Ideally, all patients with suspected bone tumors should be discussed in multidisciplinary team meetings, including the radiologist and pathologist involved in the diagnosis and the (orthopedic) surgeon and oncologist involved in treatment (2,3). Radiographic imaging is of utmost importance in diagnosing bone tumors, and very often, a reliable histologic diagnosis cannot be made without adequate radiographic information and correlation, emphasizing the importance of multidisciplinary team meetings to establish a final diagnosis (4). The site of the lesion is important as some bone tumors preferentially affect specific areas of the skeleton. Information about the location within the bone is also crucial because some lesions occur predominantly in the metaphysis, whereas others affect specifically the epiphysis or diaphysis (Table 8.1). Moreover, lesions can originate centrally (e.g., enchondroma), eccentrically (e.g., aneurysmal bone cyst), cortically (e.g., osteoid osteoma), or juxtacortically. Multifocality is observed for enchondromas, osteochondromas, fibrous dysplasia, vascular tumors, Langerhans cell histiocytosis, multiple myeloma, and metastases. The formation of matrix (calcification), the destructive pattern, the appearance of tumor margins, cortical damage, and presence or absence of periosteal reaction or soft tissue extension are crucial for interpretation and for narrowing the differential diagnosis (5). One should always start with a conventional radiograph in two planes, which can be followed by magnetic resonance imaging (MRI) or computed tomography (CT) to further define the extent of the lesion. Imaging should always be performed before biopsy.
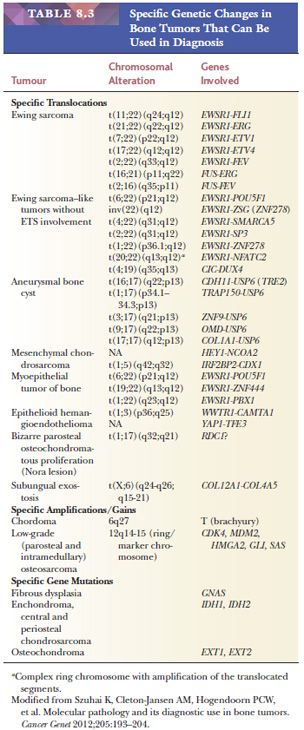
Although most bone sarcomas arise de novo, they can also develop within other conditions, such as Paget disease of bone, or in preexisting benign lesions such as enchondromas and osteochondromas. Also, bone sarcomas occur within hereditary syndromes such as Li-Fraumeni (osteosarcoma) (Table 8.2).
CLASSIFICATION OF BONE TUMORS
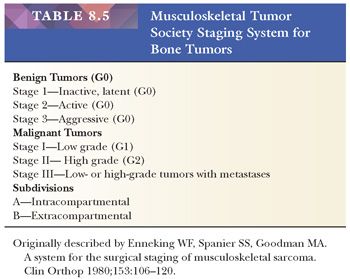
The classification of bone tumors is based on the differentiation of the tumor cells and the extracellular matrix that is being deposited. Main categories include chondrogenic tumors (depositing cartilaginous matrix), osteogenic tumors (depositing osteoid), and osteoclastic giant cell–rich tumors. In addition, fibrogenic, fibrohistiocytic, hematopoietic, notochordal, vascular, myogenic, and lipogenic tumors are distinguished based on their line of differentiation. In addition, there are tumors of undefined neoplastic nature (including, e.g., fibrous dysplasia or aneurysmal bone cyst) and miscellaneous tumors (including Ewing sarcoma, adamantinoma, and undifferentiated pleomorphic sarcoma of bone). Immunohistochemistry plays a limited role in diagnosing bone tumors, whereas molecular diagnostics can be helpful in a subset of tumors (Table 8.3).
The fourth edition of the WHO Classification of Tumours of Soft Tissue and Bone (1) set out to classify bone tumors in four categories, similar to their soft tissue counterparts: benign, intermediate locally aggressive, intermediate rarely metastasizing, and malignant. Benign tumors have a limited capacity for local recurrence, and if they do, recurrence will be nondestructive. Intermediate locally aggressive bone tumors such as atypical cartilaginous tumor (previously called chondrosarcoma grade I) often recur locally and display an infiltrative locally destructive growth pattern, although they do not have an evident potential to metastasize. They require wide excision or application of a local adjuvant (e.g., phenol or cryotherapy) for local control. Intermediate rarely metastasizing bone tumors, such as giant cell tumors of bone, bear, in addition to their locally aggressive behavior, a very small risk (<2%) of distant (lung) metastases. Such behavior cannot be predicted on histologic grounds. Malignant bone sarcomas carry a significant risk of distant metastases, ranging from 20% to 100% depending on the histotype and grade.
METHODS OF BIOPSY
After complete radiologic workup, the biopsy should be performed by the surgeon who will also perform the final surgery (2,6). The location and the technique used for the biopsy will severely affect decisions on the final surgery as the biopsy tract should be considered contaminated in the case of a sarcoma and should be removed. Unwanted contamination of unaffected tissue may therefore require radical resection or amputation, although previously, limb salvage surgery may have been an option. Prior imaging studies should be used to determine the most optimal biopsy tract and to identify the most representative part of the tumor to sample. The choice for open biopsy or core needle biopsy is dependent on the experience and preference of the surgeon and the pathologist as well as the location of the tumor. Core needle biopsy can be performed with ultrasound or CT guidance to sample the most appropriate areas. In experienced hands, core needle biopsy results in an adequate diagnosis in 80% to 90% of patients (7,8).
Frozen sections are recommended to determine whether the biopsy is adequate and representative. Moreover, frozen sections will enable the immediate request of special techniques without the loss of time due to calcification. If an infectious disease is suspected based on the frozen section, one may ask for tissue to be sent for bacterial cultures. If the frozen tissue is not representative, a second biopsy can be taken within the same procedure. The frozen tissue is also valuable for molecular diagnostics, for instance, in cases of Ewing sarcoma.
SPECIMEN HANDLING
Tissue specimens should be sufficiently formalin-fixed and adequately decalcified before embedding in paraffin. Decalcification should be used to dissolve the calcium hydroxyapatite crystals and to keep them out of the tissue, which is usually performed with an acidic solution. Various methods may be used, each with advantages and disadvantages. Proper decalcification should not compromise immunoreactivity. These paraffin sections constitute the cornerstone of histopathologic diagnosis. In the case of curettage, adequate and extensive sampling is required, e.g., to exclude dedifferentiation in chondrosarcoma, to identify an underlying primary lesion in aneurysmal bone cyst, or to identify characteristic areas of giant cell tumor within extensive reactive changes. For handling resection specimens, first, adequate orientation and identification of possibly contaminated areas or compromised resection margins are required. Correlation with preoperative radiographs may be helpful. If they are not available, one could consider preparing a specimen radiograph for orientation purposes and to visualize the relationship of the tumor to resection margins. Then, slices can be cut using a band saw, the tumor can be documented, and macroscopic images should be made. The slice with the largest tumor diameter can be divided in parts and, as such, fully submitted for microscopy (9), which is important, for example, to evaluate response to neoadjuvant treatment or to identify areas with higher histologic grade or dedifferentiation. Alternatively, one could submit one section per centimeter of maximum dimension (10). Extra sections should be obtained of the tumor in relation to vital structures (synovium, vessels, nerves, margins). A small portion of tissue should be snap frozen whenever possible, for molecular assays.
GRADING OF BONE SARCOMAS
Bone tumors vary considerably in their clinical and biologic behavior. Histologic grading is an attempt to predict the behavior of a malignant tumor based on histologic features (1). There is no widely accepted grading scheme for bone sarcomas. The 2010 College of American Pathologists (CAP) Bone Tumor Protocol advocates a pragmatic approach based mainly on histologic type or subtype (Table 8.4) (1,10). Conventional osteosarcoma and most osteosarcoma subtypes are classified as high-grade tumors with the exception of low-grade intramedullary (central) osteosarcoma and parosteal osteosarcoma (both low grade) and periosteal osteosarcoma (intermediate-grade). Grading has not proven useful in predicting the behavior of conventional giant cell tumor of bone, but malignancy in giant cell tumor is considered high grade. Certain chondrosarcoma histologic subtypes are considered high grade (mesenchymal and dedifferentiated chondrosarcoma) or low grade (clear cell chondrosarcoma).

