The design of most bone marrow biopsy needles is similar and the principle and technique for using them is basically the same (6). Needles with various-sized lumina are available, including those of small gauge designed specifically for pediatric patients. Users must have a detailed knowledge of the anatomy of the iliac crest and be well trained in the recommended technique for a specific biopsy needle. Inexperienced individuals should perform trephine biopsies under the supervision of a physician thoroughly familiar with the procedure until they have gained adequate experience. The marrow biopsy is a safe procedure in the hands of persons adequately trained who exercise good judgment and proper caution.
Several methods for processing the marrow specimen provide excellent material for microscopic examination. The techniques used are determined by individual preference and suitability for a specific laboratory. The methods described here are those preferred by the authors (4,7).
The biopsy procedure may be performed in a patient’s hospital room or in an outpatient clinic using sterile technique. Local anesthesia is used on the skin and periosteum over the intended biopsy site. Prebiopsy sedation is often helpful in pediatric patients and adults who have a high level of anxiety about the procedure. The posterior iliac crest is the preferred anatomic site for the biopsy; the anterior iliac crest may be used as an alternative. The sternum should never be the site for trephine biopsies. Marrow aspirates can be performed on the sternum with a specially designed needle equipped with a guard device to prevent advance of the needle through the sternum. Bilateral iliac crest biopsies are recommended to assess diseases in which diagnostic lesions are likely to be distributed focally, such as lymphomas and metastatic tumors (8–12).
Immediately after obtaining the trephine biopsy specimen, and prior to placing it in fixative, it should be imprinted on several glass slides. In cases where a marrow aspirate is not obtainable, the touch preparations will provide the only material for cytologic study on Romanowsky-stained slides. A variety of fixatives may be used for trephine biopsies, although the quality of cytologic detail obtained varies among them (5). Following fixation, the specimen is decalcified and processed by routine histologic techniques. Sections should be no thicker than 4 μm. In cases where lesions are expected to be small and distributed focally, sections should be mounted on the slides at multiple levels of the specimen. Samples from the ribbon can be saved temporarily and used if additional sections are required for histochemical or immunohistochemical (IHC) stains. In most instances, well-prepared paraffin sections are adequate for diagnosis when combined with marrow aspirate smears and appropriate special studies, but some laboratories prefer plastic-embedded biopsies, which often provide superior cytologic detail (7,13,14).
The marrow aspirate specimen is preferably obtained a few millimeters from the site of the trephine biopsy through the same skin incision. The major portion of the aspirated specimen can be placed in ethylenediaminetetraacetic acid (EDTA) anticoagulant and the remainder used to make smears at the patient’s bedside. Once smears are made at the bedside, the bone marrow aspirate and trephine biopsy specimens should be transported to the laboratory for processing. In the laboratory, the anticoagulated portion of the bone marrow is separated into fluid and particle portions. Smear preparations can be made from the nucleated cell layer (buffy coat) of centrifuged marrow fluid and by particle crush techniques. The remaining particles are then aggregated and used to make clot sections.
SPECIAL PROCEDURES
When the bone marrow is examined with thorough knowledge of the clinical findings, a strategy for optimal use of the available specimens for morphologic assessment and special studies can be devised. In most hematopathology practices, a host of special techniques are available to aid in diagnosis. These include histochemistry, IHC, flow cytometry, cytogenetics/fluorescence in situ hybridization (FISH), and various molecular analyses. Use of a multitechnique approach is not always necessary when morphologic findings are diagnostic but, increasingly, supplemental studies provide needed information not only for diagnosis but also for assessment of prognosis and optimal patient management. When flow cytometry or molecular analysis is required, aspirated marrow is collected in EDTA, sodium heparin, or acid-citrate-dextrose (ACD) tubes and sent for immediate processing. For cytogenetic analysis, heparin anticoagulant is preferred. In cases where a bone marrow aspirate cannot be obtained, cells teased from a fresh trephine biopsy may be used for flow cytometry, cytogenetics, and molecular studies. For infectious disease assessment, microbiologic cultures may be performed on freshly aspirated marrow or trephine biopsies.
HEMATOPOIESIS AND BONE MARROW HISTOLOGY
For most individuals, the bone marrow comprises 3.5% to 6% of total body weight. It is the major organ of hematopoiesis and is both a primary and secondary lymphoid organ that provides an environment for cell development and immunologic interaction (15). The marrow consists of hematopoietic cells and adipose and stromal tissues. The stroma is composed of connective tissue and vascular structures that include arterioles, venules, capillaries, and a system of sinusoids.
Hematopoiesis in the bone marrow begins at the third and fourth months of gestation and is the major site of hematopoiesis after 6 months (16). In the first year of life, hematopoiesis occurs throughout both the axial and radial skeleton. By the age of 25, hematopoiesis is confined to the flat bones of the central skeleton and the proximal quarter of the shafts of the femora and humeri (17). Hematopoiesis consists of two major cell lineages, myeloid and lymphoid. Their common precursor is a bone marrow pluripotent stem cell. Differentiation and maturation of the four types of myeloid cells (granulocytes, monocytes, erythrocytes, and megakaryocytes) and some lymphoid cells occur in the bone marrow; most lymphocytes, however, differentiate and mature primarily outside of the bone marrow.
All types of myeloid cells have a common progenitor. The earliest morphologically recognizable precursor of each of the four myeloid lineages is a blast. Neutrophils may be divided into two functional groups in the marrow: the mitotic and storage pools. The earlier precursors (myeloblasts, promyelocytes, and myelocytes) are present in the mitotic pool for 2 to 3 days, where they multiply and mature. The cells of the mitotic pool are mostly found in paratrabecular and perivascular locations. Additional maturation occurs in the storage pool, which is composed of metamyelocytes, bands, and segmented neutrophils. Normally, cells remain in the storage pool for approximately 5 to 7 days. Neutrophils move from the storage pool in a unidirectional fashion to the blood and finally to the tissues. Monocytes have an origin closely related to that of neutrophils, but maturational stages are less well defined. Histiocytes, the tissue form of monocytes, are commonly observed in bone marrow. As erythroid precursors mature in the bone marrow, they become smaller and richer in hemoglobin. Early precursors are found in randomly distributed cellular islands that are generally perivascular. The most mature nucleated erythroid precursors extrude their nuclei to become reticulocytes, which then spend a final 1 to 2 days in the marrow while their cytoplasm continues to mature. Megakaryocytes are normally present as single cells that are essentially randomly distributed throughout the bone marrow, although they do not normally occupy a paratrabecular location. As they mature, they become progressively larger and increase their nuclear lobes.
The bone marrow provides an environment for lymphocyte development and immunologic interaction. Variable numbers of B-cell precursors (hematogones) are present in bone marrow, most notably in children (18). However, most lymphocyte development occurs in extramedullary sites, especially in lymph nodes. Immunologic markers can characterize lymphoid maturational stages, but a morphologic developmental sequence is not recognized in lymphocytes to the degree that it is in myeloid cells. Lymphocytes comprise approximately 10% of marrow cells in normal adults. Plasma cells are found primarily in a perivascular distribution and account for approximately 1% to 2% of marrow hematopoietic cells.
Osteoblasts and osteoclasts are found along the endosteal surface of bone trabeculae in trephine biopsies but are uncommon in normal aspirate smears except in children; they may be found in aspirates in several pathologic conditions. Mast cells are found adjacent to endosteal cells and in perivascular locations and are a minor component of normal bone marrow.
BONE MARROW CELLULARITY
Assessment of marrow cellularity is important for diagnosis and treatment in several clinical conditions. These include evaluation of blood cytopenias, following patients on chemotherapy, and assessing engraftment in marrow transplant recipients. Biopsy sections are preferable to aspirate smears for assessment of cellularity. Aspirate smears are affected by hemodilution and variations in technique. Biopsies from the posterior iliac crest generally reflect the cellularity of the overall hematopoietic marrow (3). However, local marrow insults, particularly prior radiotherapy, may leave the iliac crest unrepresentative of overall marrow cellularity (19).
In neonates, nearly 100% of the ilium bone marrow consists of hematopoietic cells. In the first decade of life, after the neonatal period, approximately 80% is hematopoietic and 20% fat, a ratio of 4:1. There is gradual change to a ratio of approximately 1:1 to 1.2:1 by age 30. The cellularity remains relatively stable until the seventh decade, when hematopoietic marrow and bone trabeculae decrease and adipose tissue increases (20–22). There is significant variation in cellularity in different individuals of the same age (3). The proportion of hematopoietic marrow may also vary in different areas of the ilium; in adults, the immediate subcortical marrow is often less cellular than deeper areas, a finding that is accentuated with aging (3).
PANCYTOPENIA
There are many causes of pancytopenia (Table 16.2), and marrow cellularity and composition differ in relationship to the cause. The marrow is generally hypocellular in cases of pancytopenia caused by a primary production defect. Cytopenias resulting from ineffective hematopoiesis, increased peripheral utilization or destruction of cells, and bone marrow invasive processes are usually associated with a normocellular or hypercellular marrow.

HYPOCELLULAR BONE MARROW
The marrow in severe aplastic anemia is markedly hypocellular with a profound decrease in hematopoietic tissue and a corresponding increase in marrow fat (Fig. 16.1). Macrophages containing hemosiderin are often prominent, and increased proportions of lymphocytes, plasma cells, and mast cells are observed. Lymphocytes may be found in loose aggregates or nodules (23). Scattered erythroid islands are sometimes present, and occasional granulocytes and megakaryocytes may be observed. The quantity of residual hematopoietic cells varies with the severity of the aplastic anemia, and in milder cases, the cellularity may be patchy. Careful evaluation of the marrow sections may help in predicting the likelihood of spontaneous recovery; generally, the more severe the aplasia, the less likelihood of recovery (23). When a single blood cell type is diminished due to a primary production defect (e.g., pure red cell aplasia or amegakaryocytosis), cells of that lineage are decreased or absent but the overall cellularity of the marrow may be only slightly altered.
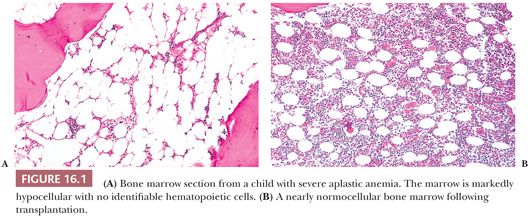
HYPERCELLULAR BONE MARROW
In a hypercellular bone marrow, there is increased hematopoietic tissue and decreased fat. In most cases, hypercellularity is a normal physiologic response to increased demand for blood cell production. Physiologic causes include increased granulopoiesis in systemic infections and increased erythropoiesis in hemolytic anemias or following blood loss. Pathologic hypercellularity results from ineffective hematopoiesis, dysplastic hematopoiesis, leukemia, myeloproliferative syndromes, and invasive neoplasms. The cause of the hypercellularity is generally reflected in the composition of the marrow cells.
