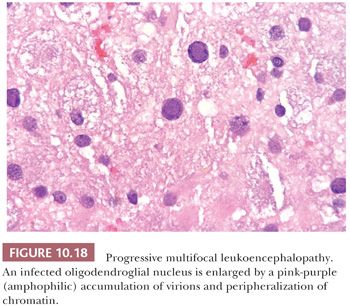
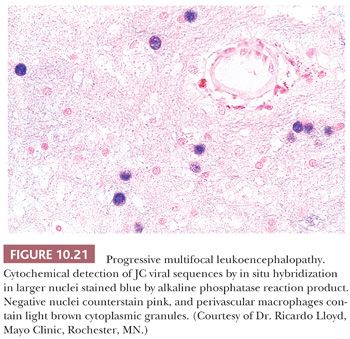
PML manifests as multiple discrete foci of demyelination. Radiographically, it may simulate MS or a mass (Fig. 10.17). Brain biopsy frequently shows an active to subacute process, (a) typically with abundant foamy macrophages, many of which, on LFB-H&E stain, contain myelin; (b) marked reactive gliosis with many astrocytes manifesting bizarre, anaplastic nuclei that can be confused with glioma; and (c) glassy nuclear inclusions in oligodendrocytes that enlarge the nucleus and marginate the chromatin (Figs. 10.18 and 10.19). Perivascular infiltrates of mature lymphocytes are prominent in some cases. The histologic findings of PML are similar in patients with and without AIDS, although bizarre astrocytes are less frequent, and perivascular inflammatory cells more frequent, in AIDS patients. Diagnosis of PML can be confirmed by IHC, ISH (Fig. 10.19), or EM (1,28,48).

Subacute Sclerosing Panencephalitis/Measles
SSPE is a rare disease, with a relatively high incidence in Asia and the Middle East. This slowly progressive disease is caused by a persistent infection of immune-resistant measles virus, usually in unvaccinated or late-vaccinated children. Optimal testing involves identification of antimeasles antibodies, both IgG and IgM in both the CSF and serum. Brain biopsy is rarely performed. When undertaken, viral encephalitis-type changes predominate and there is loss of myelin in subcortical white matter (28). Nuclear inclusions may be detected in neurons and oligodendroglial cells by H&E. IHC or ISH evaluation detects measles virus antigen.
HIV/AIDS
The CNS is significantly affected by HIV- and AIDS-related diseases. Such diseases can be classified as primary (due directly to HIV) and secondary, with opportunistic diseases resulting from the immunodeficiency. Primary and secondary CNS disease were very common in the early era of AIDS, with neurologic symptoms at clinical presentation and CNS abnormalities noted in more than half of patients (3,49). Highly active antiretroviral therapy (HAART) has markedly altered the disease course but not eliminated CNS diseases (50,51).
The primary and secondary disease processes in HIV infection and AIDS produce a spectrum of neuropathology, beginning with cerebritis, meningitis, encephalitis, and vascular pathology and ending with degenerative-metabolic changes and neoplasia.
Primary Manifestations of Human Immunodeficiency Virus
HIV causes several primary CNS diseases, including (a) HIV encephalitis, (b) leukoencephalopathy and vacuolar myelopathy, and (c) lymphocytic meningitis (51). These changes can be seen in biopsies of secondary lesions (e.g., toxoplasmosis or lymphoma) or in cerebral biopsies carried out to assess the etiology of unanticipated cognitive decline and detect a treatable cause of disease (see previous section “High-Risk Infectious Disease Case Considerations” for tissue handling).
The hallmarks of HIV encephalitis, considered pathognomonic of the disease, are multinucleated giant cells in the setting of microglial nodules, which form multiple foci of various sizes within white and gray matter (Fig. 10.20). Nonspecific viral encephalitis changes are also noted, including microglial nodules without giant cells. By IHC, the giant cells can contain abundant HIV markers, including p23.
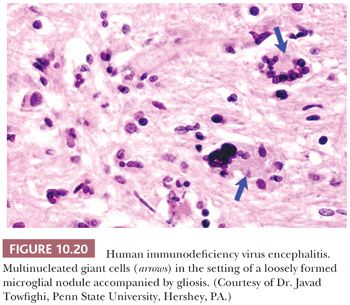
HIV leukoencephalopathy is characterized by diffuse damage to the white matter with loss of myelin, reactive astrogliosis, multinucleated cells, and macrophage infiltrates with scant lymphocytic infiltrates.
Lymphocytic meningitis is yet another primary manifestation of HIV infection and is remarkable for heavy lymphocytic infiltrates within the leptomeninges and perivascular spaces. HIV cerebral vasculitis and granulomatous angiitis may occur with lymphocytic or lymphoplasmahistiocytic multinucleated giant cell infiltration of cerebral vessel walls, occasionally accompanied by necrosis.
Since the onset of HAART, the severity and incidence of primary HIV encephalitis and leukoencephalopathy have decreased (50,51). However, a new form of HIV encephalitis, with severe leukoencephalopathy and intense perivascular macrophage and lymphocyte infiltrates, has been described—postulated to be a response of the revived immune system (50).
Secondary Manifestations of AIDS and Other Immunosuppression: Infections and Neoplastic Processes
A wide range of opportunistic diseases occur secondary to the immunosuppression caused by AIDS as well as in the context of therapy-related immunosuppression (3). The organisms of note include (a) fungi, including C. neoformans (Fig. 10.10) and Candida; (b) viruses, including the JC virus (Figs. 10.17 through 10.21) and CMV; (c) parasites, including toxoplasmosis (Figs. 10.12 and 10.13); and (d) bacteria, including Listeria monocytogenes, H. influenzae, Streptococcus pneumonia, Staphylococcus epidermidis, and Pseudomonas (28). Many viral and parasitic diseases likely represent reactivation of the infectious agent, which had been encountered in the past and held in check by the normal immune system.
Primary CNS lymphoma is a well-recognized neoplasm in AIDS (see “Lymphoma” later in this chapter). Other neoplasms can (rarely) involve the CNS in the setting of immunosuppression, including Kaposi sarcoma and leiomyosarcoma.
HAART allows recovery of the immune system in many patients with AIDS and prevents the immunodeficiency with early treatment. This has markedly decreased the incidence of some secondary, opportunistic disease processes. However, the incidence of PML and lymphoma has not been clearly reduced, and it is possible that PML may shift toward a more chronic disease in treated patients (50).