Conventional chondrosarcoma and sarcomas of soft tissue type are the two exceptions to this rule. In conventional chondrosarcoma, the grading system as proposed by Evans et al. (11) is now universally accepted (1). Under this system, conventional chondrosarcoma is divided into three grades based on cellularity, cytologic atypia, and mitotic figures (see the following discussion). Soft tissue tumor–type sarcomas such as leiomyosarcoma of bone can be graded according to the Fédération Nationale des Centres de Lutte Contre le Cancer (FNCLCC) grading system, as is commonly used for soft tissue sarcomas.
STAGING OF BONE SARCOMAS
Although staging systems have been described for both benign and malignant bone tumors, they are more useful in the management of malignant bone tumors. Bone sarcomas are staged using either the Musculoskeletal Tumor Society (MSTS) system, first described by Enneking et al. (12), or the American Joint Committee on Cancer (AJCC) system. The MSTS and AJCC system have many features in common.
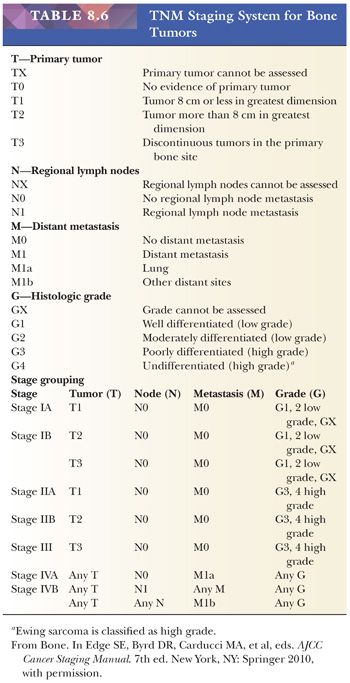
The MSTS staging system also includes benign tumors (Table 8.5). The sarcomas are classified as stage I, II, or III based on their histologic features (stage I: low grade, stage II: high grade) or metastatic spread to regional lymph nodes or distant sites (e.g., the lung) (stage III). For staging purposes, grade 1 is considered low grade (G1), whereas grade 2 and grade 3 are considered high grade (G2) (10). In addition, tumors are also classified on the basis of whether they are confined to the bone (intracompartmental; type A) or whether they extend outside the bone (extracompartmental; type B).
The AJCC recommends four histologic grades based on differentiation, which cannot be readily applied to bone tumors as discussed earlier. Therefore, a grade 1 or 2 tumor in the AJCC system is equivalent to a stage I tumor in the MSTS system; grade 3 or 4 is equivalent to MSTS stage II. In addition, the TNM staging system includes histologic subtype, size, continuity, and grade as well as local and distant spread to estimate the prognosis of the patient (Table 8.6). Lymph node metastases are, however, rare in bone sarcomas.
CHONDROGENIC TUMORS
Osteochondroma
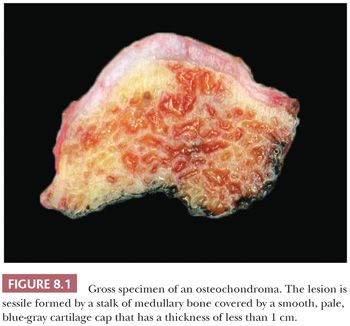
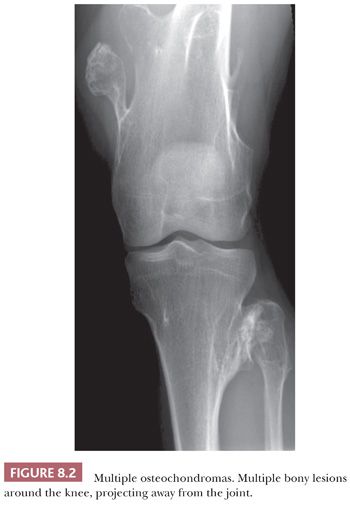
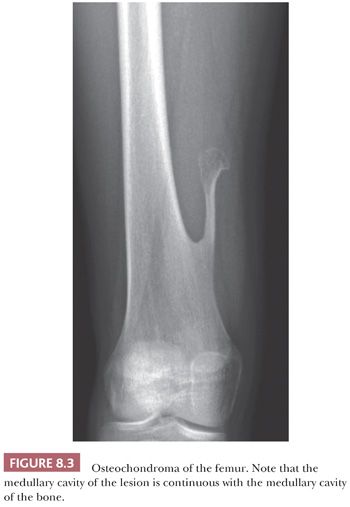
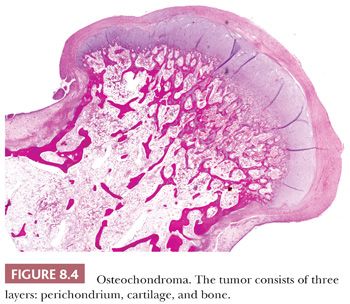
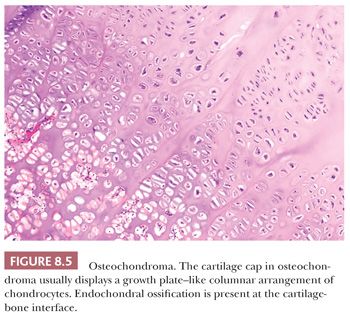
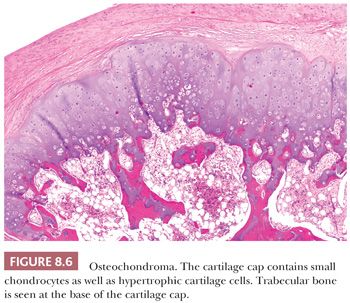
Osteochondroma is a benign cartilage-capped bony projection arising on the external surface of bone containing a marrow cavity that is contiguous with that of the underlying bone (1). The base of osteochondroma can be broad (sessile) or small (pedunculated) (Fig. 8.1). The lesion is entirely surrounded by periosteum. Osteochondromas are the most frequent cartilaginous lesions, most often found on the femur (21%), humerus (17%), and tibia (11%) (13). Osteochondromas develop and increase in size during the first decade of life and stop growing when growth plates close during puberty. The majority of osteochondromas are asymptomatic. Sporadic (solitary) osteochondromas are six times more common than those occurring within the hereditary multiple osteochondroma syndrome (previously called hereditary multiple exostoses) (Fig. 8.2). Multiple osteochondromas is an autosomal dominant condition caused by mutations in EXT1 or EXT2 (14). Radiologically, the osteochondroma is characterized by its typical localization at the transition of the metaphysis to the diaphysis, projected away from the joint, the contiguity of the cortex of the underlying bone with the cortex of the osteochondroma stalk, and the presence of spongiosa within the stalk (Figs. 8.1 and 8.3). Histologically, three layers are seen: perichondrium, cartilage, and bone (Fig. 8.4). The organization of chondrocytes in columns, as is normally observed in the growth plate, can usually also be recognized in the cartilaginous layer of osteochondroma (Fig. 8.5). Endochondral ossification is seen at the cartilage-bone interface. Cellularity is variable, depending on the age of the patient. Binucleated cells, calcification, necrosis, nodularity, and cystic changes can be seen (Fig. 8.6). Malignant transformation to secondary peripheral chondrosarcoma occurs in 1% to 5%. There are no accepted histologic criteria to distinguish osteochondroma from low-grade secondary peripheral chondrosarcoma (4); the imaging studies and gross macroscopic documentation of the thickness of the cartilaginous cap are extremely important, as a cartilage cap exceeding 1.5 to 2 cm in adults is suggestive of malignancy (1,4).