CELLULARITY IN NEOPLASTIC DISEASE
Determination of marrow cellularity is important at diagnosis and in monitoring patients with leukemia and other hematopoietic neoplasms. The estimated tumor load (percentage of marrow space replaced) is a staging criterion for some malignancies (24–26). Assessment of changes in marrow cellularity in response to chemotherapy is useful in managing treatment. In successful induction chemotherapy for acute leukemia, there is marked early marrow hypocellularity caused by the cytotoxic effect of the drugs (27,28). In this stage, there is predominantly necrotic tissue and proteinaceous debris in the marrow space. Other findings include sinusoidal dilation, degenerative stromal changes, multiloculated fat cells, and increased reticulin. In the blood, there is marked pancytopenia. If a significant reduction in cellularity is not achieved in the first week of induction chemotherapy, it signals resistance to the therapy and portends a worse prognosis (29,30). With successful induction, regeneration of normal bone marrow begins in the first week or two after the initiation of therapy. If growth factors are not administered, erythroid regeneration is generally first, followed in sequence by granulocyte and megakaryocyte regeneration. In patients treated with granulocyte colony-stimulating factor (G-CSF), granulocyte production occurs most rapidly. Hematopoietic marrow regeneration accelerates in the second and third weeks, with blood counts showing recovery and in some patients approaching normal within 28 days. Slow or asynchronous recovery of blood counts toward normal may indicate resistant disease. However, retarded bone marrow regeneration may also be caused by medications, viral infections (especially cytomegalovirus [CMV], human herpesvirus-6 [HHV6], and parvovirus B19), marrow stromal damage, and extraordinary sensitivity to chemotherapy (28,31). With remission and regeneration of normal hematopoietic tissue, the marrow returns to normal or near-normal cellularity, although cellularity may be more variable than normal; the bone marrow may remain mildly hypocellular in patients on maintenance chemotherapy. If blood cytopenias appear following complete remission of the disease, a bone marrow examination is indicated to determine whether the leukemia has relapsed or the marrow is suppressed by maintenance chemotherapy or other causes (31).
CHANGES IN CELLULARITY FOLLOWING BONE MARROW TRANSPLANT
Assessment of marrow cellularity is important when following hematopoietic stem cell or bone marrow transplant recipients for engraftment and rejection (32). Generally, the sequence of changes is similar for bone marrow and cord blood or peripheral blood stem cell transplants; the changes are similar to regeneration following chemotherapy described earlier (27,28). There is usually evidence of significant hematopoiesis in the bone marrow prior to any changes in blood counts, but, for practical reasons, assessment of engraftment is usually done by monitoring blood counts (33). A bone marrow examination may be performed if blood counts do not recover in the expected time and sequence. In allogeneic transplants, the first good evidence of engraftment of transplanted marrow is found 1 to 2 weeks posttransplant. Growth factor therapy, such as erythropoietin and G-CSF, accelerates the process of regeneration and may alter its sequence. Many patients receive G-CSF to stimulate granulopoiesis immediately following transplantation, and left-shifted granulopoiesis with numerous large promyelocytes and myelocytes are observed early in the engraftment phase; circulating myeloblasts are often observed (34). There is usually obvious engraftment by 3 weeks posttransplantation, and clusters of erythroid precursors are observed along with an abundance of maturing granulocyte precursors; there may be only scattered megakaryocytes. Between 4 and 8 weeks, the marrow cellularity increases and blood counts rise progressively to normal or near-normal levels. The rate of engraftment and return to normocellularity varies from patient to patient. In some cases, engraftment of one or more cell lineages may be unusually retarded. Patients who receive autologous transplants generally recover their blood counts more rapidly than do allogeneic marrow recipients. Declining blood counts after the fourth week posttransplant may be indicative of an infectious complication or graft failure. Recurrence of a neoplastic disease generally occurs somewhat later.
The usual first indication of graft rejection of an allogeneic bone marrow transplant is a decline in erythroid precursors. This may precede, by several days or weeks, the rejection of all of the marrow cell lineages and return to aplasia. Patients with graft-versus-host disease (GVHD) may have increased numbers of lymphocytes, plasma cells, and eosinophils in their marrow; lymphocytic aggregates and granulomas have been noted in some cases (28).
GROWTH FACTORS
Erythropoietin, G-CSF, and granulocyte-monocyte colony-stimulating factor (GM-CSF) are commonly used to enhance hematopoiesis in patients with marrow suppression following chemotherapy or a marrow or stem cell transplant and in some cases of primary or secondary anemia and neutropenia. Patients treated with G-CSF or GM-CSF exhibit an early predominance of granulopoiesis, which often evolves to granulocytic hyperplasia. There are increased large, toxic-appearing promyelocytes and myelocytes that may occupy much of the marrow space. The blood smear may show a leukemoid reaction with increased and immature neutrophils, eosinophils, and monocytes. Myeloblasts and neutrophils with hyposegmentation or hypersegmentation of nuclei, Döhle bodies, and atypical granulation are frequently observed on blood smears (34). In patients with acute myeloid leukemia (AML), the presence of strikingly left-shifted granulopoiesis, circulating myeloblasts, and dysplastic features in some of the maturing neutrophils may be confused with residual AML in the early post–G-CSF–treated marrow. In a small number of patients, treatment with GM-CSF has been reported to induce increased marrow fibrosis (35). G-CSF and GM-CSF theoretically have no effect on red cell and megakaryocyte recovery, although megakaryocyte hyperplasia has been described in patients receiving G-CSF (36). Erythropoietin stimulates erythropoiesis and may induce erythroid hyperplasia in a marrow in early recovery. The erythropoietin-stimulated erythroid compartment may exhibit left shift, increased binucleation, megaloblastoid changes, and terminal dyserythropoiesis. Thrombopoietin administration may result in marrow changes resembling myeloproliferative neoplasms (37).
SEROUS ATROPHY
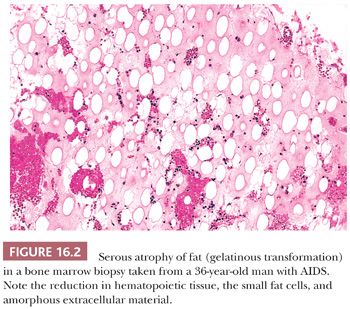
Serous atrophy, sometimes referred to as gelatinous transformation of marrow adipose tissue, is usually associated with severe malnutrition (38–42). It may be found in kwashiorkor, anorexia nervosa, cachexia, starvation from other causes, advanced malignancy, and AIDS. Patients are generally anemic and may have leukopenia or thrombocytopenia. The bone marrow is hypocellular with decreased hematopoietic tissue. Hematopoietic cells are found in clusters within areas of the degenerating fat or in focal uninvolved areas of the marrow. Fat cells are decreased and smaller than normal. The histologic appearance may resemble edema, necrosis, or amyloid (38) (Fig. 16.2). The extracellular gelatinous material is amorphous and faintly eosinophilic. It consists primarily of hyaluronic acid in patients with anorexia and starvation (39). In patients with AIDS, the gelatinous material contains large amounts of sulfated glycosaminoglycan in addition to hyaluronic acid (41). Sulfated glycosaminoglycan has been shown to adversely affect erythropoiesis and may contribute to the anemia in patients with AIDS.
BONE MARROW NECROSIS
Bone marrow necrosis is seen occasionally in patients with infectious diseases, leukemia, lymphoma, metastatic tumor, systemic lupus erythematosus, sickle cell anemia, and miscellaneous other disorders (43–47). The necrosis may involve extensive areas of marrow or may affect only focal areas of a malignant tumor or granuloma. In cases of generalized marrow necrosis, the underlying primary disorder is frequently complicated by infection and sepsis.
If necrosis is of recent origin, the individual cells are recognizable in the sections but show early nuclear and cytoplasmic degenerative changes (e.g., pyknosis, granular cytoplasm). With more advanced necrosis, karyolysis occurs; nuclei are not clearly visualized and the cytoplasm is uniformly eosinophilic. With advanced degenerative changes, only amorphous debris remains. Macrophages containing phagocytosed material are often present at this stage. Occasionally, patients with lymphoblastic leukemia, chronic myelogenous leukemia, Burkitt lymphoma, and metastatic tumors have extensively necrotic marrow that precludes a diagnosis. An additional marrow biopsy in another anatomic site may reveal viable diagnostic tissue. If not, the biopsy may be repeated after a few days or weeks when marrow and the neoplastic tissue have regenerated. It is generally important to characterize a bone marrow neoplasm as quickly as possible. Therefore, if at least a few viable cells can be obtained, special studies such as immunophenotyping or molecular analysis should be performed. Some IHC stains are useful on necrotic tissue. Involved tissue in other anatomic sites should always be sought.
INFLAMMATORY DISORDERS OF THE BONE MARROW
GRANULOMAS
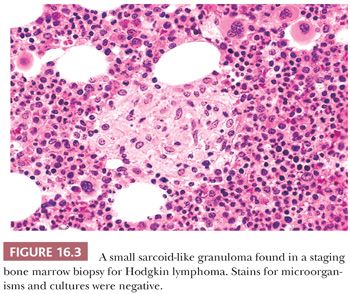
Granulomas may be incidental findings in bone marrow trephine biopsies or discovered during the course of evaluation for infectious disease or a fever of unknown origin. The majority of bone marrow granulomas have no demonstrable infectious etiology. Despite this, appropriate special stains and microbiologic cultures should generally be performed. Most of the granulomas without an infectious etiology consist of epithelioid histiocytes, lymphocytes, occasional giant cells, and eosinophils. They are usually small, focal, and well circumscribed. These nonspecific granulomas have been reported in several conditions, including sarcoidosis, Hodgkin disease, non-Hodgkin lymphoma, other malignancies, and miscellaneous other conditions (48–54) (Fig. 16.3).
Bone marrow granulomas with an infectious etiology are found in mycobacterial and fungal infections, brucellosis, typhoid fever, Q fever, and viral infections including infectious mononucleosis, CMV infections, and herpes zoster (53,55–57). There are no morphologic features that are pathognomonic of a particular infection. Necrosis, commonly present in mycobacterial and histoplasma infections, may also be found in other infections and in granulomas resulting from immune vasculitis. Intracellular yeast can often be identified in hematoxylin and eosin (H&E)–stained sections of granulomas in cryptococcosis and histoplasmosis but are often few in number and difficult to recognize (56). Appropriate cytochemical stains and cultures for mycobacterium and fungi should always be performed. Cultures of bone marrow aspirates may be positive in several types of infections when the special stains on trephine sections are negative.
Lipid granulomas are a relatively common finding in marrow sections (58). They are usually small and composed of macrophages with lipid vacuoles, lymphocytes, plasma cells, eosinophils, and occasionally giant cells. Large, extracellular lipid deposits may be observed. Hemosiderin deposition may also be present. Lipid granulomas are frequently associated with lymphoid aggregates of variable size; these may have infiltrative borders and reactive atypia in the lymphocytes. The association with a lipid granuloma helps identify the benign nature of such aggregates.
Bone marrow aspirate smears are often normal, even when several granulomas are identified on the marrow sections. Occasionally, clusters or sheets of histiocytes are observed on the smear. In some mycobacterial and yeast infections, histiocytes may contain intracellular microorganisms.
NONSPECIFIC INFLAMMATORY REACTIONS
Nonspecific inflammatory changes accompany several marrow disorders, including acute and chronic infections, malignant tumors, and collagen vascular diseases. In acute inflammation, an exudative reaction with increased mature granulocytes, edema, and necrosis is observed. In chronic infections or malignancies, decreased hematopoiesis with increased lymphocytes, plasma cells, and mast cells is more common (59). When the disease process is prolonged, hematopoiesis is diminished and reticulin fibrosis and alterations of vascularity may be present.
REACTIVE MYELOFIBROSIS
Reactive myelofibrosis can occur in infectious and metabolic disorders, neoplastic disease, secondary to various physical and chemical agents, and in patients with autoimmune disorders (60–64). The underlying disease process is generally identified in the biopsy sections or is known from the patient’s medical history. The fibrosis is confined to the areas involved by the primary disease and may consist of increased reticulin fibers only or variable amounts of collagen. The bone trabeculae may be normal or manifest increased osteoblastic or osteoclastic changes.