PRION DISEASES
“PRION” was coined from PROteinaceous INfectious particle with the vowels transposed (4). The counterpart of the pathogenic prion protein is a normal constituent of neurons, white blood cells, and other cell membranes with a still-undefined function (4). When an abnormal tertiary structure is acquired, the protein both (a) becomes resistant to normal protein degradation and (b) has the autocatalytic capacity to convert the tertiary structure of other prion proteins to the pathogenic conformation. As this cycle continues, an increasing percentage of normal protein is converted into the pathogenic prion configuration.
Several forms of prion disease affect man, including (a) the most common sporadic form, CJD, in which no germline mutation is present; (b) familial forms, with germline mutations, including Gerstmann-Sträussler-Scheinker, familial CJD, and fatal familial insomnia; and (c) acquired forms, including kuru (passed from person to person by the eating of the human brain of dead relatives), variant CJD (see the following texts), and iatrogenic CJD (patients receiving growth hormone derived from pooled cadaver pituitaries, contaminated neurosurgical and ophthalmologic surgical equipment, depth electrodes, or contaminated transplanted tissue [dura, cornea]) (28). Sporadic CJD is the most common of these. Prion diseases, known as “transmissible spongiform encephalopathies,” occur sporadically in humans, cows, sheep (scrapie), goats, mink, cats, deer, and elk. Variant CJD appears to have arisen in cows in the setting of use of sheep tissue, including scrapie-containing brain as cattle feed, and was perpetuated by inclusion of affected cow nervous tissue in feed. In man and animals, these diseases are uniformly fatal.
Clinical diagnosis of CJD is difficult and imprecise, especially at early stages. Detection of the 14-3-3 protein in CSF is a valuable but imperfect tool (52). Because many CNS diseases can increase 14-3-3, its utility for CJD may tilt toward a negative 14-3-3 ruling out CJD, rather than a positive 14-3-3 confirming CJD. Ultimately, histopathologic and molecular evaluations of brain tissue are the gold standard for diagnosis (53).
Key considerations for biopsy of a “possible CJD” case include a reasonable chance to find a treatable cause of the cognitive deficits and safety of operating room and pathology staff. Biopsy for the identification of a prion-related disease is not warranted if the clinical suspicion is very high, based on clinical features (rapid progression of the disease), brain imaging studies, electrodiagnostic studies (electroencephalogram), and confirmatory serologic and CSF studies that exclude other explanations (e.g., stroke, vasculitis, meningoencephalitis).
Histologic Appearance
Several histologic changes are evidence of prion disease, including a combination of neuropil vacuolation and gliosis (54). Histologic changes in CJD include (a) vacuolation of the neuropil, with involvement of all six cortical layers in cerebrum or with diffuse involvement of the cerebellar molecular layer (Figs. 10.22 and 10.23); (b) reactive gliosis that is particularly prominent in late stages of the disease (Fig. 10.24); and (c) occasional plaques of amyloid-like material in the neocortex in new-variant CJD. By H&E, a few vacuoles in the neuropil are glassy or lightly eosinophilic rather than clear. Microglial cells are increased in number, but perivascular inflammatory infiltrates are negligible.
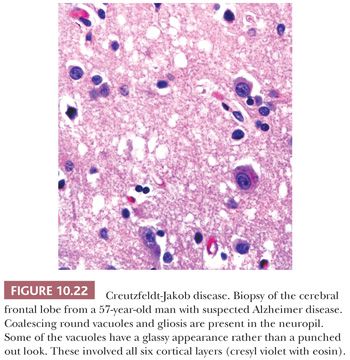
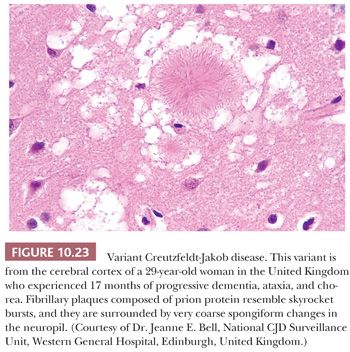
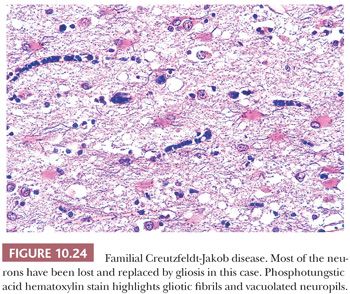
Lack of identification of diagnostic histologic changes from a single biopsy site does not rule out CJD because (a) different subtypes of the disease exist, variable with the mutation in the prion protein, with variably cortical-predominant, basal ganglia–predominant, thalamus-predominant, and hippocampal-predominant disease and (b) considerable variation can be seen from gyrus to gyrus in CJD and at different stages of the disease (55,56). A diagnostic pitfall—diffuse cortical vacuolization—is a common finding in advanced Alzheimer disease. Superficial cortical vacuolization occurs in ischemia and in frontotemporal dementia; the vacuoles in these cases are clear, punched out, and different from those in CJD. Processing artifacts can produce vacuoles that encircle neurons, vessels, and glia; these are larger than CJD vacuoles. Nonetheless, because many vacuoles are not related to prions, molecular diagnosis of prions is needed to confirm suspicions based on vacuoles.
Immunohistochemistry
IHC after protease predigestion of normal prion proteins helps identify protease-resistant prion protein. However, there is a fairly high background labeling among the IHC techniques, challenging their sensitivity and specificity (54,55). Specifically, most available antibodies manifest some degree of background labeling of neurons in control cases, and they can stain neuritic plaques. These should be interpreted only by an experienced observer.
Molecular Diagnosis
The gold standard for identification of the prion protein is molecular evaluation for the protease digestion–resistant protein by Western blot, performed in specialized medical centers and at the NPDPSC in Cleveland, Ohio (53) (see “High-Risk Infectious Disease Case Considerations” section earlier in this chapter). Sporadic CJD lacks a germline mutation, which can be confirmed by sequencing of the prion protein gene to rule out known familial CJD variants.
Variant Creutzfeldt-Jakob Disease/“Mad Cow Disease”
In the mid-1990s, “new-variant” CJD (vCJD) emerged in Great Britain and France, originally called “mad cow disease.” It has been linked by molecular analysis to the eating of meat from cows with bovine spongiform encephalopathy (BSE), in most cases through contamination of the meat with brain and other highly infected tissue during slaughter and butchering. The previously postulated “species barrier” to transmission was broached (54). By early 2003, approximately 135 cases of vCJD had been reported, most cases diagnosed in the United Kingdom.
In the United Kingdom and Europe, the BSE epidemic is now ending and the number of cases is decreasing, thanks to the strict control of animal foodstuff that was the main source of prion contamination (57). BSE in North America (two cases to date) may be at its beginning. Significant measures have been adopted to reduce the risk from recycling of infection via feed, but it remains to be seen whether they are watertight (58).