Enchondroma
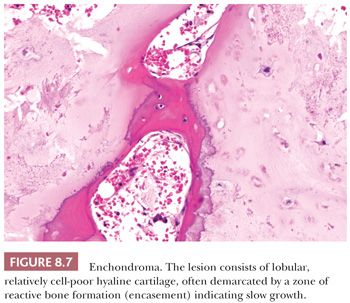
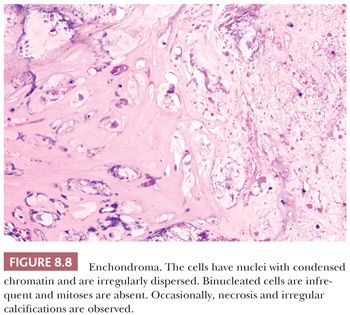
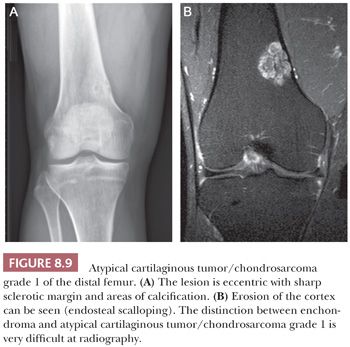
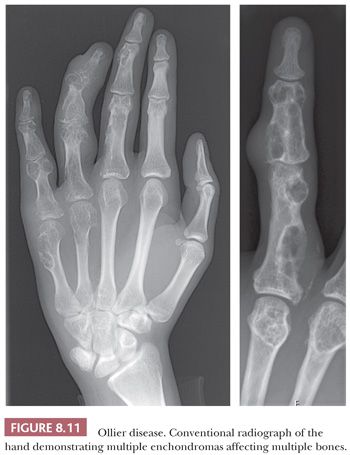
Enchondroma is a benign hyaline cartilage neoplasm of medullary bone. Most tumors are solitary; however, they occasionally involve more than one bone or site in a single bone (1). Enchondromas are relatively common and have a wide age range. Enchondromas are located in the long bones, and 50% of all cases are located in the bones of the hands and feet (15). Chondrosarcomas are only rarely seen at this location (16). The size of an enchondroma is usually less than 3 cm. Radiologically, the presence of matrix mineralization in the form of “popcorn” calcifications is most typical. The edge of the lesion is most often lobular and sharply circumscribed. Histologically, enchondroma consists of lobular, relatively cell-poor hyaline cartilage, often demarcated by a zone of reactive bone formation (encasement) (Fig. 8.7). The chondrocytes have nuclei with condensed chromatin and are evenly dispersed. Binucleated cells are infrequent and mitoses are absent. Occasionally, degenerative features such as ischemic necrosis or calcifications are found (Fig. 8.8). The distinction between an enchondroma and atypical cartilaginous tumor/chondrosarcoma grade 1 can be difficult radiologically (Fig. 8.9) (17). Moreover, this distinction is also difficult on histology (18), causing high interobserver variability (19,20). Mucomyxoid matrix degeneration and entrapment of preexisting host bone are most predictive of atypical cartilaginous tumor/chondrosarcoma grade 1 (Fig. 8.10) (20). Immunohistochemistry or molecular diagnostics cannot help in this differential diagnosis. Radiologic presentation, localization, and age should also be taken into account. For instance, in the phalanges, the matrix of enchondromas can contain more myxoid features, and the lesion may be more cellular. At this location, the main characteristics of malignancy are the presence of mitoses, cortical breakthrough, and soft tissue involvement (16). Malignant transformation of enchondromas is thought to be extremely rare (<1%) (13). Multiple enchondromas are seen in the context of nonhereditary Ollier disease (multiple enchondromas with a unilateral predominance) (Fig. 8.11) and Maffucci syndrome (multiple enchondromas combined with [spindle cell] hemangiomas) (21) caused by somatic mosaic mutations in the IDH1 or IDH2 gene (22,23). In these conditions, the risk of malignant transformation is increased to 18% to 46%, depending on the extent and localization of the lesions (24).
Periosteal Chondroma
Periosteal chondroma is a rare, benign hyaline cartilage neoplasm of bone surface that arises from the periosteum and is often located on the fingers or proximal humerus (1). On radiographic imaging, a small (<5 cm), sharply demarcated tumor is seen on the surface of bone, with erosion of the exterior cortex, whereas marrow invasion is absent. In contrast to enchondroma, the mixture of low and highly cellular areas, the presence of binucleated cells, hyperchromasia of the nuclei, myxoid change of the matrix, and nuclear pleomorphism are histologically tolerated, whereas these features would lead to a diagnosis of chondrosarcoma if the tumor were located outside the periosteal area (25,26). The distinction between a periosteal chondroma and a periosteal chondrosarcoma is mainly based on the presence of cortical destruction in the case of a periosteal chondrosarcoma. Furthermore, periosteal chondrosarcomas are usually more than 5 cm in size (27). Approximately 70% of periosteal chondromas harbor mutations in IDH1 (28).
Osteochondromyxoma
Osteochondromyxoma is an extremely rare, benign, sometimes locally aggressive chondroid and osteoid matrix–producing tumor with extensive myxoid change, occurring in approximately 1% of patients with Carney complex (29). Carney complex is an autosomal dominant condition with multiple neoplasms characterized by cardiac, endocrine, cutaneous, and neural myxomatous tumors as well as a variety of pigmented lesions of the skin and mucosae.
Subungual Exostosis and Bizarre Parosteal Osteochondromatous Proliferation of Bone (Nora Lesion)
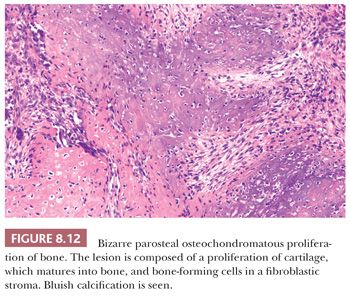
Bizarre parosteal osteochondromatous proliferation (BPOP) is an osteochondromatous proliferation involving the surface of the small bones of the hands and feet (30,31). There is a wide age range with no sex predilection. In contrast to osteochondroma, the lesion is not contiguous with the underlying cortex and medulla. Histologically, bone and spindle cells are seen, with hypercellular cartilage with enlarged chondrocytes and irregular bone-cartilage interfaces (Fig. 8.12). A characteristic, strikingly blue color can be found, even after decalcification (“blue bone”). There is marked proliferative activity. In contrast to chondrosarcoma, the cartilaginous matrix is typically fibrillary. The maturation to trabecular bone with spindle cells in the intertrabecular spaces that is often present may lead to confusion with parosteal osteosarcoma (30), although in contrast to parosteal osteosarcoma, BPOP never shows atypia of the fibrocytes (31). The lesion has a pronounced tendency to recur (about 55%), whereas metastases have not been reported. A recurrent chromosomal translocation t(1;17) has been found (32).
Subungual exostosis is a benign osteochondromatous proliferation involving the distal phalanx (1). The histology resembles BPOP, although the classic blue bone is lacking, and the lesion is more organized with a gradual maturation from peripheral spindle cell proliferation, to hyaline cartilage, to trabecular bone. Subungual exostosis, as the name implies, is typically located at the distal phalanx, whereas BPOP is not located at that site. In contrast to osteochondroma, which is extremely rare on the digits, the cartilaginous matrix is highly fibrillary. Subungual exostosis is characterized by a recurrent chromosomal translocation t(X;6), rearranging the COL12A1 and COL4A5 genes in chromosome bands 6q13-14 and Xq22 (33), upregulating expression of the IRS4 gene (34).
Synovial Chondromatosis
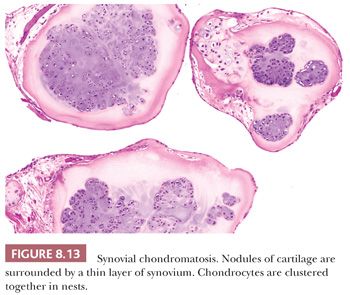
Synovial chondromatosis is a rare benign neoplasm presenting as multiple hyaline cartilage nodules in the subsynovial tissue (1). There is a male predominance, with an average age of 41 years (35). The condition is monoarticular, with the knee most often affected (35). At radiography, the presence of opaque bodies and/or stippled calcification is found (35). At macroscopy, multiple gray-white nodules are seen. Histologically, cartilaginous nodules are seen surrounded by a thin layer of synovium. Characteristically, the chondrocytes in the nodules cluster together in nests (Fig. 8.13) (35). The cartilage is hypercellular with plump hyperchromatic nuclei, with numerous binucleate cells (35). Inflammation and ossification can be found. Malignant transformation to chondrosarcoma is extremely rare and is suggested by the presence of atypical chondrocytes not arranged in nests, mitoses, crowding and spindling of nuclei at the periphery of lobules, myxoid change, necrosis, and, most importantly, permeation of bone trabeculae (35).