ACQUIRED IMMUNODEFICIENCY SYNDROME
Several types of pathologic changes are commonly observed in the bone marrow and blood of patients with AIDS. These include various cytopenias, changes in marrow cellularity, ineffective hematopoiesis, dyspoiesis, hyperchromatic and bare megakaryocyte nuclei, increased marrow plasma cells, increased histiocytes, serous atrophy, reactive polymorphous lymphohistiocytic lesions, reactive germinal centers, granulomas, and involvement by lymphomas (41,65–74). Most of the changes are found in the more advanced stages of HIV infection, and the frequency of these findings is therefore decreased in the era of highly active antiretroviral therapy. Although none is specific for AIDS, a combination of several is strongly suggestive of the diagnosis. The reactive and other nonneoplastic changes in the marrow can resemble findings in myelodysplastic syndromes, myeloproliferative neoplasms, or lymphomas. A thorough history and familiarity with the histologic features typical of HIV infection are essential to avoid an erroneous diagnosis of a hematopoietic neoplasm (73).
A bone marrow biopsy may be performed in patients infected with HIV who have blood cytopenias, infectious disease symptomatology, fever of unknown origin, or suspected lymphoma. The marrow cellularity in patients with blood cytopenias may be normal, increased, or decreased depending on the cause; the majority have a normocellular or hypercellular bone marrow (71). A single-cell lineage may be hyperplastic in patients with hemolytic anemia or immune thrombocytopenia (75). The etiology of blood cytopenias, ineffective hematopoiesis, and myelodysplasia is often multifactorial. A direct effect of HIV infection on bone marrow stem cells may contribute, but altered cytokine regulation of hematopoiesis and autoimmune phenomenon are probably more important (73,76,77). In addition to these factors, many patients are chronically infected, malnourished, and receiving several drugs, some of which may suppress myelopoiesis and cause dysplasia. For example, azidothymidine (AZT) causes myelosuppression, which may lead to macrocytic anemia and neutropenia. Pancytopenia and a hypocellular bone marrow are uncommonly related to AZT (78). Many agents used to treat secondary infections and neoplasms also cause marrow suppression.
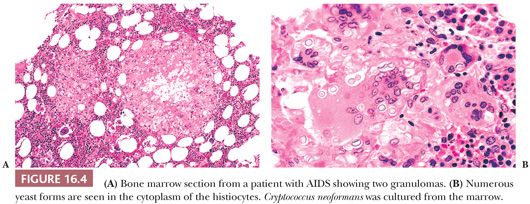
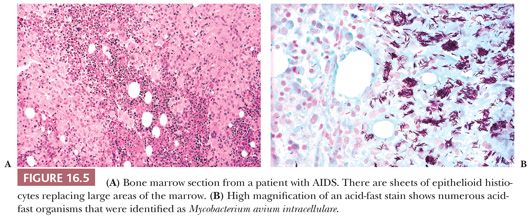
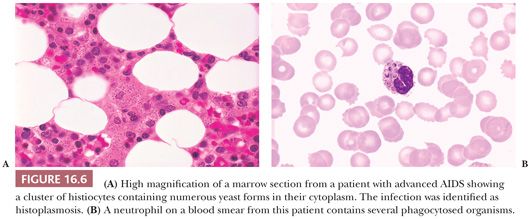
Mycobacterial, histoplasma, and cryptococcal infections are particularly likely to involve the marrow of patients with AIDS; rarely, Pneumocystis carinii is found in bone marrow (79) (Figs. 16.4 through 16.6). Granulomas, histiocytic clusters, diffuse histiocytic infiltrates, and marrow necrosis may be observed (66,69,70,72,80). In some cases of Mycobacterium avium intracellulare infection, there is no clear evidence of disease in routine sections, but organisms are identified in scattered histiocytes with acid-fast stains. Cultures and special stains for microorganisms may be indicated in patients with AIDS, even when granulomas or necrosis are not observed in the biopsy sections.
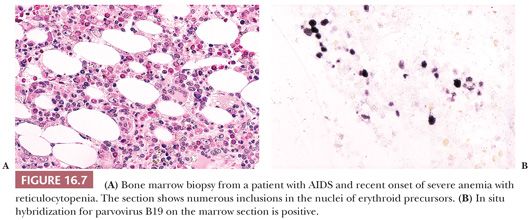
Persistent red cell aplasia in patients with AIDS may be caused by chronic parvovirus B19 infection (81–83). The bone marrow may exhibit red cell aplasia with scattered giant erythroblasts or normal numbers of variably dyspoietic erythroid precursors with intranuclear inclusions that are most apparent on biopsy sections (Fig. 16.7). Documentation of suspected parvovirus B19 infection should be attempted by polymerase chain reaction (PCR), IHC, or in situ hybridization techniques on the bone marrow because serologic studies for antibodies to parvovirus are often negative.
Polymorphous reactive lymphohistiocytic lesions are commonly found in marrow sections from patients with AIDS (69,70,72,84). They are composed of a polymorphous population of lymphocytes, plasma cells, epithelioid histiocytes, eosinophils, and endothelial cells. In some cases, these lesions replace large areas of the marrow (66,72,84,85). Diffuse histiocytic proliferations such as those seen in the infection (virus)-associated hemophagocytic syndrome (see the section “Histiocytic Proliferations” later in the chapter) may be observed in very ill or terminal patients (86).
Patients with AIDS have an increased incidence of both Hodgkin and non-Hodgkin lymphoma, and the bone marrow is frequently involved. However, the incidence of lymphomas in HIV has decreased since the institution of highly active antiretroviral therapy (87). The most common types of non-Hodgkin lymphomas are Burkitt and diffuse large B cell, but other types are less frequently encountered, including peripheral T-cell lymphomas (88–90). Occasionally, the marrow is the primary or only diagnostic tissue (88). If the marrow lesions are in any way equivocal in patients with AIDS, caution should be exercised in considering a diagnosis of marrow lymphoma. The florid reactive polymorphous lymphoid infiltrates described previously can be confused with lymphoma, particularly peripheral T-cell lymphoma or Hodgkin lymphoma (72,73,84,85). Immunophenotyping by flow cytometry or IHC and molecular gene rearrangement studies may help in the differential diagnosis. Hodgkin lymphoma involves the marrow in about 50% of cases in patients with AIDS and marrow is occasionally the diagnostic tissue. The lesions are usually typical of Hodgkin lymphoma, as described later in the chapter (see “Bone Marrow Lymphoid Disorders”), but should always be confirmed with appropriate IHC stains.
ACUTE LEUKEMIAS AND MYELODYSPLASTIC SYNDROMES
The diagnosis of acute leukemia or a myelodysplastic syndrome (MDS) is usually considered because of abnormal blood counts or findings on a blood smear. A bone marrow examination should always be done for confirmation of the diagnosis and to obtain material for supplemental studies.
Optimal morphologic evaluation for leukemia and MDS includes examination of well-prepared blood and bone marrow smears and biopsy sections. Most of the errors in the diagnosis of these disorders result from inadequate specimens or technically poor blood and marrow preparations. In cases of suspected leukemia, the pathologist interpreting the blood and marrow slides should always be equipped with complete clinical information before rendering an opinion.
ACUTE LEUKEMIAS
In cases of AML in which the leukemic blasts exhibit features of myeloid differentiation, the diagnosis is commonly made by morphologic examination of blood and marrow smears and selected cytochemical and IHC stains. Some cases require additional studies for diagnosis. Poorly differentiated AML and acute lymphoblastic leukemia (ALL) require immunophenotyping, preferably by flow cytometry, for diagnosis. Cytogenetic and molecular studies are necessary in the classification of both AML and ALL and for the prognostic and therapeutic information they provide.
Morphology
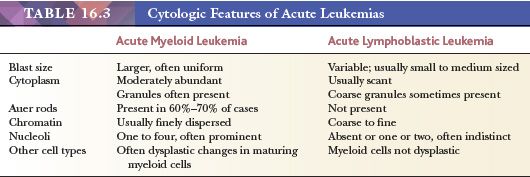
Morphologic assessment of blood and marrow smears and biopsy sections is important in the distinction of leukemias from other neoplasms and nonneoplastic proliferations, to distinguish AML from ALL, and in the classification of acute leukemia. The usual morphologic features that help distinguish AML and ALL in routine blood and marrow smears are shown in Table 16.3. Overall, there are differences between AML and ALL in each of the morphologic parameters listed in the table, but there may be overlapping features in individual cases, which show some features common to AML and others more typical of ALL. All morphologic features should be considered in composite when making an interpretation. Only the presence of unequivocal Auer rods always distinguishes AML from ALL. There are numerous descriptions of the cytologic changes in blood and marrow smears in acute leukemias and of the features that help define the various categories (91–103). These will be reviewed in the discussion of the World Health Organization (WHO) classification of acute leukemias later in this chapter.
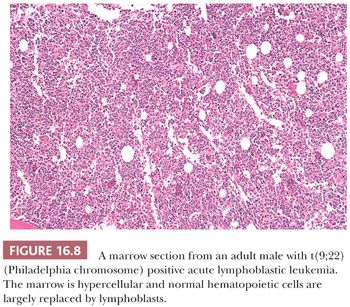
The biopsy sections are markedly hypercellular in the majority of cases of acute leukemia (Fig. 16.8), but occasionally, in AML, the marrow is normocellular or even hypocellular. The normal distribution and spectrum of maturation of hematopoietic cells is lacking, and the marrow is mostly replaced by very immature-appearing cells. Mitotic activity is variable in acute leukemias, but usually, mitotic figures are easily found. In ALL, the blasts are relatively uniform, whereas in AML, the cytologic features vary depending on the percentage of myeloblasts and the lineage differentiation patterns. Myelofibrosis is present in some cases of both AML and ALL, and it may be particularly prominent in acute megakaryoblastic leukemia (91,95,99). Reticulin fibrosis may preclude obtaining an aspirate specimen. When this occurs, the trephine biopsy sections and touch preparations may be particularly important in the diagnosis.
Cytochemistry
Cytochemical stains on blood and bone marrow smears are helpful in the distinction of AML and ALL and in the subclassification of AML (99,104). The combination of a myeloperoxidase or Sudan black stain and a nonspecific esterase stain provides the desired information in most instances. The myeloperoxidase or Sudan black reactions are most useful in establishing the identity of AML. The nonspecific esterase stain is used to identify a monocytic component in AML and to distinguish poorly differentiated monoblastic leukemia from AML, minimally differentiated and ALL.
Immunophenotype
Immunophenotyping by flow cytometry is the primary method for distinguishing ALL and AML when the morphology and cytochemistry of the leukemic blasts lack defining features (91,99,101,105). Immunophenotyping is also vitally important in distinguishing B- and T-ALL, in identifying the rare cases of mixed phenotype acute leukemia, and in detection of minimal residual disease (99,105–108). Immunophenotyping may also provide prognostic information for some leukemias. Assessment of blasts for the progenitor cell–associated antigens terminal deoxynucleotidyl transferase (TdT) and CD34 and expression of T-cell–associated antigens (CD2, surface and cytoplasmic CD3, CD5, CD7) and B-cell–associated antigens (e.g., human leukocyte antigen DR-1 [HLA-DR], CD10, CD19, CD22, CD79a) will identify and categorize nearly all cases of ALL. Likewise, assessment for myeloid-associated antigens (e.g., CD13, CD14, CD15, CD33, CD36, CD61, CD64) will identify the large majority of cases of AML. Antibodies to additional lineage-associated antigens will be necessary in some cases for accurate classification. Panels should always include several antigens associated with each of the major cell lineages. Loss of antigens or aberrant expression is common in acute leukemias. In many cases, only by analysis of panels of antigen expression can an accurate interpretation be made. If necessary, immunophenotyping with IHC stains on bone marrow smears or biopsy sections may be used as an alternative to flow cytometry.
Cytogenetics
Bone marrow cytogenetic studies are essential in the evaluation of patients with acute leukemia and MDS (99,101,104,109–112). Cytogenetic findings are an important independent indicator of prognosis, and they define several categories in the WHO classification of acute leukemia (91,113–115). They may also help distinguish between AML and ALL in selected cases and between an MDS and a nonneoplastic cause of myelodysplasia. Prognostic implications of cytogenetic findings in acute leukemias are shown in Table 16.4.