Despite vCJD being a small fraction of prion diseases, it is a major public health and economic concern. After halting all imports of U.S. beef in 2003, the Japanese government decided in December 2005 to allow importation of beef from the United States derived from young cattle from which specified risk materials are removed (59). Rapid tests for misfolded prion protein in central nervous tissue of slaughter cattle now allows hope for better monitoring (58), provided our hands are on our wallets and our heads out of the sand.
The histologic appearance of vCJD differs from sporadic CJD. In the cerebral and cerebellar cortex, (a) striking stellate, “florid” plaques composed of prion protein, easily seen by H&E, labeled by PAS and anti–prion protein antibodies; (b) coarse vacuolization rather than the fine vacuolation of CJD; and (c) marked gliosis are the most consistent neuropathologic features (Fig. 10.23) (54). These cerebral cortical plaques and coarse spongiform features have not been seen among typical sporadic CJD cases. Similar plaques can be noted in the cerebellum of sporadic CJD (4,56).
Hereditary Prion Diseases
The vacuolization, neuron loss, and gliosis in hereditary prion disorders such as Gerstmann-Sträussler-Scheinker syndrome, fatal familial insomnia, and familial CJD (Fig. 10.24) are similar to sporadic CJD and kuru (28). These diseases are rarely biopsied.
INFLAMMATORY DISEASES
Causes of inflammatory diseases of the CNS are elusive because there is considerable overlap in their clinical and histologic findings. A range of inflammatory diseases affects the brain, including connective tissue diseases, sarcoid, and rheumatoid disease. They variably affect leptomeninges, blood vessels, and brain tissue. Altered mental status and meningeal changes invite biopsy to evaluate etiology and to rule out infections. Optimal sampling that includes dura, arachnoid, and gray matter in the area of a radiologic enhancement improves the chance of reaching a lesion.
GRANULOMATOUS INFLAMMATION
Inflammatory diseases can present a granulomatous appearance, with or without distinct granuloma formation, with or without regions of necrosis. See “Reactive Changes” and “Infectious Diseases” sections earlier in this chapter.
Granulomatous inflammation can also be seen in the setting of dural, leptomeningeal, or parenchymal involvement of the CNS by Wegener granulomatosis and rheumatoid arthritis. Granulomatous meningitis has a predilection for the base of the brain. Neurosarcoidosis is always a diagnosis of exclusion in cases of granulomatous inflammation. Infection must be ruled out first. Sarcoid typically affects the leptomeninges and blood vessels of the brain or spinal cord. Lesions contain lymphocytic leptomeningeal infiltration, slight lymphocytic cuffing of intraparenchymal vessels, scattered small leptomeningeal and parenchymal noncaseating granulomas, and mild reactive astrocytosis, especially in the white matter, with only moderate loss of myelin.
SMALL VESSEL DISEASE AND VASCULITIS
Brain biopsies performed in search of small vessel disease, particularly vasculitis, frequently do not provide a satisfactory correlation between the pathologic specimen and the radiographic data, although step sectioning through the entire block of tissue may occasionally yield diagnostic material. Small vessels are primarily affected in isolated angiitis of the CNS, and both angiography and leptomeningeal plus parenchymal biopsy of a radiographic lesion are essential to the diagnosis (60–62).
Criteria for diagnosis of vasculitis include both (a) vessel wall inflammation and (b) vessel wall damage and destruction. Chronic, healed, or resolving vasculitis manifests irregular vessel wall fibrosis, often with partial damage of elastica. H&E, trichrome, type IV collagen, and elastic stains are keys to confirming vasculitis.
Temporal arteritis is a common disorder that affects small to medium-sized arteries of elderly women. It is often biopsied bilaterally (61). The disease particularly affects the intima, with giant cells a prominent part of the infiltrate. Although uncommon, SLE affects the CNS more frequently than other collagen diseases (62). Rarely, SLE produces microinfarcts and necrotizing vasculitis with thrombosis. Isolated angiitis and arteritis associated with collagen vascular disease, infection, toxins, and granulomatous disease can cause strokes in young people (63).
A multitude of diseases are responsible for individual cases of vasculitis. Cerebral angiitis can be a part of systemic vasculitis in mixed cryoglobulinemia and Wegener disease. Isolated, primary CNS angiitis may include inflammation of very small vessels (60). Angiitis also occurs in association with herpes varicella-zoster virus (64), relapsing polychondritis, and Behçet disease (65). Lymphomatoid granulomatosis is a process that spans inflammatory and neoplastic entities. It is discussed under “Lymphoma.”
Intrathecal methotrexate therapy can cause mineralizing microangiopathy (66). Meningovascular syphilis and associated vasculitis is rising in frequency associated with AIDS. Illicit drugs, such as cocaine, heroin, and amphetamines, affect not only the cardiac vessels but also cerebral vessels and can cause cerebral vasculitis associated with intraparenchymal or subarachnoid hemorrhage (67).
DEMYELINATION
Demyelination results principally from (a) presumed autoimmune etiologies such as MS or acute disseminated encephalomyelitis (ADEM; perivenous demyelination) (68); (b) infectious etiologies, most notably PML (see “Infectious Disease” section earlier in this chapter); and (c) toxic metabolic etiologies, including the leukodystrophies (e.g., Gaucher disease, adrenoleukodystrophy). Survivors of carbon monoxide poisoning and patients with long-standing hypertension and atherosclerosis can have diffuse cerebral hemisphere white matter demyelinating lesions that can histologically mimic the demyelination of MS in surgical specimens. The absence of a margin with less affected brain may help distinguish this from MS plaques, which typically have a sharp margin.
Demyelinating diseases are usually investigated clinically without biopsy (Table 10.2). Exceptions occur with unusual clinical presentations where the clinical diagnosis is unknown or unclear. For instance, active MS lesions can be mistaken radiologically for neoplasm (69). Demyelination due to a primary viral process (PML, HIV leukoencephalitis, CMV, varicella-zoster) is also occasionally seen at biopsy; these entities are considered in the “Viruses” section earlier in this chapter. The leukodystrophies are very rarely seen by the surgical pathologist.
Histology
Primary, acute demyelinating lesions histologically show destruction of myelin and abundant foamy macrophages containing myelin debris and lipid droplets (Fig. 10.25). There is relative sparing of axons in demyelinating disease, although a fraction of axons are lost in chronic MS. In contrast, axonal loss precedes myelin loss in an infarct (Fig. 10.26). The amount of gliosis increases with the chronicity of the lesion.