Chondromyxoid Fibroma
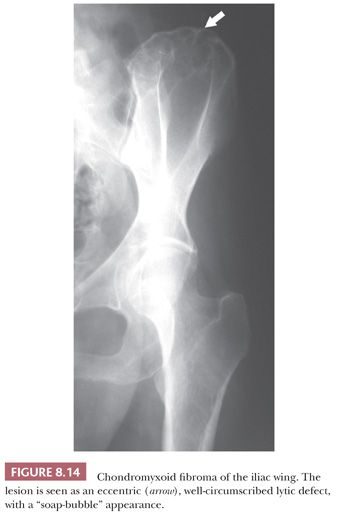
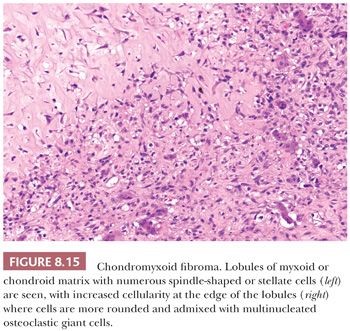
Chondromyxoid fibroma is a very rare benign cartilaginous neoplasm composed of lobules of spindle-shaped or stellate cells with abundant myxoid or chondroid intercellular material (1). The lesions can affect any bone. Patients are adolescents or young adults, predominantly males. Radiologically, the lesion presents as an eccentric, oval, radiolucent lesion in the metaphysis (Fig. 8.14) (36). Histologically, the periphery of the lobules are most cellular, containing spindle-shaped or round cells, mixed with a varying number of multinucleated giant cells (Fig. 8.15). In the center of the lobules, more stellate cells are found lying in a mucomyxoid matrix. Nuclear enlargement and hyperchromasia can be seen, sometimes causing confusion with high-grade central chondrosarcoma. Mitoses are unusual but can be seen. High-grade chondrosarcoma, however, has a different clinical and radiologic presentation, and the architecture is different. Moreover, expression of CD166, cyclin D1, and p16 would favor chondromyxoid fibroma (37). The cells in chondromyxoid fibroma express S100, and the cells at the periphery of the lobules are myofibroblastic, expressing MSA and SMA. Chromosome 6 aberrations are frequent (38). These variable rearrangements were all shown to target the GRM1 gene, as they all result in upregulation of GRM1 (39). High GRM1 expression was shown to be specific for chondromyxoid fibroma, suggesting that aberrant glutamate signalling is involved in its histogenesis (39).
Chondroblastoma
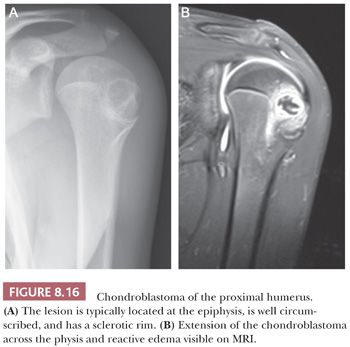
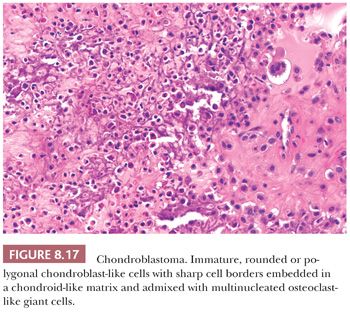
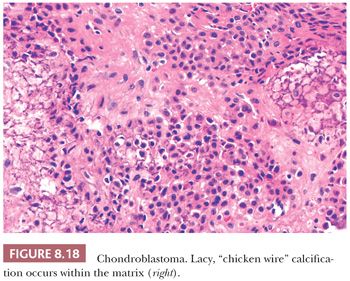
Chondroblastoma is a rare benign, chondroid-producing neoplasm composed of chondroblast-like cells usually arising in the epiphyses of skeletally immature patients. The lesion has a slight predominance in males. In general, its behavior is benign and the lesion is treated by curettage, although very rare (~1%) metastases have been described after biopsy or curettage (intermediate, rarely metastasizing category according to WHO 2013) category (1). At radiography, chondroblastoma presents as an eccentric radiolucent lesion, with a sharp sclerotic margin and areas of calcification, characteristically located in the epiphysis of the long bones (Fig. 8.16). Histologically, the tumor is highly cellular with relatively uniform, immature, rounded, or polygonal chondroblast-like cells with sharp cell borders, intermingled with multinucleated osteoclast-like giant cells, which are arranged individually or in clusters (Fig. 8.17). There is a variable deposition of extracellular matrix, both cartilaginous and osteoid, and small areas of cartilage matrix with focal net-shaped (“chicken wire”) calcification (Fig. 8.18). Occasionally, (nonatypical) mitoses are found. In about 15% of cases, the formation of a secondary aneurysmal bone cyst is observed (40). The cells express S100 and may express keratins. Distinction from giant cell tumor can sometimes be challenging (Table 8.8); the morphology of the mononuclear cells and the extracellular matrix are helpful distinguishing features. Immunohistochemistry is not particularly helpful in this distinction, although recently, the expression of DOG1 by clusters of cells in chondroblastoma has been reported (41). Very recently, a consistent mutation in H3F3B (p.Lys36Met), which is one of the two genes for histone H3.3, was found in chondroblastoma (42). Interestingly, giant cell tumor of bone harbors alterations in H3F3A, the other gene for histone H3.3 (42).
Conventional Chondrosarcoma
Chondrosarcoma is a locally aggressive to malignant cartilaginous matrix–producing neoplasm. The term chondrosarcoma is used to describe a heterogeneous group of lesions with diverse morphologic features and clinical behavior (Table 8.7) (1). Chondrosarcoma is mainly seen in adults in the third to sixth decade of life, with equal gender distribution. They usually arise in the thoracic, pelvic, and long bones. Surgery is the mainstay of treatment as chondrosarcomas are highly resistant to chemotherapy and radiotherapy (43).
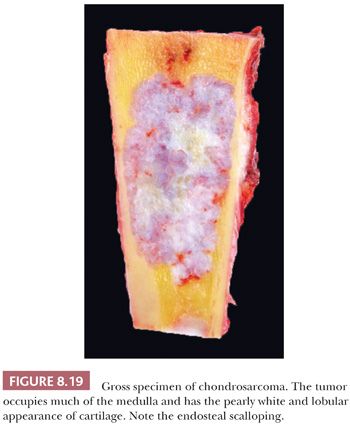
Primary central chondrosarcoma arises centrally in previously normal bone as opposed to secondary chondrosarcoma arising in a preexisting benign cartilaginous tumor. Primary central chondrosarcomas account for about 75% of all chondrosarcomas, and the majority is well differentiated. Central chondrosarcoma can be found in almost all parts of the skeleton arising from endochondral ossification. Radiologically, it can be difficult to distinguish enchondroma from chondrosarcoma (Fig. 8.9) (17). In chondrosarcomas of higher grade, the edge of the lesion is no longer lobular and sharply demarcated, and cortical destruction and soft tissue extension can be seen. At macroscopy, a typical translucent, lobular, blue-gray or white cut surface is seen (Fig. 8.19). Primary central chondrosarcoma is divided into three histologic grades, as described in the following section.
Mutations in the isocitrate dehydrogenase genes IDH1 and IDH2 are found in 38% to 70% of respectively primary central chondrosarcomas (23,28). Hot spot mutations are exclusively found at the IDH1 R132 and the IDH2 R172 positions, of which the R132C mutation is most common in chondrosarcoma. Mutation analysis can be helpful for instance in the distinction between high-grade chondrosarcoma and chondroblastic osteosarcoma (28,44). Mutations in IDH are early events because they occur in enchondromas and therefore do not distinguish between enchondroma and chondrosarcoma (23,28). The (epi)genetic events causing malignant transformation of enchondroma to chondrosarcoma are so far unknown, although upon progression, chondrosarcomas become aneuploid and karyotypes become complex with increasing histologic grade, with aberrations in the p53 and Rb pathways.
Secondary central chondrosarcoma is a chondrosarcoma arising in a preexisting enchondroma. Although the risk of malignant transformation in solitary enchondroma is estimated to be very low (<1%), patients with Ollier disease and Maffucci syndrome have a markedly increased risk of developing secondary chondrosarcomas (18% to 46%) (24). IDH mutations are found in 86% of secondary central chondrosarcomas. Radiologic, macroscopic, and histologic features are identical to primary central chondrosarcoma.