Molecular Analysis
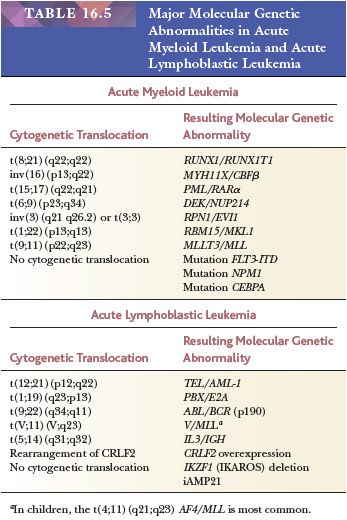
Molecular analysis contributes to the diagnosis and management of patients with acute leukemia. Molecular diagnostic studies may establish clonality, detect specific chromosome numeric and structural abnormalities, and identify gene mutations and cryptic structural rearrangements not detectable by conventional cytogenetics (91,116–119). Information provided by detection of molecular translocations and gene mutations contributes directly to the classification of leukemias and may provide valuable treatment and prognostic information (119). PCR and FISH studies for specific gene segments are also sensitive indicators of minimal residual leukemia and early relapse (120). Probes are available for detection of many of the mutations and fusion genes that identify clinically relevant categories of acute leukemia, MDSs, and myeloproliferative neoplasms. Examples of some of the most common ones found in acute leukemias are listed in Table 16.5 with corresponding cytogenetic changes, where applicable. New methods for high-resolution genome-wide analysis are providing new information on the genetic basis of leukemogenesis, identifying novel subtypes of leukemia, and providing markers to integrate into diagnostic testing and to be targeted with novel molecular-based therapy.
CLASSIFICATION OF ACUTE LEUKEMIAS
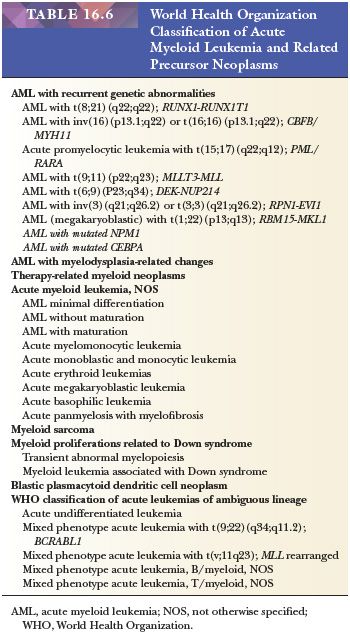
The WHO classification of hematopoietic neoplasms is the most widely used for acute leukemia and has largely replaced the French-American-British (FAB) Cooperative Group classification (96). It incorporates morphologic, immunophenotypic, genetic, and clinical features to define categories of acute leukemia that are biologically homogeneous and have clinical relevance. It provides a more precise diagnosis than do former classifications based primarily on morphology and introduces important prognostic and treatment correlations. The WHO classification of hematopoietic neoplasms was most recently revised in 2008 (91). The categories listed in Tables 16.6 and 16.7 and discussed in the text are those in the 2008 WHO classification. With the rapidity of expanding molecular genetic information, it is likely that there will be modifications to the classification in the near future.
ACUTE MYELOID LEUKEMIA
There are seven groups of AML and related precursor neoplasms in the WHO classification (91). Those with recurrent genetic abnormalities include AML with more homogeneous biologic and prognostic features. AML with myelodysplasia-related changes recognizes a group that has morphologic features or cytogenetic abnormalities shared with the MDSs. These leukemias may arise de novo or as part of the evolution of a preexisting MDS. Therapy-related AML is a group of leukemia occurring in patients who have previously received cytotoxic chemotherapy for another disease. AML not otherwise specified (NOS) includes cases of AML that lack recurrent genetic rearrangements or myelodysplasia-related changes and have not received prior cytotoxic drugs. This group is subclassified into traditional morphologic categories. Myeloid sarcoma is a designation for a tumor mass consisting of myeloid blasts occurring in an anatomic site other than bone marrow. A separate group with several distinctive features is myeloid proliferations related to Down syndrome. Blastic plasmacytoid dendritic cell neoplasm is a recently designated group derived from precursors of plasmacytoid dendritic cells with a high frequency of cutaneous and bone marrow involvement and leukemic dissemination (91). Each group of AML and related precursor neoplasm is covered in more depth in the discussion that follows.
Acute Myeloid Leukemia with Recurrent Genetic Abnormalities
There are seven categories of AML with recurrent cytogenetic abnormalities and two provisional categories with specific gene mutations. The genetic findings in this group have prognostic significance. They are described in the following paragraphs.
A defining feature of AML and one that distinguishes it from other myeloid neoplasms is the requirement for a minimum of 20% blasts or blast equivalents in the blood or bone marrow (91,121–123). Exceptions to this 20% requirement are made for three of the AML with recurrent genetic abnormality categories: AML with t(8;21), AML with inv(16), and APL with t(15;17). These myeloid neoplasms are considered acute leukemia even if the presenting blast count is less than 20% (91).
AML with t(8;21)(q22;q22);(RUNX1-RUNX1T1) exhibits characteristic morphology, immunophenotype, and clinical findings and constitutes about 8% of AML (91,124,125). The morphologic features are nearly always those of AML with maturation and include large blasts, frequent and often large Auer rods, and striking dysplasia in the neutrophil lineage. Immunophenotypically, there is usually aberrant expression of the B-lymphocyte–associated antigens CD19, PAX5, and CD79a and often expression of CD56 (125–127). AML with a t(8;21) is associated with a favorable prognosis in adult patients. Reported adverse factors in prognosis are CD56 expression and KIT mutations (128,129).
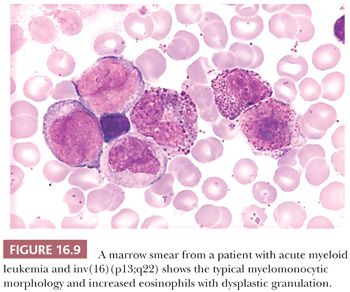
AML with inv(16)(p13:q22) or t(16;16)(p13;q22); (CBFBMYH11) generally has the morphologic features of acute myelomonocytic leukemia, with the addition of increased and dysplastic eosinophils in the marrow (130,131) (Fig. 16.9). The dysplastic eosinophils are recognized by an abundance of large, basophilic-staining granules. AML with inv(16) constitutes approximately 8% of cases of adult AML and approximately 25% of cases of acute myelomonocytic leukemia (91,132). The incidence of extramedullary disease is higher (approximately 50%) than for most types of AML; lymphadenopathy and hepatomegaly are particularly common. Myeloid sarcoma concurrent with or preceding bone marrow involvement appears to be more common than in other leukemias. Some investigators have reported a high incidence of central nervous system (CNS) relapse with intracerebral myeloblastomas (133). High complete remission rates are expected for AML with an inv(16), and the prospect for extended remission is good (134).
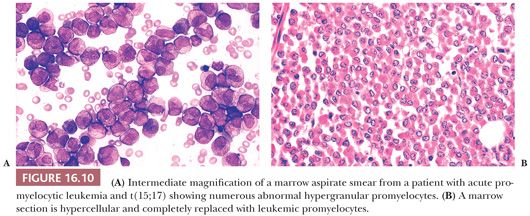
Acute promyelocytic leukemia (APL) with t(15;17)(q22;q21); (PML/RARα) comprises about 8% of AMLs and is associated with distinctive biologic and clinical features (91,132). The leukemic cell population is abnormal promyelocytes that usually contain numerous red to purple cytoplasmic granules (91,135) (Fig. 16.10). The granules are often larger and darker staining than normal and may be so numerous as to obscure the nuclear borders. In some cases, a high percentage of the leukemic cells have intensely basophilic cytoplasm. Cells containing multiple Auer rods are found in approximately 90% of cases. The Auer rods may be numerous and intertwined. Large globular inclusions of Auer-like material are found in the cytoplasm of occasional cells. The nuclei of many of the cells are reniform or bilobed. In the microgranular variant of APL, the leukemic cells have sparse or fine granulation and markedly irregular nuclei, which may obscure their identity as abnormal promyelocytes (136). Cells containing multiple Auer rods are usually present but less abundant than in typical hypergranular APL. Both typical hypergranular APL and the microgranular variant have the same characteristic clinical, ultrastructural, cytogenetic, and molecular features. They differ only in the size and number of granules in the cytoplasm, the prominence of the abnormal nuclear shape in the predominant leukemic cells, and in the magnitude of the blood leukocyte count. Hypergranular APL is generally associated with leukopenia at presentation, whereas microgranular APL often presents with marked leukocytosis (135).
Ultrastructurally, APL is distinctive by the presence of Auer rods with a specific tubular substructure, markedly dilated endoplasmic reticulum, and stellate complexes of rough endoplasmic reticulum (135,137). The immunophenotype by flow cytometry is also characteristic in most cases and is different from other types of AML, mainly by increased side scatter and lack of expression of HLA-DR and CD34. The most striking clinical feature in APL is the high frequency of disseminated intravascular coagulation (DIC). In many patients, there is severe DIC and hemorrhage prior to or during standard induction chemotherapy (138). Hemorrhage is the cause of early death in some patients.
The t(15;17) breakpoint regions are at the PML gene on band q22 of chromosome 15 and on band q21 in the first intron of the retinoic acid receptor (RAR) gene on chromosome 17 (124,139,140). The resulting PML-RARα fusion messenger RNA product inhibits maturation of the affected cells, leading to an accumulation of large numbers of abnormal promyelocytes. Treatment with all-trans retinoic acid (ATRA) can overcome the maturation blockage in most instances and lead to a temporary complete remission. Treatment with standard induction chemotherapy along with ATRA is required to sustain remission. For adult patients who achieve complete remission, the prognosis is better than for any other category of AML (91).
Promyelocytic leukemias with t(V;17)(V;q21); (V/RARα) have many of the morphologic and clinical features of APL but have a variant cytogenetic translocation that involves the RARα gene on chromosome 17 but not the PML gene on 15; t(11;17)(q23; q12) ZBTB16. RARα is one of the more common variant translocations (91,140). Morphologically, cases with t(V;17) may have morphologic features intermediate between APL and AML with maturation. As with t(15;17) APL patients with a t(V;17) APL often experience DIC. It is important to distinguish the two because most t(V;17) APL do not respond to ATRA therapy and often have an aggressive clinical course (140).
AML with t(9;11)(p22;q23); (MLLT3-MLL) is usually monoblastic, monocytic, or myelomonocytic and occurs most frequently in children and young adults (9% to 12% of cases in children and 1% to 2% in adults) (91,141,142). Translocation (9;11) may also be found in cases of topoisomerase II inhibitor, therapy-related AML, and in biphenotypic leukemias (91,143). 11q23 (MLL) is involved with several other translocations and partner genes in acute leukemia. De novo AMLs with a t(9;11) are associated with an intermediate prognosis; 11q23 (MLL) leukemia with other translocations/partner genes usually have a poor prognosis (124,144).
AML with t(6;9)(p23;q34) (DEK-CAN) is a rare type of AML that occurs in both children and adults (145,146). There is a wide range of morphologic features and frequently evidence of multilineage dysplasia; basophilia is often present. There are no distinctive immunophenotypic features. Cases with t(6;9) have an overall poor prognosis.
AML with inv(3)(q21;q26) or a t(3;3) presents either de novo or following an MDS (91,147–149). A notable feature is the frequent presentation with normal or elevated platelet counts. Megakaryocytes are increased in the marrow and exhibit dysplastic features. Multilineage dysplasia is a common finding. This is an aggressive type of AML with a poor prognosis.