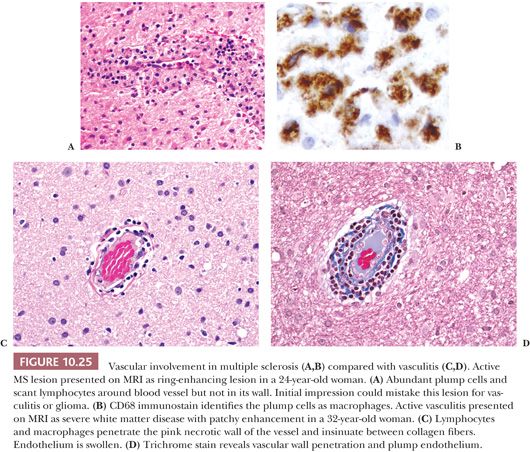
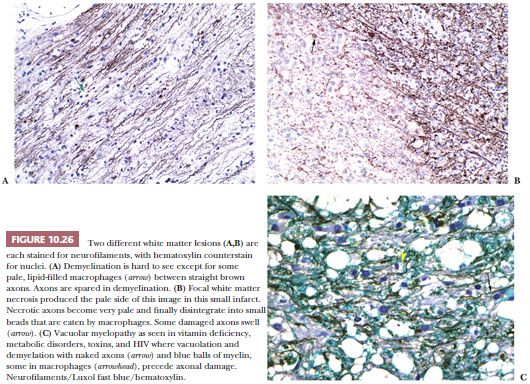
Histochemically, special stains for myelin (LFB-H&E) and axons (Bodian or Bielschowsky silver stain or neurofilament IHC) help to distinguish between (a) demyelination (foamy macrophages containing LFB-positive myelin debris present with axons mostly retained intact in brain tissue) and (b) necrosis, including infarct (axons lost), and Wallerian degeneration (myelin loss is secondary to axonal loss) (Fig. 10.26). GFAP stain helps discern gemistocytic reactive astrocytes from macrophages; CD68 and CD45/CD45RB identify macrophages and lymphocytes. If the lesion was induced by a virus, neuronal or oligodendroglial intranuclear inclusions may be found, particularly at the periphery of the lesion.
Multiple Sclerosis
The major demyelinating disorder of adults is MS. MS is likely an autoimmune disease (70). Despite an immense amount of sophisticated research spanning the past three decades, much remains unknown about this disease. It remains uncertain why some areas of myelin are attacked and others remain spared.
MS manifests acute (or “active”), subacute, and chronic lesions. The former are those that are typically seen at biopsy from a patient suspected of having something other than MS (Fig. 10.25). Due to loss of integrity of the blood–brain barrier, they can have imaging characteristics that mimic a malignant neoplasm (69). MS should be differentiated from other disorders that have similar lesions, including Behçet disease and late-onset forms of leukodystrophies.
Acute Disseminated Encephalomyelitis
Perivenous demyelination is a prominent pathologic finding in postinfectious and postvaccination encephalomyelitis, which is thought to be an autoimmune phenomenon initiated by a viral pathogen, most commonly varicella-zoster and influenza (68,71–73). ADEM is in some cases associated with hemorrhage. It is rarely biopsied. If biopsied, identification of its perivascular pattern of demyelination is important. The demyelination of MS extends beyond vessels to a confluent patch or plaque.
Differential Diagnosis
A demyelinating lesion may be confused with neoplastic disease. Gliosis in the subacute demyelinating lesion can be robust, with large astrocytes with chromosomes spread apart in their cytoplasm (so-called Creutzfeldt cells), mimicking glioma cells (69). In addition, macrophages can accumulate in large numbers in an active region of demyelination to the extent that they mimic glioma, especially oligodendroglioma, or they may mimic hemangioblastoma. Leu-7– and S-100–positive oligodendrogliomas have fewer cytoplasmic vacuoles and more central nuclei than lipid-laden CD68-positive macrophages. Hemangioblastomas are more vascular than oligodendrogliomas (see Fig. 10.5B and Figs. 10.54 and 10.120 in the “Tumors” section later in this chapter). LFB-H&E stain will reveal phagocytosis of myelin in a demyelinating lesion.
CEREBROVASCULAR DISEASES
INFARCT
Infarction of CNS parenchyma is a common pathologic process often associated with clinical stroke. Thrombosis of an atherosclerotic artery or emboli of blood clot or of atherosclerotic plaque commonly causes infarcts. Other etiologies include coagulopathy, amyloid angiopathy, local vasospasm, major changes in blood pressure, CADASIL, mitochondrial encephalopathy with lactic acidosis and strokes (MELAS), and infectious and inflammatory processes. Infarcts can occur in young adults with normal arteries as a result of a severe episode of hypotension or in association with coagulopathy or cerebral vasospasm (74).
Representative portions of the specimens should be submitted for histologic evaluation to optimize evaluation of blood vessels by sectioning perpendicular to the surface of the brain and by including arachnoid, gray matter, and white matter. Vessels are carefully examined for evidence of embolism, arteriosclerosis, atherosclerosis, amyloid angiopathy (see “Amyloid Angiopathy” section later in this chapter), and an infectious–inflammatory process (see “Small Vessel Diseases” later in this chapter). If mitochondrial disease and CADASIL have not been ruled out, then portions of gray matter and gray–white matter junction should be saved for possible EM examination.
In CNS tissue, the age of the infarct is evaluated for active changes and evidence of organization and classified as acute, subacute, or remote. Special stains for axons (Bodian or Bielschowsky silver stain, neurofilament IHC) help to distinguish between necrotic tissue (axons missing or disintegrated) and demyelination (axons retained).
INTRACRANIAL HEMORRHAGE
Clotted blood or hematoma removed from an intracranial location represents a common surgical neuropathology specimen. Such a specimen can be removed from the epidural, subdural, subarachnoid, or intraparenchymal regions. Possible causes of intracranial hemorrhage are summarized in Table 10.5 (75).

The contribution of the surgical pathologist to such specimens lies in careful macroscopic and microscopic inspection of the material and clinicopathologic correlation with (a) evaluation of potential etiologies for intracranial hemorrhage, including clinically unsuspected causes, such as a cryptic vascular malformation, vascular disease, or neoplasm; and (b) approximate dating of the hematoma as acute, subacute, or chronic.
Close macroscopic examination of aspiration collection device contents and resected specimens should be done, submitting for histologic evaluation representative brain parenchyma and other solid regions when present. At least one cassette of hematoma, or more as indicated by gross findings, should be submitted for processing. Bone fragments may represent bone dust generated during the surgery or may be secondary to traumatic injury.