Secondary peripheral chondrosarcoma arises in a preexisting osteochondroma. A minority of chondrosarcomas (up to 15% in referral centers) develop through malignant transformation within the cartilage cap of a preexisting osteochondroma (Fig. 8.20) (13,45). Secondary peripheral chondrosarcoma is mainly found in the ilium (19%), scapula (15%), tibia (12%), femur (11%), pubic bone (10%), and ribs (10%). The radiologic and gross macroscopic documentation of the thickness of the cartilaginous cap is extremely important in the distinction of osteochondroma and secondary peripheral chondrosarcoma, as a cartilage cap exceeding 1.5 to 2 cm in adults is suggestive of malignancy (1,4). Histologically, central and peripheral chondrosarcomas are similar and three grades are applied, which correlate with outcome, as described in the following section.
Although for osteochondroma formation inactivation of the EXT1 or EXT2 genes is required, most secondary peripheral chondrosarcomas arising in osteochondroma are EXT wild-type, indicating that EXT is not important for malignant transformation and that the nonmutated cells in osteochondroma are more vulnerable for as yet unknown (epi)genetic changes promoting them to become malignant (46).
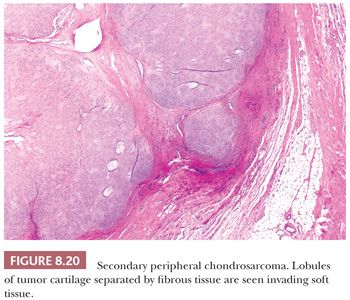
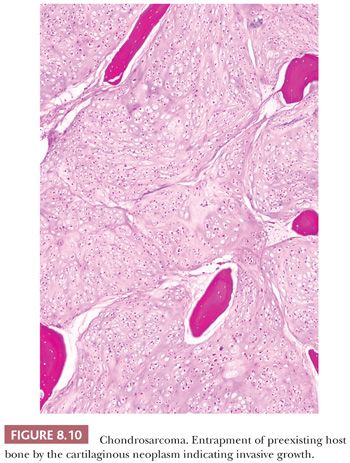
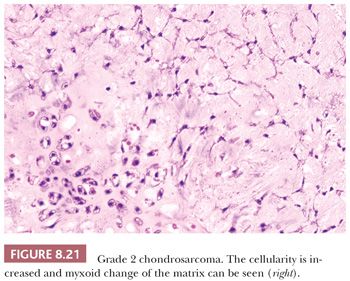
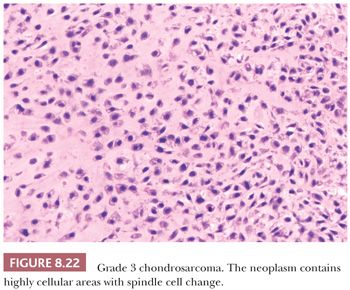
Histologic Grading of Conventional Chondrosarcoma. Both conventional central and peripheral chondrosarcomas are histologically classified in three grades using the criteria of Evans et al. (1,11). Histologic grade is thus far the best predictor of clinical behavior. What was previously called central chondrosarcoma grade 1 has now been renamed atypical cartilaginous tumor (1), and the histology shows an abundance of predominantly hyaline cartilaginous matrix in which chondrocytes with small, condensed nuclei are seen. Atypical cartilaginous tumor/chondrosarcoma grade 1 (ACT/CS1) is distinguished from enchondroma by its higher cellularity, irregular distribution of the cells, and the occurrence of binucleated cells. An important criterion is the growth pattern, in which the presence of entrapment (of preexisting lamellar bone) (Fig. 8.10) (18,20) and the absence of encasement (Fig. 8.7) favor ACT/CS1 over enchondroma (18). Also, the presence of more than 20% mucomyxoid change of the matrix is an important argument for the diagnosis of ACT/CS1 (20). For the distinction between osteochondroma and secondary peripheral chondrosarcoma, no reliable histologic criteria could be identified, indicating the diagnosis can only be made in a multidisciplinary team setting, taking the thickness of the cartilaginous cap into account (Fig. 8.20) (4). In grade 2 chondrosarcomas, the cellularity and nuclear atypia are increased, and mitoses, although scarce, can be seen. The matrix becomes more mucomyxoid (Fig. 8.21). In grade 3 chondrosarcomas, the cellularity is high, with nuclear atypia, and mitoses are even more easily identified (Fig. 8.22). At the periphery of the lobules, spindle cell change may occur (11). The 10-year survival of grade 1, 2, and 3 tumors is 83%, 64%, and 23%, respectively (11). ACT/CS1 has a relatively high chance of recurrence in case of incomplete resection but only rarely metastasizes, whereas 10% of grade 2 tumors and 71% of grade 3 tumors metastasize (10,47). However, in approximately 13% of recurrent chondrosarcomas, progression to a higher grade of malignancy is observed (11,48). Because considerable heterogeneity may occur in chondrosarcoma, it is important to select several areas to be submitted for histology. The tumor needs to be graded based on those areas demonstrating the highest grade. Also, histologic grading has been shown to be subject to interobserver variability (19,20). An absolute correlation between histology and biologic behavior is lacking. This is especially true for the chondrosarcomas of the phalanges, in which the histology can be highly malignant but the behavior indolent. Grading of phalangeal chondrosarcomas therefore does not seem useful (16).
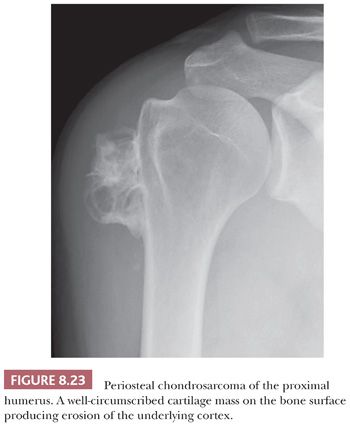
Periosteal Chondrosarcoma
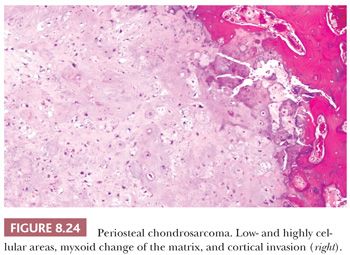
Periosteal chondrosarcoma is a malignant hyaline cartilage tumor, which occurs on the surface of bone and originates from the periosteum (1). The tumor is extremely rare (<1% of chondrosarcomas) and is mainly found in the metaphysis of the femur or humerus in young adults, preferentially in males, with the highest incidence in the fourth decade of life. Radiologically, the tumor presents as a large lobular mass (often >5.5 cm) located on the cortex, with irregular borders (Fig. 8.23) (49). The histology is very similar to conventional chondrosarcoma, displaying well-differentiated lobular cartilage with extensive areas of secondary calcifications and endochondral ossification. Osteoid or bone directly formed by the tumor cells is absent. Bone marrow invasion is rare. The demarcation with the soft tissue is indistinct, occasionally with infiltrative growth into the soft tissues. Erosion and invasion of the underlying cortex is usually seen and distinguishes periosteal chondrosarcoma from periosteal chondroma (Fig. 8.24) (26). Moreover, periosteal chondrosarcoma often has a larger tumor diameter (>5 cm) (27). Periosteal chondrosarcomas also need to be distinguished from secondary peripheral chondrosarcomas, in which the medulla of the underlying bone is contiguous with that of the stalk of the lesion, whereas in periosteal chondrosarcoma, the underlying cortex can usually still be recognized. In the differential diagnosis, periosteal (chondroblastic) osteosarcoma should also be considered. Periosteal osteosarcoma, which is often of the chondroblastic subtype, has a much worse prognosis (26,50). Within periosteal osteosarcomas, by definition, direct deposition of osteoid by tumor cells is seen. Moreover, the cartilaginous areas in periosteal osteosarcoma display considerable nuclear pleomorphism, whereas this is usually limited in chondrosarcoma.