AML with t(1;22)(p13;q13); (RBM15-MKL1) is a rare form of acute megakaryoblastic leukemia, representing less than 1% of AML and most frequently occurring in infants. It is associated with marked organomegaly and prominent myelofibrosis and may have features of a panmyelosis. The pattern of marrow and other organ involvement often resembles that of a metastatic small cell tumor (150). Some reports suggest that AML with t(1;22) may respond well to intensive treatment for AML (151).
AML with mutated NPM1 (provisional category) has mutations of the gene that result in aberrant cytoplasmic expression of nucleophosmin, which can be detected by IHC staining (152,153). Mutated NPM1 is one of the most common recurring genetic abnormalities in AML—found in at least 50% of adults with normal conventional banding chromosome analysis—but may also occur in a small number of cases with chromosomal abnormalities (152,154,155). There are no specific morphologic or immunophenotypic features, but the leukemia frequently has myelomonocytic or monocytic morphology (152). It occurs most commonly in females (152). AML with mutated NPM1 in patients with normal conventional cytogenetics seems to show a good response to therapy and favorable prognosis unless there is also FLT3-internal tandem duplication mutation, in which case it is associated with a less favorable prognosis (156).
AML with mutated CEBPA (provisional category) is found in approximately 10% of AMLs, and about 70% of these have a normal karyotype (157,158). In most cases, the leukemia has features of either AML with maturation or AML without maturation. The importance of the CEBPA mutation is its association with a favorable prognosis, similar to t(8;21) and inv(16) AML (159).
Acute Myeloid Leukemia with Myelodysplasia-Related Changes
AML with myelodysplasia-related features may arise in patients with an MDS or de novo with an MDS-related cytogenetic abnormality or multilineage dysplasia (91). These leukemias increase in incidence with age and are rare in children; they account for approximately one-third of all AMLs (160–162). The morphologic features are variable. The blast count is at least 20% of the nucleated cells in the bone marrow, and there is evidence of dysplasia in 50% or more of the maturing cells in two or more lineages, and/or an MDS-related cytogenetic abnormality is present. There is frequently an obvious panmyelosis (163). The immunophenotype varies but there is usually expression of panmyeloid markers and aberrant antigen expression is frequent. Cytogenetic findings are similar to those found in MDS and include gains or deletions of major segments or whole chromosomes (e.g., −5 or 5q−, −7 or 7q−, i(17q)/t(17p), −13/del(13q), etc.) and complex cytogenetic rearrangements. A number of translocations involving chromosomes 3, 5, or 11 are also sufficient for diagnosis of AML with myelodysplasia-related features when 20% or more blasts are present (91). The prognosis for this category of AML is generally unfavorable (160,162).
Therapy-Related Myeloid Neoplasms
Therapy-related AML and MDS occur in patients previously treated with chemotherapy or radiation therapy. Alkylating drugs and the topoisomerase II inhibitors are the agents most commonly implicated (143,164). The median onset of therapy-related AML or MDS is approximately 5 years after initiation of alkylating agents and 2.5 to 3 years after first use of topoisomerase II inhibitor drugs. In cases associated with alkylating agent therapy, patients typically present with pancytopenia and features of an MDS. The dysplastic changes in the blood and marrow cells are often severe; however, the marrow myeloblast percentage may be less than 5% (165). In some cases, the MDS is followed in a short time by progression to AML. In others, severe bone marrow failure leads to patient demise without evolution to leukemia. Myelofibrosis, hypocellularity, and ringed sideroblasts are encountered more frequently than in de novo AML or MDS. Therapy-related MDS associated with alkylating agents commonly has cytogenetic abnormalities affecting chromosomes 5 or 7 and complex cytogenetic abnormalities. Patients with AML and MDS secondary to alkylating agent drugs generally respond poorly to treatment and have a short survival. The topoisomerase II inhibitor drugs more often lead to acute monocytic or myelomonocytic leukemias and have abnormalities of chromosome 11q23 (MLL); only rarely do they present as an MDS (143,166). Although the initial response to treatment is often favorable in topoisomerase II inhibitor–induced AML, overall prognosis is poor.
Acute Myeloid Leukemia, Not Otherwise Specified
Cases of AML that are not included in one of the three major categories cited previously are classified as AML, NOS. These are classified morphologically using descriptive terminology from the FAB classification (92,96). The presence of recurrent cytogenetic abnormalities, myelodysplasia-related changes, or history of prior cytotoxic therapy all take priority over the categories of AML, NOS in the classification of AML regardless of the morphologic features.
AML, minimally differentiated exhibits no definitive evidence of myeloid differentiation by morphology and cytochemistry; the nature of the blasts is determined by immunophenotyping (91). The blasts are agranular, lack Auer rods, and are myeloperoxidase and Sudan black B negative (97). They express one or more panmyeloid antigens, such as CD13, CD33, and CD117, and may express other myeloid lineage–associated antigens. CD34 and TdT are expressed more frequently than for other types of AML (149). CD7, CD2, and CD19 are expressed in a few cases but the blasts generally lack expression of B- and T-lymphocyte–restricted antigens such as CD3 and CD22. Chromosome abnormalities, often complex, are found in most cases, but cytogenetic changes unique to AML, minimally differentiated have not been identified (167,168). The prognosis is usually poor with a shorter survival than for other types of AML.
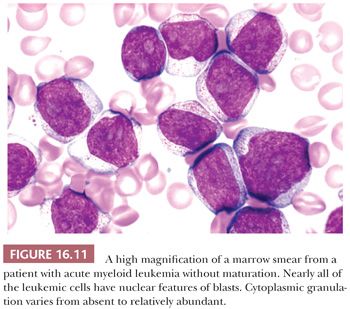
AML without maturation accounts for about 10% of cases of AML (91,132). The sum of blasts must be 90% or more of the nonerythroid cells in the bone marrow (91,92) (Fig. 16.11). Evidence of maturation to promyelocytes is variable but may be minimal or absent. The cytologic features of the myeloblasts vary considerably from case to case. The blast nucleus may be round or indented and in some cases exhibits a distinctive invagination (169). Cytoplasmic granulation varies from abundant to virtually absent; Auer rods are found in approximately 50% of cases. In those without Auer rods, at least 3% of the leukemic myeloblasts must be myeloperoxidase or Sudan black B positive.
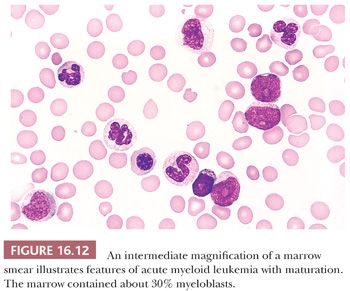
AML with maturation may be the single most common type of AML NOS. It may occur at any age but is more common in older individuals (91,92,132). The marrow blast percentage is 20% to 89% of the nonerythroid cells (Fig. 16.12). Granulocytes from promyelocytes to mature neutrophils comprise more than 10% of cells; monocytes and precursors are less than 20%. The maturing neutrophils often show dysplastic features. In about 70% of cases, Auer rods can be identified (132). Erythroid and megakaryocyte precursors may show evidence of dyspoiesis, and a frank panmyelosis is present in some cases. If the dysplastic changes in two or more lineages exceed 50% of the cells, the case should be classified in the WHO category of AML with multilineage dysplasia. Because of the obvious maturation of the leukemic cells, the myeloid nature is not often in question. The blasts and maturing granulocytes are positive for myeloperoxidase and Sudan black B. There are no distinctive immunophenotypic features that characterize AML with maturation. A majority of cases have demonstrable cytogenetic abnormalities typical of AMLs, but there is no common aberration for this category. The prognosis is variable from poor to quite favorable. Those of advanced age and with multilineage dysplasia and unfavorable cytogenetic changes generally respond less well to therapy and have a shorter survival.
Acute myelomonocytic leukemia (AMML) comprises 15% to 25% of AMLs but some with morphology have recurrent genetic abnormalities and are classified as such (91,132). Bone marrow myeloblasts and monoblasts/promonocytes number 20% or more, but the sum of myeloblasts and neutrophils and precursors is 80% or less; 20% or more of the marrow cells are in the monocyte lineage (92). If fewer than 20% of the marrow cells are monocytes, the diagnosis may still be AMML if blood monocytes number more than 5 × 109 per liter. Both granulocytic and monocytic differentiation is present in varying proportions in the bone marrow. The major difference between AMML and AML with maturation is the proportion of the promonocytes and monocytes, which must be 20% or more in AMML. Early promonocytes cannot always be distinguished from early granulocyte precursors in routine marrow smears. For this reason, an additional requirement of nonspecific esterase reactivity in 20% or more of the cells is included. Auer rods are present in the myeloblast component in approximately 60% of cases. The immunophenotype of AMML generally exhibits a spectrum of myeloid-associated antigens with some monocyte-associated antigens such as CD14, CD11, CD36/CD64, and CD4. There may be two distinctive immunophenotypic populations of granulocytic and monocytic cells. Similar to AML with maturation, cytogenetic abnormalities are common and include rearrangements typical of AML in general. Those with marrow eosinophilia and an inv(16) are classified in the AML with recurrent genetic abnormalities, described previously. AMML occurs in both children and adults. The median age at diagnosis is approximately 50 years. The blood leukocyte count is often markedly elevated. Organomegaly, lymphadenopathy, and other tissue infiltration are commonly present. The prognosis is variable and overall similar to other AML.
Acute monoblastic and acute monocytic leukemias comprise about 8% of AMLs when those with a t(9;11) chromosome abnormality are included (170). They are composed of 80% or more leukemic cells in the monocyte lineage (91,92). Acute monoblastic leukemia is seen predominantly in children and young adults. Monoblasts exhibit moderately abundant, variably basophilic cytoplasm, which frequently contains delicate peroxidase-negative azurophilic granules. The nucleus is round with reticular chromatin and one or more prominent nucleoli. Acute monocytic leukemia may be seen in all age groups. The leukemic cells are predominantly promonocytes that show more obvious evidence of monocytic differentiation and maturation (Fig. 16.13). The nuclei are folded or cerebriform with delicate chromatin. The cytoplasm is less basophilic than in monoblasts and contains a variable number of azurophilic granules. Both monoblasts and promonocytes are nonspecific esterase-positive. Monoblasts are generally myeloperoxidase negative but promonocytes may exhibit weak to moderate myeloperoxidase activity. The leukemic monocytes express variable patterns of myeloid antigens, including monocyte-associated antigens such as CD14, CD15, CD11b, CD36, and CD64. Cytogenetic abnormalities are more common in acute monoblastic (approximately 75%) than monocytic (approximately 30%) leukemias.
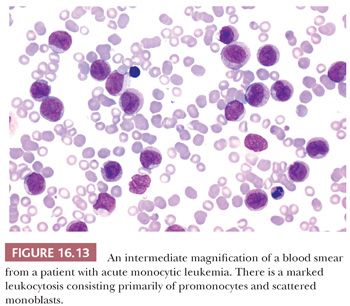
Acute monoblastic and monocytic leukemias are associated with a high incidence of organomegaly, lymphadenopathy, and other tissue infiltration (e.g., gingival, skin). There is a higher incidence of CNS involvement than for other types of AML. In a few cases, the first clinical manifestations of leukemia result from extramedullary tissue infiltrates. Prognosis is poor to intermediate, at least partly resulting from the association of high-risk clinical findings (e.g., high presenting leukocyte counts, extramedullary involvement, 11q23 chromosome abnormalities).