Microscopic examination of the hematoma can roughly distinguish among (a) acute hemorrhage (minutes to hours old, in which white cell nuclei are intact and no evidence of organization is noted), (b) subacute hemorrhage (days old, in which white cell nuclei are karyorrhectic and macrophages ingesting red cells are noted), and (c) remote or chronic hemorrhage (weeks to years old, with hemosiderin-containing macrophages and ingrowth of blood vessels and fibroblasts in the meninges) and reactive astrocytes surrounding the lesion (in parenchymal lesions). Hemosiderin is evident 4 days after hemorrhage and reactive astrocytes 2 weeks after hemorrhage. Both persist indefinitely.
Epidural and Subdural Hemorrhage
Epidural hemorrhage typically results from trauma, most frequently with temporal bone fracture and rupture of the middle meningeal artery. Most such evacuated specimens contain acute hematoma.
Subdural hemorrhages may or may not follow known trauma (76). They are most common in elderly patients with some degree of cerebral atrophy (and bridging vein susceptibility to tear) as well as in patients with systemic cancer (77) and CNS tumors in postoperative settings (76). Acute subdural hematoma is, as a rule, removed by aspiration; contents may come for evaluation in a suction collection device. Subacute or chronic subdural hematomas require excision and histologic evaluation of the subdural membranes to rule out a neoplasm.
Left in situ, subdural hematoma organization continues for weeks. Fibrotic membranes are formed on both sides of a hematoma. The membrane on the dural (outer) side forms first and is usually about 2 to 5 cells thick in a 5-day-old subdural hematoma and approximately 12 cells thick after a week; it eventually may become as thick as normal dura. Movat pentachrome stain distinguishes new membrane collagen, which stains green, from dura (which stains yellow). The membrane on the arachnoid (inner) side of the clot organizes more slowly than the dural membrane and is often not evident in a surgical specimen. Hematomas may rebleed and contain admixed acute, subacute, and chronic changes, confusing the picture. As organization proceeds, abundant hemosiderin-containing macrophages are present. In chronic membranes, eosinophils can be very numerous. Long-standing hematomas may calcify.
Subarachnoid Hemorrhage
Rupture of a saccular aneurysm of the circle of Willis causes subarachnoid hemorrhage, the contents of which may or may not be evacuated (74). Occasionally, these aneurysms also rupture into brain tissue. Any systemic disease with a bleeding tendency can elicit meningeal hemorrhage, but nonaneurysmal bleeds rarely require surgical intervention. Subarachnoid extension from an intraparenchymal hemorrhage that ruptures into the ventricular space is also common.
Hemorrhage into Brain Parenchyma
Intraparenchymal hemorrhage has many potential causes, including iatrogenic hemorrhage, and often accompanies other lesions within the CNS (Table 10.5) (74). A hemorrhage may remain relatively circumscribed within the brain or may present as a rapidly expanding mass that requires rapid removal, resulting in a specimen containing acute hemorrhage. Massive hemorrhage displaces and compresses the surrounding brain tissue. Smaller hemorrhages may be subacute or chronic. With time, rarefaction of the brain parenchyma eventually forms a slit or a cystic cavity lined by gliosis mixed with macrophages, blood pigment deposits, and occasionally partially lysed red blood cells.
The hemorrhages in hypertensive individuals occur most commonly in the putamen, thalamus, cerebrum, pons, and cerebellum. A surgical specimen from a massive hypertensive hemorrhage consists of a large amount of acute hemorrhage, although subacute changes may be seen as well, suggesting previous limited hemorrhage. Portions of edematous brain tissue with petechial hemorrhages and characteristic arterioles with sclerosis as a result of replacement of smooth muscle by collagen are occasionally evident on examination. A clue to hypertensive origin of a hemorrhage is atherosclerotic damage to intact arterioles and small arteries in any brain tissue resected with the hematoma, often with fragmentation and reduplication of elastic tissue or intramural lipidized macrophages (3).
Arteriovenous malformations and cavernous angiomas are vascular malformations with risk of hemorrhage. See the following “Vascular Malformation” section.
Subarachnoid aneurysms occasionally rupture into the brain, but radiography reveals their nature. Other causes of intracerebral hemorrhage include exposure to cold, medications (e.g., anticoagulation therapy), and surgery, particularly cardiac surgery and carotid endarterectomy (75,76).
VASCULAR MALFORMATION
Most vascular malformations are developmental in origin, likely dating to embryonic maldevelopment, with some familial cases identified (76,78). Vascular malformations can occur anywhere in the brain, spinal cord, and associated leptomeninges (Fig. 10.27).
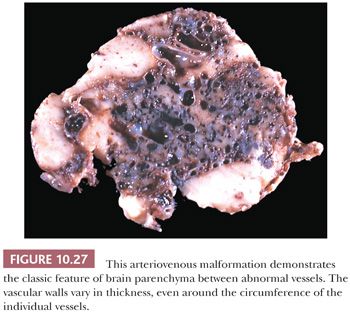
Four types of vascular malformations are generally recognized: capillary telangiectasia, cavernous angioma, arteriovenous malformation, and venous malformation (Table 10.6) (78,79). A dilation of a venous structure, or varix, is one type of venous malformation; the most common example is the great vein of Galen malformation. Sturge-Weber disease (cerebrofacial or cerebrotrigeminal angiomatosis) typically manifests a “port wine”–type hemangioma of the face and an underlying complex vascular malformation that involves the leptomeninges and cortex. The vessels are often mineralized, and cortical mineralization by CT scan is a useful marker of the lesion. Resection is carried out in the setting of intractable seizures.
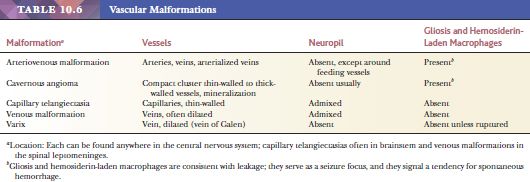
Histologic distinction among these malformations takes into account (a) the size and nature of the vessels involved, including the presence or absence of large arterialized vessels; (b) the presence or absence of nervous tissue interposed among the vessels; (c) the presence or absence of hemosiderin-laden macrophages suggesting leakage of vessels; and (d) reactive gliosis surrounding and intermixed with the vessels (Table 10.6).
Arteriovenous malformations (AVMs) and cavernous angiomas are the two vascular malformations most relevant to the surgical pathologist. Because their vessels tend to leak and induce gliosis, cavernous angiomas and AVMs can present with seizure disorder. Both lesions can also cause spontaneous hemorrhage. Best seen by trichrome or elastic stains, AVMs manifest artery–vein connections with associated arterialization of veins. Arterialization is thickening of a venous wall to the point where it resembles an artery, possibly due to fibrotic reaction to shunted arterial blood. Arteries show irregular duplication or loss of elastin (Fig. 10.28). Thrombi are common. In AVMs discovered without hemorrhage, the risk of hemorrhage has been estimated to be 1.3% to 4.0% yearly, with morbidity following hemorrhage of 53% to 81% and mortality of 10.0% to 17.6%. Surgical treatment offers good prognosis and seizure and hemorrhage control (78,79).