Dedifferentiated Chondrosarcoma
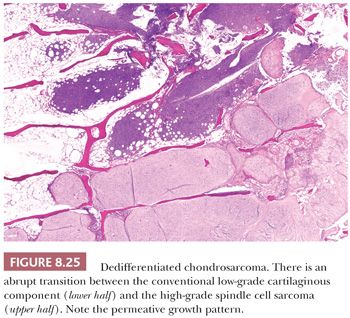
Dedifferentiated chondrosarcoma is a highly malignant variant of chondrosarcoma, characterized by the occurrence of two clearly defined components: a well-differentiated cartilaginous tumor, either an enchondroma, ACT/CS1, or grade 2 chondrosarcoma, juxtaposed to a high-grade noncartilaginous sarcoma. There is a histologically abrupt transition between the two components (Fig. 8.25) (1). Dedifferentiated chondrosarcoma comprises about 10% of all chondrosarcomas. Dedifferentiated chondrosarcomas are usually central, as dedifferentiated chondrosarcoma arising in osteochondroma is extremely rare (51). Patients are often older than 50 years, and males and females are equally affected. The lesion is most often located in the pelvic bones, proximal femur or humerus, distal femur, or the ribs. The prognosis is unfavorable, irrespective of treatment (52). The metastases typically only show the high-grade anaplastic component. The sharp transition between the two components is an important criterion in the distinction between grade 3 chondrosarcoma (in which the cells at the edge of the lobules show a gradual transition toward more cellular spindle-shaped cells) and dedifferentiated chondrosarcoma. The noncartilaginous, anaplastic component can show characteristics of undifferentiated pleomorphic sarcoma, osteosarcoma, fibrosarcoma, leiomyosarcoma, rhabdomyosarcoma, or angiosarcoma. The demonstration of the presence of a low-grade cartilaginous component is crucial for the distinction; however, on biopsy specimens, one of the components can be absent. Radiologic correlation is therefore crucial in the diagnosis. Radiology of the resection specimen can be of help, and extensive sampling for histologic examination is required. Macroscopically, the different components can often clearly be identified at the cut surface of the resection specimen. The rare genetic reports on dedifferentiated chondrosarcoma demonstrate that both components share identical genetic aberrations with additional genetic changes in the anaplastic component (53), indicating a common precursor cell with early diversion of the two components. Approximately 50% of dedifferentiated chondrosarcomas carry mutations in IDH1 or IDH2 in both components (23,28,54). The demonstration of an IDH mutation may confirm the diagnosis of dedifferentiated chondrosarcoma in case the biopsy specimen only shows the anaplastic component, whereas the radiology suggests the presence of a low-grade cartilaginous component as well.
Mesenchymal Chondrosarcoma
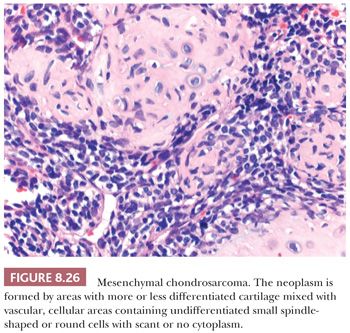
Mesenchymal chondrosarcoma is a rare malignant neoplasm characterized by a biphasic pattern that is composed of poorly differentiated small round cells and islands of well-differentiated hyaline cartilage (1). Mesenchymal chondrosarcoma comprises about 2% of all chondrosarcomas. The tumor can be found at all ages, with a peak in the second and third decades, without a gender predilection (55). The majority (65% to 86%) of mesenchymal chondrosarcomas is located in the skeleton, especially in the lower extremities such as the femur; facial bones; and the pelvis (55). A minority of cases (14% to 34%) are of extraosseous origin, in which the meninges and lower extremities are most often affected (55). Mesenchymal chondrosarcomas are in general of high-grade malignancy, with a 10-year survival of less than 30% (56). In contrast to conventional chondrosarcoma, metastases to lymph nodes and other bones are common (56). Radiologically, a lytical and destructive lesion is seen, with spotlike calcifications, resembling a conventional chondrosarcoma (55). Histologically, mesenchymal chondrosarcoma is characterized by the presence of areas with usually well-differentiated cartilage mixed with highly vascular, cellular areas containing undifferentiated small spindle-shaped or round cells with scant cytoplasm, with a hemangiopericytoma-like vascular pattern (Fig. 8.26) (55). Mitoses can be observed. The cartilaginous areas are often relatively small, sharply demarcated, and cytologically benign or low grade (55). The small cell component is positive for SOX9 and variably for CD99 and can show expression of desmin. The undifferentiated small cell areas may morphologically resemble Ewing sarcoma, and both tumors contain cytoplasmic glycogen (57). However, by definition, cartilage is absent in Ewing sarcoma. Furthermore, small cell osteosarcoma and dedifferentiated chondrosarcoma should also be considered in the differential diagnosis. Irregular fine trabecular deposition of osteoid, characteristic of small cell osteosarcoma, and a sharp demarcation between the two components, characteristic of dedifferentiated chondrosarcoma, are absent in mesenchymal chondrosarcoma (55). A recurrent HEY1-NCOA2 fusion has been identified in mesenchymal chondrosarcoma (58). When the cellular, undifferentiated component predominates, the tumors can be sensitive to chemotherapy and radiation (56).
Clear Cell Chondrosarcoma
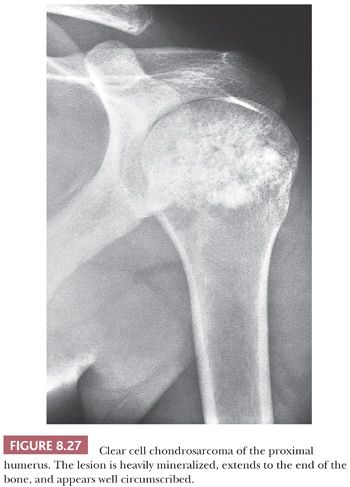
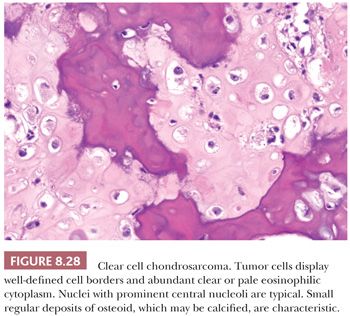
Clear cell chondrosarcoma is a rare, low-grade variant of chondrosarcoma with a predilection for the epiphyseal ends of long bones. It is characterized histologically by bland clear cells, resembling the hypertrophic cells in the growth plate, in addition to hyaline cartilage (1). The tumor comprises about 2% of all chondrosarcomas and is often found in adults, with a slight male predominance and a peak in the third and fourth decades (59). Similar to chondroblastoma, the proximal parts of the femur, humerus, or tibia are most often affected. Radiologically, the lesion is located in the epiphysis, can be lytic, and demonstrates expansile growth, often with a sharp border (Fig. 8.27) (59,60). Macroscopically, the tumor is distinguished by the absence of the typical glassy white-gray aspect of conventional chondrosarcoma but is red and granular, occasionally with cavities. Histologically, round cells are seen with large round nuclei with centrally located nucleoli and clear empty cytoplasm with sharp cell borders; the tumor cells are positive for S100 (Fig. 8.28) (59,61). Throughout the lesion, metaplastic woven bone is regularly deposited, and osteoclast-like giant cells are present. Mitoses are rare. Focally, a hyaline cartilaginous matrix or even areas of more conventional chondrosarcoma can be observed. Areas of cystic degeneration resembling aneurysmal bone cyst are often seen. Owing to its epiphyseal location, chondroblastoma is often raised in the differential diagnosis, although clear cells are rare or absent in chondroblastoma. Metastasis of (renal) clear cell carcinoma should be considered in the differential diagnosis, in which case immunohistochemistry for keratin and PAX8 may be helpful. IDH mutations are absent (23). Clear cell chondrosarcomas often recur after curettage or marginal excision, whereas metastases are rare.
OSTEOGENIC TUMORS
Osteogenic tumors are characterized by the deposition of osteoid or bone by the tumor cells. Malignant transformation of benign bone-forming lesions is extremely rare. The production of bone by the tumor cells should be distinguished from metaplastic or reactive bone formation in nonosteogenic tumors or lesions (e.g., clear cell chondrosarcoma) or from growth plate–like endochondral ossification (e.g., in osteochondroma, or fracture callus).
The aspect of the bone matrix plays an important role in the classification of osteogenic tumors. Histologically, using polarized light, bone matrix can be divided into lamellar, woven, and combinations of lamellar and woven bone. In lamellar bone, the lamellae of bone are organized in parallel layers. Lamellar bone is a sign of slow growth rate characterizing mature bone tissue. Woven bone is disorganized and the collagen matrix is irregularly arranged in a random fashion. Woven bone is primitive and immature and much weaker than lamellar bone. This type of mineralization is seen during development, in fracture callus, in bone-forming tumors, and in conditions characterized by a highly accelerated rate of bone turnover (such as Paget disease and hyperparathyroidism). The bone matrix of lamellar and woven bone can be produced by osteoblasts, showing prominent lining by active (swollen) or inactive (flat) osteoblasts (such as in osteoblastoma), or can be directly formed through fibro-osseous metaplasia in which the collagen fibers of the background are contiguous with the metaplastic bone (such as in fibrous dysplasia) (62).