Acute erythroid leukemias are characterized by a predominance of leukemic cells that are erythrocyte precursors. There are two subtypes, erythroleukemia (erythroid/myeloid) and pure erythroid leukemia (91,92). In erythroid/myeloid leukemia, 50% or more of all nucleated marrow cells are in the erythroid lineage and 20% or more of the remaining cells (nonerythroid) are myeloblasts; dyserythropoiesis is prominent. The predominant leukemic cells are erythroid precursors including the least mature erythroblasts. The erythroid component is characterized by abnormalities of nuclear development, including megaloblastoid changes, karyorrhexis, and occasional giant erythroblasts with multiple nuclei. The leukemic erythroblasts may contain confluent cytoplasmic vacuoles, which correspond to cytoplasmic glycogen and react positively with the periodic acid-Schiff (PAS) stain. There is often evidence of a panmyelosis with striking megakaryocytic and platelet abnormalities. Some of these cases might preferably be classified as AML with multilineage dysplasia by WHO classification criteria. Auer rods are present in myeloblasts in 50% to 60% of cases (132). The blood smear in erythroid/myeloid leukemia may show striking erythroblastemia. Progression of the disease is frequently marked by an increase in myeloblasts and decrease in erythroblasts.
Pure erythroid leukemia is very rare. The only obvious neoplastic cells are erythroid; a myeloblast component is not apparent (171). The leukemic erythroid cells are predominantly or exclusively proerythroblasts and early basophilic erythroblasts. These cells may constitute 90% or more of the marrow elements. Despite the lack of myeloblasts, these cases should be considered acute leukemia.
The immunophenotype reflects the mixture of myeloid and erythroid precursors in erythroid/myeloid leukemia. The erythroid cells in both subtypes typically express CD36, CD71, hemoglobin A, E-cadherin, and glycophorin A. The least mature erythroblasts are often negative for hemoglobin A and glycophorin A; E-cadherin appears to be the most sensitive and specific marker for immature erythroblasts (172). No specific cytogenetic abnormalities are associated with the erythroleukemias, but complex structural rearrangements are common and chromosomes 5 and 7 are frequently involved.
The changes in the revised 2008 WHO classification of AML have profoundly affected the number of cases classified as acute erythroid leukemia. Many cases previously classified as acute erythroid leukemia are now considered AML with myelodysplasia-related changes (172–174).
Acute megakaryoblastic leukemia (AMKL) comprises 3% to 5% of AMLs and is found in both adults and children. Fifty percent or more of the leukemic blasts are of megakaryocyte lineage (91). The category excludes cases of AML with myelodysplasia-related changes with megakaryoblasts and cases with a t(1;22) or inv(3) and Down syndrome–related cases. Blasts are usually identified as megakaryocytic by expression of antigens specific for megakaryocytes. In blood and marrow smears, megakaryoblasts are usually medium-sized to large cells with a high nuclear-to-cytoplasmic ratio. Chromatin is dense and homogeneous. There is scanty, variably basophilic cytoplasm, which may be vacuolated. An irregular cytoplasmic border is often noted, and occasionally, there are projections resembling budding of atypical platelets. Transition between poorly differentiated blasts and recognizable micromegakaryocytes is often observed. In many cases, the majority of the leukemic cells are small blasts with features similar to lymphoblasts. Megakaryoblasts are negative for myeloperoxidase. Often, a marrow aspirate is difficult to obtain because of myelofibrosis. The biopsy sections may reveal morphologic evidence of megakaryocytic differentiation that is not appreciated in the smears.
Clues to the lineage of poorly differentiated megakaryoblasts include the presence of circulating micromegakaryocytes, atypical platelets, projections on the surface of the blasts, zoning of the cytoplasm, myelofibrosis, and clusters of small megakaryocytes in sections. More precise identification can be accomplished by immunophenotyping. Megakaryoblasts variably express CD13, CD33, HLA-DR, and CD71 and mostly express CD36. Some of the blasts express the megakaryocyte-specific antigens CD41 and CD61 in virtually all cases (175). The more differentiated megakaryocytes express factor VIII antigen. There are no distinctive cytogenetic abnormalities in adults with AMKL. AML t(1;22)(p13;q13) is a category of AML with recurrent genetic abnormalities (described earlier) that occurs in infants (150). AMKL in Down syndrome is discussed in the following texts.
Acute basophilic leukemia is a rare form of AML (91,176). Cases may show obvious basophil differentiation or be cytologically undifferentiated with only ultrastructural evidence suggesting the basophil lineage. The poorly differentiated acute basophilic leukemias would most likely be classified as AML minimally differentiated without confirmation by electron microscopy. Acute basophilic leukemias are myeloperoxidase negative on light microscopy. The granules stain metachromatically with toluidine blue. Myeloid antigens such as CD13 and CD33 are usually expressed. There are no specific cytogenetic findings but a t(9;22) (Philadelphia chromosome) is frequently found. In some cases of AML with t(6;9), basophils are a predominant component. There are no clinically distinguishing features of acute basophilic leukemia, but it may be more common in children and young adults and carry a poor prognosis (176).
Acute panmyelosis with myelofibrosis is an uncommon form of acute leukemia occurring primarily in adults and rarely in children (91). Patients present with pancytopenia and panmyelosis. Cases of acute panmyelosis with myelofibrosis do not meet the criteria for AML with myelodysplasia-related changes. There are usually megakaryocytic abnormalities but fewer than half of blasts are megakaryoblasts. The degree of fibrosis varies. In most cases, there is marked reticulin fibrosis; collagen fibrosis is less common. The disorder shares features with other myeloid disorders with prominent myelofibrosis. At initial presentation, it may be difficult to exclude primary myelofibrosis, AMKL, or an MDS with myelofibrosis as diagnostic considerations. The spleen is generally normal or only minimally increased in size. No distinctive immunophenotypic or cytogenetic features have been described for acute panmyelosis with myelofibrosis. The disease has an aggressive course.
Myeloid Sarcomas
Myeloid sarcoma is a tumor mass of neoplastic immature myeloid cells in an extramedullary site. Usually, the patient has evidence of myeloid leukemia in the bone marrow and blood but, in some instances, myeloid sarcomas occur without obvious leukemia. The most common sites for myeloid sarcomas are subperiosteal bone, skin, lymph nodes, orbit, spinal canal, and mediastinum (91) (Fig. 16.14). Most types of AML may occasionally present in an extramedullary site, but the monocytic and myelomonocytic leukemias have the highest propensity.
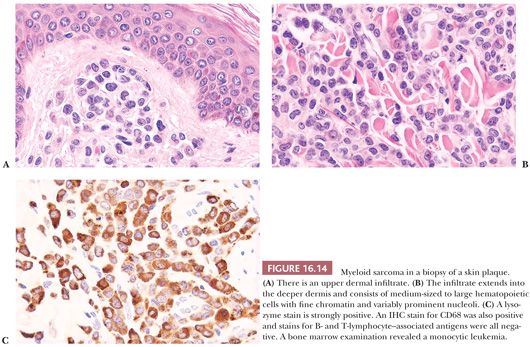
Myeloid Proliferations Related to Down Syndrome
There is a striking increase in the incidence of acute leukemia in young children with Down syndrome. Unlike non–Down syndrome children, there is an approximately equal distribution between AML and ALL and furthermore, there is a markedly increased incidence of AMKL. Approximately 10% of neonates with Down syndrome manifest a unique transient abnormal myelopoiesis (TAM) (177). TAM is an abnormal proliferation of myeloid blasts and other immature myeloid cells in the blood. In TAM, total leukocyte and blast counts vary from mildly to markedly increased, rarely reaching several hundred thousand per cubic millimeter. There is typically a prominent megakaryoblast component to the proliferation, and the blast percentage is usually higher in the blood than in the bone marrow. The process usually resolves without therapeutic intervention in 2 to 14 weeks. In 20% to 30% of patients with TAM, an AML evolves 1 to 3 years later and is most commonly AMKL. The AMKL shows similar morphologic features to the TAM but does not resolve without therapeutic intervention. There are clinical differences between the two processes, one being age of onset. AMKL rarely occurs in Down syndrome in the first month of life. Blasts in TAM only rarely have cytogenetic abnormalities other than constitutional trisomy 21, whereas those in AMKL often exhibit additional abnormalities. Evidence indicates that TAM is a clonal disorder. Mutations in the transcription factor GATA1 have been found in children with TAM and the mutation is nearly always present in Down syndrome patients with AMKL, which suggests that GATA1 mutagenesis represents a very early event in Down syndrome leukemogenesis and that TAM and AMKL are related disorders (178–181). AMKL may represent clonal evolution of a spontaneously remitted and dormant TAM (175). The prognosis for young Down syndrome patients with AMKL is very favorable relative to non–Down syndrome patients with AMKL.
BLASTIC PLASMACYTOID DENDRITIC CELL NEOPLASM
This rare tumor is a highly aggressive, blastic neoplasm derived from plasmacytoid dendritic cells that almost always presents initially in skin. Blood and bone marrow involvement is variable at presentation, but as a rule, overt leukemia develops over time (91,182–187). Marrow involvement ranges from low-level interstitial involvement to diffuse effacement. The cells are typically primitive appearing, medium-sized blasts with irregular nuclei; fine chromatin; small nucleoli; and scanty, agranular cytoplasm, closely resembling lymphoblasts. The neoplastic cells express CD123, CD56, CD4, CD45, and CD43. They express TdT in a substantial minority of cases but are CD34(−). They may express CD7 and/or CD33 but usually lack any other lymphoid and myeloid antigens. Expression of CD56 and TdT (when present) differentiate this entity from proliferations of mature plasmacytoid dendritic cells, which may be seen in reactive states and in association with other myeloid neoplasms, particularly chronic myelomonocytic leukemia (CMML) (188). The differentiation from AML may be challenging, but the characteristic clinical and immunophenotypic features allow confident diagnosis in most cases. Recently, expression of BDCA-2 and BDCA-4 have been suggested as useful features for the diagnosis of blastic plasmacytoid dendritic cell neoplasm (186).
ACUTE LEUKEMIAS OF AMBIGUOUS LINEAGE
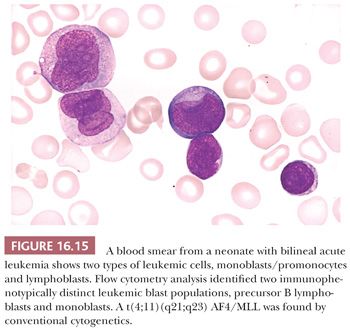
In these leukemias, the morphologic, cytochemical, and immunophenotypic features of the blasts lack sufficient specificity to classify the leukemias as either myeloid or lymphoblastic (91). These may be acute, undifferentiated leukemias in which there is lack of expression of lineage differentiation antigens. Other cases may exhibit morphologic and immunophenotypic features of both myeloid and lymphoblastic cells. These may be either bilineal leukemias with separate blast populations—one expressing myeloid characteristics and the other lymphoid—or biphenotypic, in which the blasts express characteristics of both myeloid and lymphoid cells to a degree that it is not possible to assign specific lineage (120,189,190) (Fig. 16.15). In addition to mixed immunophenotypes, there may be a mixture of morphologic, cytochemical, and ultrastructural features in these cases. Bilineal leukemias may be synchronous with simultaneous, distinct populations of leukemic cells of more than one lineage or metachronous (lineage switch) in which one lineage is expressed following the other. In the latter case, reappearance of the original clone must be demonstrated to distinguish from the emergence of a secondary (therapy-related) leukemia. There is an increased incidence of chromosome translocations involving 11q23 or a t(9;22) in mixed phenotype acute leukemias (189,190). The WHO classification of acute leukemias of ambiguous lineage and the requirements for assigning more than one lineage to a single blast population are shown in Tables 16.6 and 16.8 (91).