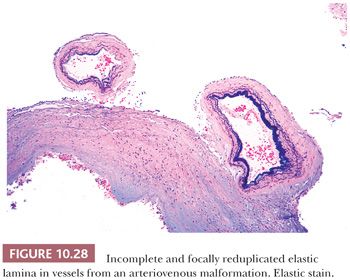
In a small biopsy sample with abundant hemosiderin within a highly cellular matrix, an AVM or cavernous angioma may be confused with the edge of a hemorrhage, infarct, inflammatory focus, or neoplasm. Demonstrate the abnormal vessels with Movat or other elastic and collagen stains, plus the reactive macrophages and glia with CD68 and GFAP, to avoid confusion. Inflamed vessels are common in vascular malformations.
SUPERIOR SAGITTAL SINUS THROMBOSIS
Thrombosis of the superior sagittal sinus is occasionally encountered. Sinus thrombosis followed by venous infarction may occur, usually as a complication of a preexisting infectious disease, connective tissue disease, or in the setting of hypercoagulability. Occasionally, surgical decompression is done. The specimen of clotted blood is treated identical to any hematoma, with histologic evaluation and evaluation for tumor, infectious agents, and other possible etiologic entities.
AMYLOID ANGIOPATHY
Amyloid deposition restricted to the CNS is a frequent form of localized amyloidosis (3). It is due to deposition of a CNS type of beta-amyloid rather than the proteins seen in systemic amyloidosis. Cerebral amyloid (congophilic) angiopathy is seen in elderly patients with or without dementia and in cases of familial (hereditary) amyloidosis (80). Leukoencephalopathy is a feature of amyloid angiopathy. Microinfarcts or small foci of early ischemic damage with hemosiderin deposits may be associated findings in cases of amyloid angiopathy of leptomeningeal and superficial cortical arteries.
Cerebral amyloid angiopathy (CAA) results from deposition of beta-amyloid in the cerebral vessels and associated vessel wall injury. Although CAA may be suspected by H&E stain, based on a smudgy appearance of vessel walls and loss of smooth muscle nuclei, confirmation is required. Amyloid fibrils can be identified by (a) histochemical stains, including Congo red stain (salmon orange) with confirmation by polarization (“Granny Smith apple green”) or thioflavin T stain (requires fluorescence microscopy), or (b) immunostaining for beta-amyloid (Fig. 10.29A–C). CAA can be graded using the Vonsattel criteria as absent, mild (focal deposition of amyloid in vessel walls), moderate (circumferential deposition of amyloid, loss of smooth muscle cell nuclei, and vessel-in-vessel appearance), and severe (vessel wall damage, aneurysm formation). Only severe CAA and fibrinoid necrosis of vessel walls appear to be associated with “lobar” hemorrhage (80). A granulomatous reaction to the amyloid is seen in some cases.
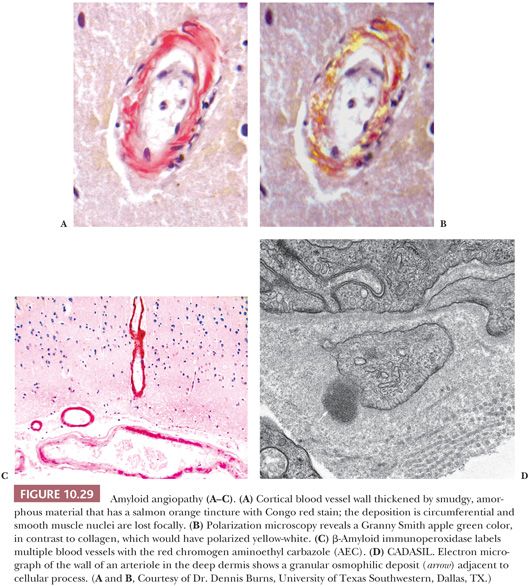
Cerebral parenchyma from hematoma specimens from adult patients must be submitted for careful histologic evaluation for amyloid deposition in blood vessels because amyloid angiopathy may provide an unanticipated etiology for the hemorrhage, and this is an important finding for patient management.
SMALL VESSEL DISEASES
Most of the small vessel disease entities have inflammatory etiologies (vasculitis) and are discussed in the “Inflammatory Diseases” section. CADASIL and arteriosclerosis are vasculopathies but are not inflammatory.
Cerebral Autosomal Dominant Angiopathy with Subcortical Infarcts and Leukoencephalopathy
Presenting with migraine headaches; strokes; and generalized, progressive neurologic deficits including dementia, CADASIL is a rare disorder due to Notch3 gene (chromosome 19) mutations. Mutation evaluation can be performed at commercial laboratories on DNA extracted from white blood cells. Imaging identifies infarct-like lesions and white matter disease (leukoencephalopathy). Although biopsy of the brain has been undertaken for diagnosis, the characteristic vascular changes can be identified in biopsies of more accessible skin and muscle (81,82). By light microscopy, the affected vessels have a thickened appearance with basophilic, granular material seen by H&E stain. This granular material is PAS-positive and it displaces smooth muscle cells, seen best when stained for smooth muscle actin. EM reveals dark, granular osmophilic deposits immediately adjacent to smooth muscle cells (Fig. 10.29). Most of our work has used EM on skin biopsies (82). This approach is adequate when a positive case is identified, but the sampling problem of EM risks missing the lesion.
IHC staining for Notch3 protein deposition has been described. Sampling should be less of a problem with this approach than with EM. In a study of 200 patients, specificity was a greater problem than sensitivity, and family history was needed to increase specificity (81).
Arteriosclerosis
Causes of CNS arteriosclerosis are hypertension, diabetes, and aging. Walls of small arteries are thickened with hyalinized collagen. Perivascular macrophages may be present. They may contain light brown, iron-negative phagolysosomes or dark brown, iron-positive hemosiderin.
VASCULAR DEMENTIA, MULTI-INFARCT VASCULAR DEMENTIA
The concept of so-called vascular dementia is controversial, and detailed discussion is beyond the scope of this chapter (2,4). A review of the topic from the neuropathology perspective concludes that vascular dementia has perhaps 7% to 10% prevalence (83). Vascular dementia is a diagnosis that should not be rendered on a surgical specimen.
TUMORS
The neoplasms of the brain that predominate in adults differ from those seen in children (Table 10.7). More pediatric neoplasms occur in the posterior fossa than in the anterior fossa, whereas the opposite is true of adult neoplasms.