Osteoma
Osteoma is a benign tumor composed of compact mature bone arising on the surface of the bone. When developing in the medullary cavity, it is known as enostosis (1). It typically occurs in bones formed by membranous ossification, such as calvarial, facial, and jawbones, with a predilection for the paranasal sinuses. Osteoma is rare outside the skull. It predominantly affects adults between the ages of 30 and 50 years. Multiple osteomas are seen in the setting of the autosomal dominantly inherited Gardner syndrome (characterized by adenomatous polyposis coli, osteomas/enostosis, impacted supernumerary teeth, odontomas, desmoid-type fibromatosis, and epidermoid cysts) (63). Gardner syndrome is a variant of familial adenomatous polyposis, caused by heterozygous mutation in the APC gene.
Osteoma is usually asymptomatic. However, when large or in the paranasal sinuses, osteomas can cause obstruction or local swelling. Radiologically, osteoma generally appears as a sharply marginated ossified mass projecting from a bony surface. Histologically, predominantly lamellar mature bone is seen, varying from compact to more trabecular. The trabeculae are lined by active (plump and rounded) and inactive osteoblasts within a well-vascularized moderately cellular fibrous stroma. If complicated by inflammation, usually due to an obstruction sinusitis, bone remodeling and less mature bone can be found. Outside the craniofacial skeleton, parosteal osteosarcoma should be considered in the differential diagnosis. Asymptomatic lesions do not require treatment because they are slow growing and pursue an indolent clinical course (64).
Osteoid Osteoma
Osteoid osteoma is a benign bone-forming tumor characterized by small size (<2 cm), limited growth potential, and disproportionate pain, usually responsive to nonsteroidal anti-inflammatory drugs (NSAIDs) (1). In general, osteoid osteoma occurs in children and adolescents. However, it can occasionally be seen in older individuals. It is more common in males. Characteristically, patients complain of localized and progressive pain that is usually worse at night and often relieved by NSAIDs. Although every bone can be affected, the metaphysis or shaft of long bones and the cortex, particularly of the proximal femur, are most often affected (1).
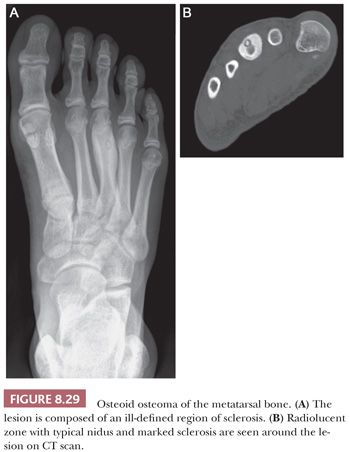
Imaging studies are usually diagnostic because osteoid osteoma consists of dense cortical sclerosis surrounding a radiolucent nidus. The nidus is best seen on CT scan because it can be obscured by the sclerosis in plain radiographs (Fig. 8.29). The lesion is usually less than 1 cm and, by definition, less than 2 cm; for lesions larger than 2 cm, osteoblastoma should be considered.
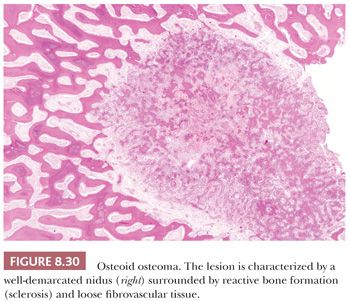
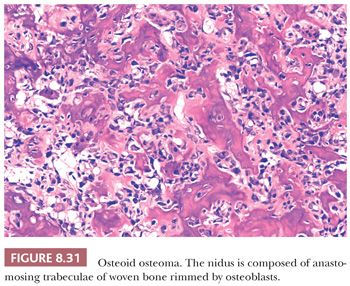
The gross appearance of osteoid osteoma is typical, as the nidus is small, red, and granular and therefore softer than the surrounding sclerotic tissue. However, nowadays, when the clinical presentation and the radiology are typical, minimally invasive techniques are used to ablate the tumor, which is not always preceded by a successful and representative core needle biopsy. The interface between the nidus and the surrounding bone is very sharp (Fig. 8.30). Histologically, the nidus is composed of small, often closely packed irregular trabeculae of woven bone, lined by swollen osteoblasts and occasional small, osteoclast-like giant cells. The stroma is loose and well vascularized and contains swollen fibroblasts, osteoblasts, and osteoclast-like giant cells (Fig. 8.31). The nuclei are not atypical but rather uniform with an open chromatin pattern and a nucleolus (active cells). The prognosis is excellent and recurrences are uncommon. Some lesions have been reported to disappear without surgical therapy (1).
Osteoblastoma
Osteoblastoma is a benign bone-forming neoplasm, greater than 2 cm by definition, which produces woven bone spicules, bordered by prominent osteoblasts (1). It is rare, accounting for about 1% of all bone tumors and is more common in males. Although osteoid osteoma is most common in the long bones, osteoblastoma preferentially affects the posterior elements of the spine and sacrum. At appendicular sites, the proximal femur, distal femur, and proximal tibia are most common (1). The vast majority of cases are intraosseous (medullary), but a small percentage can occur at the surface of the bone in a periosteal (peripheral) site. In the spine, osteoblastomas and osteoid osteomas have similar symptoms, such as back pain, scoliosis, and nerve root compression. However, for osteoblastoma, the pain is usually not worse at night and is less likely to be relieved with NSAIDs (65).
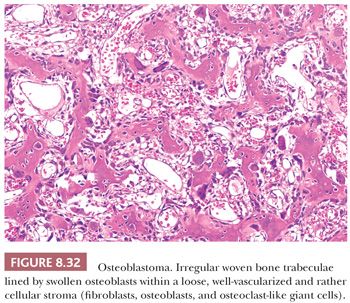
Radiologically, osteoblastomas are lytic and well-circumscribed lesions, greater than 2 cm (generally 3 to 10 cm). In those cases with secondary aneurysmal bone cyst, the lesions are often much larger. At macroscopy, osteoblastoma appears red or red-brown, due to its high vascularity, with a pushing border. Histologically, osteoblastoma is identical to osteoid osteoma, comprising interconnected trabeculae of woven bone; the distinction is made based on size. The trabeculae are haphazardly arranged, lined by a prominent, single layer of osteoblasts (Fig. 8.32). Osteoblasts may have mitoses, but they are usually not atypical (1). A highly vascularized stroma is present between the trabeculae. Diffusely scattered osteoclast-type, multinucleated giant cells are often observed. Sometimes, large, plump osteoblasts with a larger nucleus containing a prominent nucleolus can be seen, accompanied by mitoses, causing confusion with osteosarcoma. These lesions are referred to as epithelioid osteoblastoma (previously known as aggressive osteoblastoma). Distinction from osteosarcoma can be made by careful examination of the interface between the lesion and preexisting bone, which is not infiltrated in the case of osteoblastoma. Moreover, cartilage is usually absent in osteoblastoma unless there is (micro)callus formation. There is no evidence that epithelioid osteoblastoma has a worse prognosis than conventional osteoblastoma (1). In addition, degenerative nuclear atypia can be seen in the so-called pseudomalignant variant in the absence of mitoses. Moreover, the osteoid matrix can sometimes merge to form larger masses, possibly causing confusion with sclerosing osteosarcoma, in which atypia can be limited. Again, entrapment of preexisting bone should be absent in osteoblastoma. Osteoblastoma has a higher rate of recurrence than osteoid osteoma and requires surgical management.
Osteosarcoma
Osteosarcoma is a collective name given to a group of sarcomas in which the hallmark is the production of osteoid directly by the malignant tumor cells. In addition to osteoid, the malignant cells may also produce cartilage and collagen. Osteosarcoma can be subdivided according to the site of origin (intraosseous, surface or soft tissue), grade of malignancy, and differentiation (e.g., fibroblastic, chondroblastic, etc.).