PRECURSOR LYMPHOID NEOPLASMS
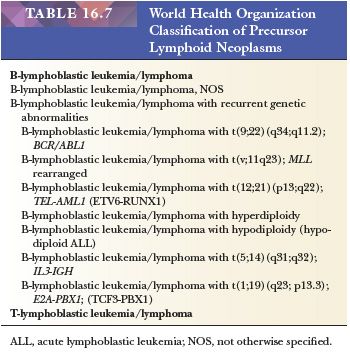
The WHO classification of lymphoblastic neoplasms is based on immunophenotypic and cytogenetic/molecular findings and recognizes the most important treatment and prognostic groups (91) (Table 16.7). There are two major immunophenotypic types, B-lymphoblastic leukemia/lymphoma and T-lymphoblastic leukemia/lymphoma. Within the B-lymphoblastic group, there are six categories determined by recurrent cytogenetic/molecular abnormalities.
B-Lymphoblastic Leukemia/Lymphoma
B-lymphoblastic leukemia/lymphoma is the most common of the immunophenotypic categories, accounting for about 85% of cases in children and 75% in adults. B-lymphoblastic neoplasms are acute leukemias in the large majority of cases and only rarely present as lymphoma without bone marrow and blood involvement (191,192). The presenting signs and symptoms of B-lymphoblastic leukemia (B-ALL) usually relate to blood cytopenias resulting from bone marrow failure. Physical findings may include pallor, ecchymoses or petechiae, lymphadenopathy, and organomegaly. In a minority of patients, the presenting clinical symptoms are caused by extramedullary leukemic infiltrates. Lymph node, CNS, skin, gonadal, renal, bone, and joint involvement are most frequent sites of extramedullary involvement; the CNS and testicles are major sites of extramedullary relapse, often independent of bone marrow relapse.
The morphologic features on blood and marrow smears that distinguish ALL and AML are listed in Table 16.3. The lymphoblasts in B-ALL are small to medium-sized, approximately twice the size of normal small lymphocytes, with sparse cytoplasm and a high nuclear-to-cytoplasmic ratio (Fig. 16.16A). The nucleus is generally round or oval, but a variable number of cells have an indented or convoluted nuclear outline. The chromatin is usually coarsely reticular and quite homogenous. In most cases, nucleoli are small and indistinct or not visualized. The cytoplasm is variably basophilic. Vacuoles are often present, and cytoplasmic granules are found in a small number of cases. In a minority of cases, the predominant lymphoblasts are large. In these, the blast size may be relatively uniform or quite heterogeneous, but most of the lymphoblasts exceed twice the size of a normal small lymphocyte. In the large blasts, nucleoli are often prominent and vary from one to four; these cells may be difficult to distinguish from myeloblasts.
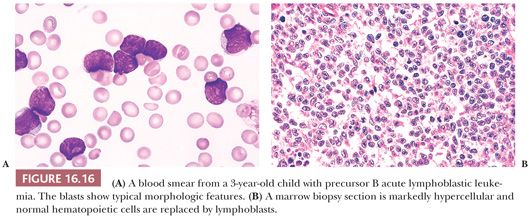
In biopsy sections, the marrow is usually markedly hypercellular (Figs. 16.8 and 16.16B) but may be normocellular and, in rare cases, hypocellular. Normal hematopoiesis is markedly reduced, and the marrow is replaced by a uniform, diffuse proliferation of lymphoblasts. Occasionally, there is only partial marrow involvement in an interstitial pattern. A focal pattern of involvement is rare in ALL at diagnosis but may be observed at relapse (193). Cytoplasm is barely discernible and nuclei are mostly medium sized with evenly dispersed to moderately dense chromatin and inconspicuous nucleoli. Nuclear contour is often heterogeneous; convoluted cells are usually present and may predominate. Mitotic activity is variable but usually brisk, and mitotic figures are always easy to find.
Immunophenotyping should be performed in all cases of ALL to differentiate it from AML and to distinguish B-ALL and T-ALL. The profile of antigen expression on the leukemic blasts also serves as an important fingerprint for later assessment for minimal residual disease (105,194). The lymphoblasts in B-ALL express various combinations of the B-lymphocyte–associated antigens CD19, CD22, CD79a, CD24, CD10, and CD9 and several lineage-nonspecific antigens, including CD34, TdT, HLA-DR, CD38, and CD45 (106,108,195). Mu immunoglobulin heavy chains (cytoplasmic immunoglobulin [CIg]) are present in the cytoplasm of lymphoblasts in 20% to 25% of B-ALL but, except in rare cases, lymphoblasts lack expression of surface immunoglobulin. More than 95% of cases of B-ALL are TdT positive and the progenitor cell–associated antigen CD34 is found in about 80%. TdT is useful in differentiating ALL from lymphoproliferative disorders of mature lymphocytes, which are TdT negative, but more than 90% of precursor T-ALL and 5% to 10% of AML also express TdT (196). In virtually all cases of B-ALL, the lymphoblasts exhibit incomplete maturation and immunophenotypic asynchrony and aberrancy that deviate from the spectrum of antigen expression typical of normal B-lymphocyte stages of maturation (18,195). In 30% to 80% of cases, one or more myeloid-associated antigens are detected on the neoplastic lymphoblasts; CD15, CD13, and CD33 appear to be most common (18,195).
Conventional cytogenetic analysis shows 80% to 90% of cases of precursor B-ALL have demonstrable chromosome abnormalities (197,198). The incidence is even higher when FISH techniques are used to supplement conventional studies (199). The major recurrent cytogenetic abnormalities define categories of B-ALL in the WHO classification. Chromosome numeric changes are found in about 50% of children and 15% of adults with B-ALL. In children, ploidy defines important prognostic groups, but in adults, it has little effect on prognosis except in a small group of hypodiploid cases (200,201). The five major chromosome numeric groups of B-ALL are hyperdiploid with more than 50, hyperdiploid with 47 to 50, diploid, hypodiploid, and pseudodiploid. Structural changes are always present in pseudodiploid ALL and may be found in the other numeric groups except diploid. Translocations are most important because several recurrent ones are independent indicators of prognosis.
Molecular technologies provide capability for identifying additional prognostic groups of ALL and may provide markers to integrate into diagnostic testing and to be targeted with novel molecular-based therapy. New methods for high-resolution genome-wide analysis have identified new subtypes of high-risk B-ALL (202). One of these is B-ALL associated with a deletion of IKZF1, a gene that encodes the lymphoid transcription factor IKAROS. Deletion of IKZF1 is seen with high frequency in BCR-ABL1–positive ALL but also is found in cases of BCR-ABL1–negative B-ALL and is associated with high-risk disease in both groups (203).
B-Lymphoblastic Leukemia/Lymphoma, Not Otherwise Specified
B-lymphoblastic leukemia/lymphoma, NOS is a precursor B neoplasm presenting as acute leukemia, or rarely as lymphoma, without any of the designated recurrent genetic abnormalities used to define specific categories of B-lymphoblastic neoplasms (91). There are no distinctive clinical morphologic, immunophenotypic, or genetic findings in B-lymphoblastic leukemia/lymphoma, NOS.
B-Lymphoblastic Leukemia/Lymphoma with Recurrent Genetic Abnormalities
B-ALL with t(9;22)(q34;q11), BCR/ABL1 (Philadelphia [Ph1] chromosome) arise from a reciprocal translocation involving the cytoplasmic tyrosine kinase gene ABL on chromosome 9q34 and the BCR (breakpoint cluster region) on chromosome 22q11 (204). The t(9;22) is found in the lymphoblasts of 3% to 5% of children with ALL and approximately 30% of adults, making it the most common structural abnormality in adults with ALL (197,201,205). Translocation (9;22) cases span the spectrum of morphology for ALL. There are no defining cytologic features, but there appears to be a higher proportion of cases with a predominance of large blasts with prominent nucleoli than for other B-ALL, and cytoplasmic granules are more commonly observed (206,207). Translocation (9;22) ALL is characterized by an older age and high presenting leukocyte counts and, in some reports, more frequent organomegaly and CNS involvement (206,208,209). The prognosis is unfavorable in both children and adults.
B-ALL with t(v;11q23);MLL rearranged is found in up to 80% of infants with ALL and approximately 10% of older children and adults (201,210,211). Most of the translocations at 11q23 involve the MLL gene (197,212). There are numerous partner genes in MLL translocations; the AF4 gene at 4q21, which partners in the t(4;11)(q21;q23)-AF4/MLL, is the most frequent, occurring in about 60% of infants, 2% of other children, and 3% to 6% of adults with ALL (113,201). The leukocyte count is typically markedly increased in t(4;11) ALL. In blood and marrow smears, there are no defining cytologic features. In cases of bilineal t(4;11) leukemia, both lymphoblasts and neoplastic myeloid cells (usually monoblasts and promonocytes) are observed (113). The immunophenotype is characteristically that of early precursor B-ALL: CD10(−), TdT(+), CD34(+), CD19(+), HLA-DR(+). The myeloid-associated antigen CD15 is present in the majority of cases and CD13 and CD33 are commonly expressed (113). The presence of an MLL rearrangement is significantly associated with high-risk clinical features: age under 1 year, markedly elevated leukocyte counts, and a relatively high frequency of CNS involvement. The prognosis is among the worst for ALL, with a high rate of relapse and poor overall survival (113,200,210).
B-ALL with t(1;19)(q23;p13), PBX1/E2A is the most frequent translocation identified by conventional cytogenetics in children with precursor B-ALL. It is found in 5% to 6% of patients, 25% of CIg-positive cases, and approximately 1% of CIg-negative cases; it is less common in adults (111,197). There are no distinctive morphologic findings associated with t(1;19) ALL. The immunophenotype of the lymphoblasts is characterized by homogeneous expression of CD19, CD10, CD9, absent CD34, and absent or underexpression of CD20 (213).
High-risk features have been reported in ALL with t(1;19), including high leukocyte counts, more frequent CNS involvement, and black race (201,214). Recent studies, however, have not always corroborated the high frequency of these adverse features (215). The poor prognosis once ascribed to CIg-positive ALL, and attributed specifically to the t(1;19), appears to have been overcome by contemporary therapies (216).
B-ALL with t(12;21)(p13;q22), TEL/AML-1 (ETV6/RUNX1) is a cryptic translocation generally not found by conventional cytogenetic karyotyping because the rearranged segments are too small to be recognized. The translocation is identified by molecular techniques (e.g., PCR or FISH). The TEL/AML-1 fusion is found in 16% to 39% of children and 3% to 4% of adults with precursor B-ALL, making it the most common chromosome structural abnormality in childhood ALL (201,217,218). In some patients, t(12;21) is the only cytogenetic aberrancy and the karyotype appears normal. In others, additional abnormalities are present, but high hyperdiploidy (>50 chromosomes) is virtually never observed with t(12;21) (218). There are no distinctive morphologic features associated with t(12;21) ALL, and the immunophenotype is generally that of common precursor B-ALL. Bright CD10 and HLA-DR, lack of expression of CD9 and CD20, and frequent expression of the myeloid-associated antigens CD13 and CD33 are characteristic.
Most studies suggest that t(12;21) ALL has an excellent prognosis (217,218). Response to conventional antimetabolite-based therapy is excellent, with remission rates approaching 100% and high event-free and overall survival.
B-ALL with hyperdiploidy with more than 50 chromosomes is found in 25% to 30% of children and about 5% of adults with precursor B-ALL. There are no unique morphologic or immunophenotypic findings in this category. In children, hyperdiploidy greater than 50 is associated with favorable clinical features and an excellent prognosis; long-term, event-free survival is more than 80% (219). There are prognostic variables within hyperdiploidy greater than 50 ALL. Duplication of chromosomes 4 and 10 appears to impart a particularly favorable prognosis (220). Assessment for duplication of these chromosomes can be done by interphase FISH analysis if conventional cytogenetic studies are not available.