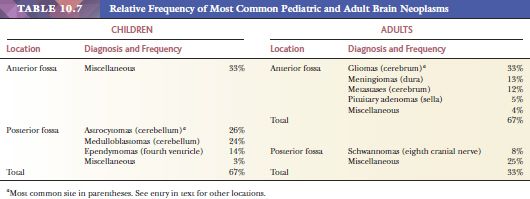
GRADING MALIGNANT POTENTIAL OF TUMORS
The World Health Organization (WHO) sponsors a uniform terminology and grading system of brain tumors (8). Histologic criteria of malignancy are applied by WHO to gliomas and some other tumors. Starting with the most benign as grade I, numeric grades II, III, and IV represent increasing malignancy (Tables 10.8 and 10.9). Some prefer using grades 1, 2, 3, and 4 because these are not affected by double vision. Numeric grades assigned by WHO are used in this chapter.

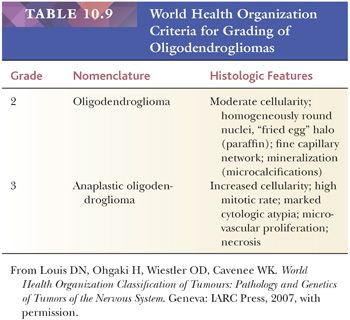
ASSAYS OF PROLIFERATIVE CAPACITY
Proliferation antigens such as Ki-67 epitopes (Fig. 10.30) and proliferating cell nuclear antigen (PCNA) are nuclear antigens that appear during one or more proliferative phases of the cell cycle. Positive nuclei stain brown, and percentage of positive nuclei divided by total nuclei [brown plus blue] can be counted and calculated. This is called a labeling index (LI) or proliferation index (PI). The MIB-1 antibody was the first to detect Ki-67 epitopes in paraffin sections of formalin-fixed tissue. Antibodies that detect Ki-67 in paraffin sections are preferred because they cover all proliferative phases of the cell cycle, and they provide crisp distinction between positive and negative cells. Now, there are antibodies other than MIB-1 that detect Ki-67 in paraffin, and Ki-67 is a more common term. This PI is usually sampled in regions of highest proliferation called “hot spots.” A simple way to do this is to dot hot spots with a pen under a 10× objective and count these regions under a 40× objective (84).
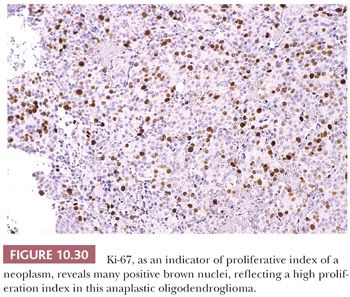
Antibodies to Ki-67 have greater potential in diagnostic pathology than thus far delivered largely due to wide variations in sampling and counting techniques. Even when hot spots are counted, the pathologist may count each hot spot in a field of 100 cells, 1000 cells, or the number of cells seen in a given objective lens. Standardization of the method of counting is needed.
I argue that a standard number of 100 cells is better than larger numbers for brain tumors because the smaller denominator around hot spots accentuates differences in proliferation among low-grade tumors. Low-grade tumors have few or inconspicuous mitoses, and this is where the Ki-67 is most useful. Most high-grade tumors show enough mitoses to estimate proliferation. However, in low-grade tumors, a spectrum of proliferative capacity can be exposed with Ki-67. If I look for hot spots with Ki-67 like I look for single or multiple mitoses in tumors, a denominator of 1000 cells diminishes the impact of the hot spot in judging proliferation. In fact, the argument could be made to count only the number of cells within the periphery of the hot spot. That ratio of positive nuclei to all nuclei would maximize distinctions between high and low proliferative capacities.
There are rare situations where a uniquely large series of patients’ tumors have been processed and counted in a specific manner; these PI correlated with outcome, and a PI of a certain percentage separates most good from bad outcomes. In such cases, reproducing the counting method on your own case makes sense. Distinguishing between central neurocytomas may be one example that applies (8).
Here, we have sufficient tumors of many kinds to establish our own baseline. Ki-67 predicts survival among fibrillary astrocytic neoplasms (85,86). These PIs augment prognostication by standard histopathologic criteria.
Studies have revealed the prognostic value of MIB-1 labeling indices. In one analysis, MIB-1 PI was the only independent predictor of survival (87). Among only grade II astrocytoma patients, MIB-1 distinguishes tumors with good prognosis by their low labeling indices (85). Lower MIB-1 PIs are found in younger patients with glioblastomas, and they have a significantly better prognosis than do older patients (84).
As valuable as it is to assessment of proliferative capacity, the Ki-67 PI is a semiquantitative assay based on a qualitative staining method. Thus, Ki-67 staining methods and counting criteria vary among laboratories. A published value of PIs may not be relevant to a different laboratory. This means that individual laboratories should set their own standards for PIs based on their previous cases. Organizations that standardize assays worldwide might contribute to standardization of Ki-67 staining methods and counting criteria.
Basic points of information about Ki-67 include the ability of reactive and inflammatory changes to produce conspicuous PIs, easily as high as most gliomas of low or intermediate grade (in our laboratory, up to 20%). Thus, a good pathologist and H&E stain is essential for setting the context for interpretation of the Ki-67 PI. A check on nuclear features of Ki-67–positive cells can reveal very important data: rounded nuclei of reactive cells or leukocytes, elongated nuclei in astrocytoma cells mixed with other glioma cells, and more mitoses than seen on other stains (they look like dark granola). Always consider structure and context when interpreting this and other stains.
MOLECULAR EVALUATION
Molecular data regarding tumors can have diagnostic, prognostic, and therapeutic implications (20). For example, combined 1p and 19q deletions in oligodendrogliomas are associated with enhanced chemoresponsiveness and improved prognosis (88). In addition, several other molecular markers are providing useful diagnostic and prognostic information. For example, N-Myc amplification has prognostic significance in medulloblastomas, and SMARCB1 (INI1) studies are useful in the diagnosis of the atypical teratoid/rhabdoid tumor.
A new molecular marker to show prognostic relevance is isocitrate dehydrogenase (IDH) (89). IDH enzymes have a normal function in cells. There are several IDH enzymes. They catalyze the oxidative carboxylation of isocitrate to alpha-ketoglutarate, which results in the important antioxidant: reduced nicotinamide adenine dinucleotide phosphate. IDH mutates frequently in diffuse gliomas. IDH1 mutations occur in gliomas about 10 times more often than IDH2 mutations, and the R132H mutation occurs four-fifths of the time (18). This most common IDH1 mutation is a CGT–CAT transition causing a specific amino acid change from arginine to histidine at codon 132. An antibody developed to this IDH1 mutation shows that about half of oligodendrogliomas and grade II to III astrocytomas are positive by IHC. Positivity for an IDH mutation is associated with better prognosis in diffuse gliomas of intermediate grade, particularly grade III oligodendrogliomas (90).