Low-Grade Central Osteosarcoma
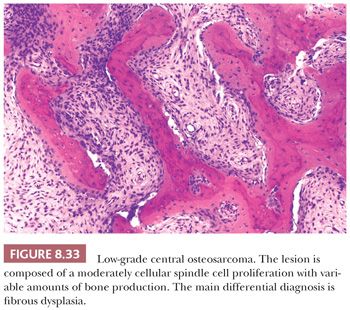
Low-grade central osteosarcoma is defined as a low-grade, malignant, bone-forming neoplasm that arises within the medullary cavity of bone (1). It is rare and accounts for approximately 1% to 2% of all osteosarcomas. In general, it is located in the long bones, predominantly in the distal femur and proximal tibia. Radiologically, a large lytic lesion with focal aggressive features may be seen. At macroscopy, a gray-white, firm, and gritty tumor is seen in the medulla of bone. Low-grade central osteosarcoma is histologically composed of an abundant hypocellular to moderately cellular fibrous tissue proliferation with variable amounts of osteoid production (Fig. 8.33). The spindle-shaped tumor cells show little mitotic activity and only slight nuclear atypia and are arranged in fascicles or interlacing bundles. Giant cells as well as foci of cartilage can be seen. The main differential diagnosis is fibrous dysplasia and desmoplastic fibroma. Bone formation is, however, absent in the latter. In the majority of low-grade central osteosarcomas, some degree of cortical disruption and/or periosteal reaction and soft tissue extension are observed, which is helpful in the distinction from its benign mimics. Moreover, low-grade central osteosarcoma may permeate surrounding cortical and/or cancellous bone, which is not seen in benign bone tumors. These features can, however, be difficult to assess on biopsy specimens, in which case molecular diagnostics may be helpful. Low-grade central osteosarcomas have a simple genetic profile with supernumerary ring chromosomes comprising amplification of chromosome 12q13-15, including the cyclin-dependent kinase 4 (CDK4) and murine double-minute type 2 (MDM2) gene region. Immunohistochemical MDM2 or CDK4 amplification can be detected by fluorescence in situ hybridization (FISH). In addition, expression of MDM2 and CDK4 is useful to differentiate low-grade osteosarcomas from benign fibrous and fibro-osseous lesions including fibrous dysplasia (66,67). Additionally, the overall lack of chromosomal aberrations and the low frequency of TP53 mutations differentiate low-grade central osteosarcoma from the other osteosarcoma subtypes. Low-grade central osteosarcoma has an excellent prognosis, although progression to high-grade osteosarcoma may occur (in 10% to 36%).
Parosteal Osteosarcoma (Low Grade)
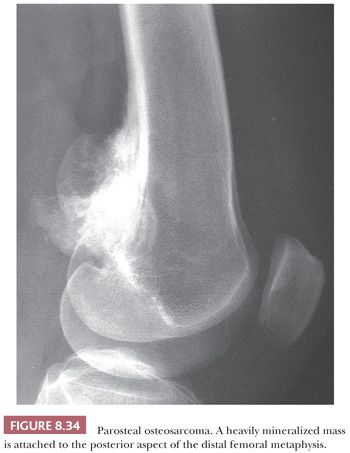
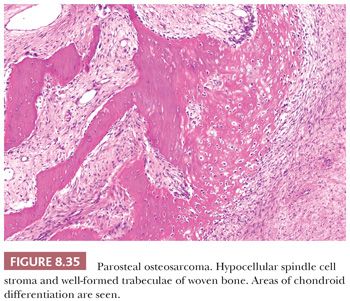
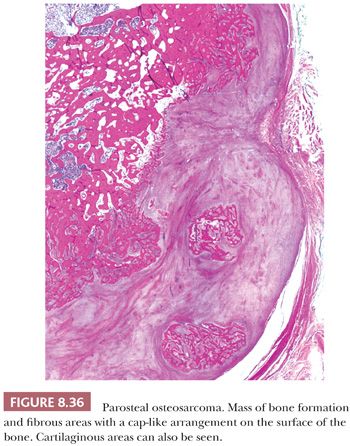
Parosteal osteosarcoma is a rare, low-grade, bone-forming lesion that arises on the surface of a long bone. It accounts for approximately 4% of all osteosarcomas (1). Around 70% of parosteal osteosarcomas are found on the distal posterior femur. The tumor often presents with a long history of a localized, slow-growing, painless mass in young adults, with a slight female predominance. The radiologic presentation can be very typical with a heavily mineralized mass attached to the cortex of the femur, tibia, or humerus (Fig. 8.34). Parosteal osteosarcoma is histologically very similar to low-grade central osteosarcoma, being composed of a moderately cellular fibrous stroma with formation of long, thin, and irregular bone trabeculae that are rather evenly spaced, oriented in a parallel fashion, and show ramifications (Figs. 8.35 and 8.36). The structure varies from fine lamellar to woven. The fibroblast-like cells are usually arranged in long fascicles and have rather uniform nuclei with coarse granular chromatin. Mitotic activity is low. Cartilaginous differentiation can be seen in approximately 50% of the tumors, either as nodules or with a cap-like arrangement. The latter causes confusion with osteochondroma, although osteochondroma has a characteristic columnar arrangement of chondrocytes, whereas malignant spindle cells at the periphery should be absent (68). If cartilaginous areas predominate (>50%), a diagnosis of periosteal osteosarcoma should be considered. Other lesions in the differential diagnosis are reactive parosteal or periosteal lesions (e.g., bizarre parosteal osteochondromatous proliferation, fracture callus) or soft tissue lesions adherent to the bone surface (e.g., myositis ossificans, desmoid-type fibromatosis). Similar to low-grade central osteosarcoma, more than 85% of parosteal osteosarcomas contain supernumerary ring chromosomes involving amplification of the 12q13-15 region (69), and immunohistochemistry or fluorescence in situ hybridization (FISH) detecting MDM2 or CDK4 amplification may be helpful in the diagnosis (66).
Progression to a high-grade sarcoma (dedifferentiation) occurs in approximately 15% to 25% of the tumors (70). These areas resemble conventional osteosarcoma or a high-grade spindle cell or undifferentiated pleomorphic sarcoma. Dedifferentiation may occur at the time of diagnosis (synchronous) or at recurrence (metachronous) and worsens the prognosis.
Periosteal Osteosarcoma (Intermediate Grade)
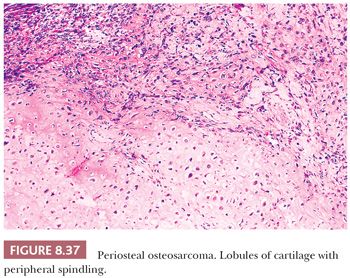
Periosteal osteosarcoma is an intermediate-grade, malignant cartilage and bone-forming neoplasm arising on the surface of bone (1). It accounts for less than 2% of all osteosarcomas. Periosteal osteosarcoma is predominantly located in the diaphysis or diaphysis/metaphysis. It is usually seen as a sessile, anterior, medial lesion, which may almost wrap around the circumference of the bone, especially the distal femur and proximal tibia. Symptoms of pain and swelling are usually of short duration. Radiologically, the lesion is predominantly radiolucent but often contains focal areas of mineralization in the soft tissue component. There are spicules of reactive bone oriented perpendicular to the underlying bone cortex, which may be focally eroded or thickened. At macroscopy, the glistening gray-bluish cartilaginous matrix can be found, with ossification at the base of the tumor. The outer margin is usually well circumscribed by a pseudocapsule. Invasion in the underlying medullary cavity is highly unusual. Histologically, nodules of atypical cartilage predominate, occasionally with myxoid change, separated by malignant spindle-shaped tumor cells (Fig. 8.37). Lacelike and woven bone is formed by the tumor cells and intermixed with cartilaginous elements and can sometimes be difficult to recognize (Fig. 8.38). In addition, reactive periosteal bone formation, extending as spicules from the base of the tumor to its outer edge (periosteal surface), can be found in between lobules of tumor but may be destroyed by tumor growth. Secondary endochondral ossification can also be seen. It can be difficult to distinguish periosteal osteosarcoma from periosteal chondrosarcoma, which is important as periosteal chondrosarcoma has a better prognosis. The radiologic presentation is important in the distinction as well as the fact that in periosteal chondrosarcoma, the cartilage is usually more lobular and well differentiated, whereas osteoid deposition by spindle cells is absent. Moreover, periosteal chondrosarcoma may contain IDH mutations, whereas for periosteal osteosarcoma, no consistent genetic alterations have been reported. Reported overall 5- and 10-year survival is 89% and 83% respectively, and local recurrence is associated with an increased risk of metastases (71).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