GLIOMAS
Glioma is a term that embraces astrocytoma, glioblastoma, ependymoma, and oligodendroglioma and their various subtypes and combinations. An important general rule is that gliomas tend to contain GFAP and lack collagen, reticulin, and fibronectin in their parenchyma, distinguishing them from nonglial neoplasms (Fig. 10.31) (89,90). However, oligodendroglioma cells are more variable in their GFAP expression; like other gliomas, they contain less specific glial proteins such as Leu-7 and S-100 protein (89–92). Uncommon variants like xanthoastrocytomas may have parenchymal reticulin. The term glioma is too vague for a final diagnosis of properly sampled permanent histologic sections. However, the term is useful when frozen sectioning obscures the subtype of a glioma or when the central tumor is missing.
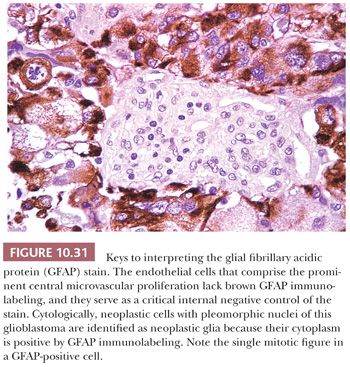
Diffuse gliomas are gliomas that conspicuously invade CNS tissue. Most diffuse gliomas are astrocytomas, oligodendrogliomas, or mixed tumors called oligoastrocytomas. Reflecting their diffuse nature, their grades start at grade II. At grade III, these are considered high-grade gliomas. Highly malignant astrocytomas are grade IV, which most doctors call glioblastomas. It is common for clinicians to reserve the term glioma for only diffuse gliomas, which occasionally causes confusion. I find that knowing I may need to translate “glioma” to mean “astrocytoma and/or oligodendroglioma, grades II to IV,” although not accurate, is sufficient for daily work.
Clinical needs are expanding the pathologist’s role in the interpretation of gliomas. The particular effectiveness of combined procarbazine, chloroethylcyclohexylnitrosourea, and vincristine (PCV) chemotherapy for gliomas with an oligodendroglial component has increased the value of recognizing this component (91). Moreover, molecular methods of defining oligodendrogliomas have been proposed (93).
Postoperative systemic thromboses are a major complication of brain tumor surgery. The pathologist may be able to predict patients likely to encounter this difficulty by noting in the report tumors (usually malignant gliomas) that contain thrombosed vessels (94).
This chapter emphasizes grading consistent with the WHO international classification of tumors (8). There are other grading systems, each with its own merit (95). To avoid confusion, the grade number should state the grading system used. Because it is felt that tumors get worse (progress) via second, third, or multiple focal changes, the highest grade identified in the specimen is the one reported, even if this grade does not predominate. The histologic term low grade applied to astrocytomas and other gliomas does not necessarily imply a benign neoplasm or even a favorable prognosis. Grade II is generally a slow-growing but diffuse glioma that cannot be totally removed and will eventually kill the patient, assuming no other major health issues. The designation benign, implying that, once removed, the neoplasm will not recur, is frequently used only for pilocytic astrocytomas and for certain neuronal tumors and ependymomas. Most of these are considered grade I. Even these tumors need to be in favorable locations and have distinct margins to have a good chance for cure.
Tumor and Tumor Margin
It is important to recognize two types of specimens from a potential glioma (96–100). The first type is the tumor epicenter (Tables 10.10 through 10.12), which has cellular density exceeding that of surrounding brain (Figs. 10.4B and 10.32). To obtain optimal tissue, it is best to monitor each glioma biopsy with cytologic and frozen sections and to indicate clearly specimens which are not satisfactory (Table 10.1; see also the “Intraoperative Consultation” section). Unless the glioma is an unequivocal glioblastoma, grading of malignancy is best saved for permanent sections after optimal evaluation of the entire sample (Tables 10.10 through 10.12). Some gliomas are highly vascular (97). The most confusing gliomas are multicentric, raising the question of metastatic disease (see “Lymphoma” and “Carcinoma” later in this chapter) (99).
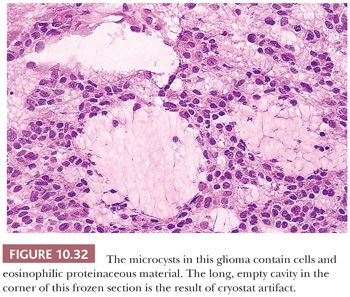
The second type of specimen is CNS parenchyma infiltrated by the margin of the glioma and is a product of the infiltrative nature of many gliomas (8). It is often impossible to determine the histologic grade and type of glioma giving rise to an infiltrative margin of neoplastic glia when only the margin is available for examination. This should be explained to the surgeon during the operation, affording the surgeon the opportunity to obtain a better specimen.
Farther from the glioma itself, neoplastic glia in CNS parenchyma are difficult to find among the gliosis. Often called gliosis versus glioma, this is actually “gliosis with glioma versus gliosis without glioma” because gliosis accompanies glioma cells. Although the short phrase is simpler to say, the problem is finding confirmatory glioma cells or finding some other reason for gliosis.
Features that distinguish glioma cells from gliosis and normal parenchyma include pleomorphism, nuclear hyperchromasia, nuclear cluster formation (Fig. 10.33), nuclear molding, mitoses, and calcifications. Mitoses are rare in low-grade gliomas and are exceptionally rare in gliosis, even when associated with demyelination (69). Mitoses suggest not only that the tumor is a glioma but also that it is a high-grade malignancy. Abnormal variations in size and shape of glial nuclei are more frequent than mitoses in margins of gliomas. In margins of gliomas that overexpress epidermal growth factor receptor (EGFR), this protein can reveal the neoplastic cells and their structure (Fig. 10.33).
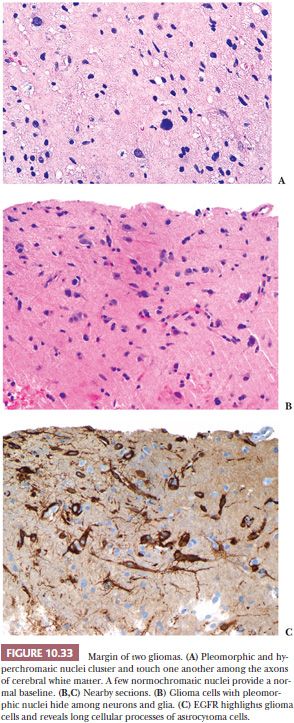
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