B-ALL with hypodiploidy is present in 2% to 9% of cases. Most of these have 45 chromosomes; chromosome 20 is commonly lost in children (111). There are no specific presenting clinical, morphologic, or phenotypic features associated with this group. Hypodiploidy with less than 45 chromosomes is considered a poor prognostic finding in children, and adults with hypodiploidy do very poorly; it is the one numeric abnormality with independent prognostic value in adults (171). Hypodiploidy with a near-haploid number of chromosomes, found in only about 1% of cases of ALL, has a particularly bad prognosis regardless of age (221).
B-ALL with t(5;14)(q31;q32), IL3/IGH is a rare category of B-ALL characterized by hypereosinophilia (91,222,223). The translocation joins the interleukin-3 gene (IL-3) at 5q31, with the immunoglobulin heavy chain gene at 14q32 forming a fusion gene (IL-3/IgH) that results in excess IL-3 messenger RNA and elevated serum IL-3 levels. The excess IL-3 presumably triggers increased production of eosinophils in the marrow. This rare type of ALL occurs in both children and adults (median age ~14 years). Patients frequently present with symptoms of the hypereosinophilic syndrome; cough, dyspnea, chest pain, skin rash, pulmonary infiltrates, CSF eosinophilia, splenomegaly, and lymphadenopathy are all common. In approximately half of the patients, eosinophilia is recognized for some time prior to the diagnosis of ALL; the leukemia is obscured by the marked eosinophilia. The eosinophilia typically resolves if a complete remission is achieved only to return as the leukemia relapses. B-ALL with t(5;14) is a high-risk disease with poor prognosis. Although about 90% of patients respond initially to therapy, most relapse and die of ALL or complications of hypereosinophilia; death due to cardiac failure is not uncommon.
The differential diagnosis of B-ALL includes several reactive processes and other neoplastic disorders that may manifest clinical or morphologic similarity to ALL. These include increased bone marrow hematogones (normal B-lymphocyte precursors), reactive lymphocytosis, aplastic anemia, AML, leukemic Burkitt lymphoma (Burkitt cell leukemia) and other non-Hodgkin lymphomas, chronic lymphoproliferative disorders, and metastatic small cell tumors. The distinctive features of these are discussed in other sections of this chapter.
T Acute Lymphoblastic Leukemia
Most of the clinical, morphologic, and cytochemical findings in T-ALL are quite similar to those of B-ALL, described previously. Only features that are distinctive for T-ALL will be discussed here.
The median age of children with T-ALL is higher than for B-ALL, and a greater proportion of patients are adults; approximately 80% of patients are male. There is more often significant extramedullary disease; approximately 50% of patients present with a mediastinal mass, and other lymphadenopathy and organomegaly are more frequent as well. T-lymphoblastic neoplasms more commonly present as a lymphoma with minimal or no bone marrow involvement (91,102). Relative to B-ALL, there is a higher incidence of CNS disease at presentation (12%) and relapse; the median leukocyte count is significantly higher; and there is more often marked leukocytosis ( >100 × 109 per liter) (224). Morphologic findings in blood and marrow smears in T-ALL are generally similar to B-ALL.
Marrow biopsy sections also appear similar but, in some cases of T-ALL, most of the lymphoblasts are convoluted and the mitotic rate is generally higher, averaging about twice the number of mitotic figures per high-power field found in B-ALL (225).
Immunophenotypically, the lymphoblasts in T-ALL express variable combinations of T-cell–associated antigens that include CD1a, CD2, CD3, CD4, CD5, CD7, and CD8, as well as several lineage-nonspecific antigens, especially CD45, CD34, and TdT (91,106,108,226,227). In children, nearly all cases of T-ALL express one or more of the antigens CD7 (approximately 97%), CD5 (approximately 95%), and CD2 (approximately 90%); concurrent expression of these three antigens is found in nearly 85% (224,227). CD3 is the most specific marker for T-lineage leukemia but is lacking on the cell surface in more than half of cases of T-ALL; however, most cases express cytoplasmic CD3 (cCD3). TdT is present in about 90% of cases, but CD34 is found less commonly than in B-ALL in children and in only about one-third of adult cases (228). TdT is valuable in distinguishing T-lymphoblastic leukemia/lymphoma from peripheral T-cell neoplasms, which lack TdT but may express any of the other T-cell–associated antigens except CD1a.
One immunophenotypic type of T-ALL, early T-cell precursor leukemia, is a very high-risk ALL (229). This leukemia is thought to arise from early T-cell precursors, a subset of thymocytes that retain stem cell–like features, and accounts for 10% to 15% of T-ALL. Early T-cell precursor leukemia is characterized by a distinctive immunophenotype, CD1a (−), CD8 (−), CD5weak, with stem cell–/myeloid-associated antigens and an early T-cell precursor–related gene expression profile. Patients with this leukemia have a very high risk of remission failure or relapse. There appears to be no specific morphologic or karyotypic features for recognizing this leukemia, and the diagnosis relies on flow cytometry immunophenotyping. It is important to recognize this subset of T-ALL at the time of diagnosis for planning the most effective clinical management (229).
Cytogenetic abnormalities are found in 50% to 60% of cases of T-ALL. In about one-third of cases of T-ALL, translocations involve the α/δ T-cell receptor loci at 14q11-q13 or the β/γ loci at 7q34; a variety of partner genes may be involved (112). In contrast to B-ALL, there is a relative lack of prognostic importance of chromosomal abnormalities in T-ALL, and no cytogenetic categories of T-ALL are included in the WHO classification (91,112). The recurring translocations of B-ALL are rarely observed in T-ALL; hyperdiploidy greater than 50 is also relatively uncommon and is not associated with survival advantage. Conversely, the two most common translocations in T-ALL, (t[11;14]) and (t[10;14]), are virtually never observed in B-ALL (112).
The differential diagnosis includes most of the same entities as listed for B-ALL and several peripheral T-cell proliferations, such as T-prolymphocytic leukemia, adult T-cell leukemia, and Sézary syndrome. These disorders are discussed in the section “T-cell and Natural Killer Cell Neoplasms.”
The remission rate and survival for patients with T-ALL has improved significantly with contemporary therapies. Despite this, the overall prognosis for T-ALL is less favorable than for B-ALL in children.
MYELODYSPLASTIC SYNDROME
MDS is a bone marrow stem cell disorder resulting in disorderly and ineffective hematopoiesis manifested by irreversible quantitative and qualitative defects in hematopoietic cells (Fig. 16.17). Most patients are over age 50 at diagnosis, but MDS may affect young adults and children. Some patients present with profoundly dysplastic changes and increased myeloblasts, allowing for an immediate diagnosis of MDS. In others, changes are subtle and the diagnosis may be delayed until the patient has been followed for several months and other causes of the cytopenias have been thoroughly evaluated and ruled out. The clinical and morphologic distinction of the more aggressive types of MDS and AML is frequently difficult. The criterion for distinguishing the two is the percentage of myeloblasts in the blood and marrow; a diagnosis of AML is made if there are 20% or more myeloblasts (91,230). Approximately one-third of patients with MDS develop AML within months.
The features used to define MDS are listed in Table 16.9. There are numerous descriptions of the cytologic features of the MDS in blood and marrow smears (91,94,96,165,229–239). These will be reviewed briefly in the discussion of the WHO categories of MDS. Examples of dysplastic changes for each of the lineages are listed in Table 16.10. The marrow sections in MDS are generally hypercellular (234,240). However, a normocellular or even hypocellular marrow is more common than in acute leukemia (237,241). The proliferative cells are obviously myeloid, with most showing maturation beyond the earliest stages. A panmyelopathy may be apparent or abnormalities of one cell type may predominate. In the refractory anemias, there is usually striking erythroid hyperplasia, and increased storage iron is common. In aggressive forms of MDS, all bone marrow cell lineages may be affected and there is often an increase in the most immature cells. In some cases, myeloblasts and promyelocytes are found in clusters remote from their usual paratrabecular location (Fig. 16.18). This abnormal localization of immature precursors (ALIP) is a useful diagnostic criterion and has been shown by some investigators to be indicative of an increased likelihood of leukemic transformation (242,243). Increased and dysplastic megakaryocytes are present in many cases. In a minority of patients, the biopsies show increased reticulin; rarely, severe myelofibrosis is observed (244–246). With progression of the MDS, the marrow usually becomes increasingly cellular and more severe dysplastic changes are noted. There is frequently a concurrent increase in myeloblasts and progressively severe bone marrow failure (229–233).

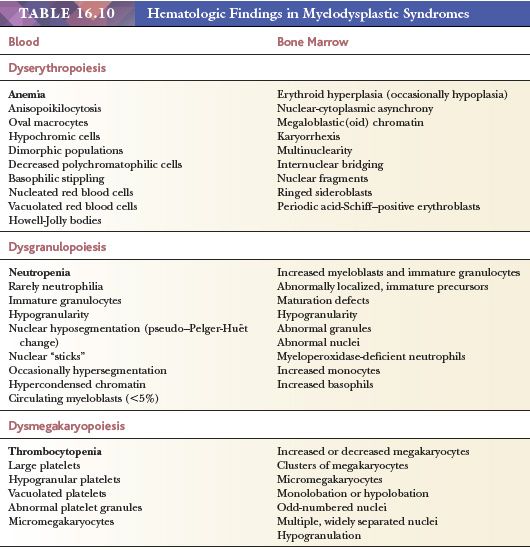
Immunophenotype and Cytogenetics
Immunophenotyping contributes to the diagnosis of MDS in some cases. On flow cytometry analysis, there is often aberrant antigen expression on the maturing leukocytes, and side scatter is abnormally diminished because of hypogranularity. IHC on marrow biopsies can aid in determining the blast percentage in some cases where there is a suboptimal aspirate.
As with the acute leukemias, bone marrow cytogenetic studies are important in the diagnosis and as an indicator of prognosis (123). The finding of bone marrow clonal abnormal (nonconstitutional) cytogenetics is very strong evidence in support of MDS in a patient with cytopenias and dysplasia (247,248). There are no cytogenetic changes that are specific for MDS. The majority of patients show a recurrent loss of chromosome material but reciprocal translocations or inversions may also be found, although less commonly than in AML. Frequent recurring chromosome defects in patients with MDS include complete or partial loss of chromosome 5 or 7, +8, 20q-, and complex chromosome abnormalities (230). 5q- alone or in combination with other chromosome abnormalities appears to be the most common defect. The type of cytogenetic abnormality has prognostic significance (123).
Classification of Myelodysplastic Syndrome
The WHO classification of MDS is shown in Table 16.11 (91,230). The categories are discussed in the following texts.

Refractory Cytopenia with Unilineage Dysplasia. The major manifestation of refractory cytopenia with unilineage dysplasia (RCUD) is ineffective hematopoiesis and dysplasia of a single lineage, erythropoiesis, granulopoiesis, or megakaryopoiesis. Refractory anemia is the most common and is characterized by anemia, reticulocytopenia, and erythroid hyperplasia in the bone marrow (91,94). The anemia may be normocytic or macrocytic with anisopoikilocytosis; oval macrocytes are common. There is no evidence of dysplastic changes in the neutrophils or platelets. Myeloblasts are not identified in the blood smear, and monocytes are less than 1.0 × 109 per liter.
The bone marrow is hypercellular or normocellular with erythroid hyperplasia in most cases; occasionally, the marrow is hypoplastic. Dyserythropoiesis may be noted but is usually not severe. Ring sideroblasts are occasionally observed but they number less than 15% of the erythroblasts. Dysgranulopoiesis and dysmegakaryopoiesis are absent. Myeloblasts are less than 5% in the marrow. A thorough evaluation for other causes of anemia must always be performed before a diagnosis of refractory anemia (RA) is made. In some cases, the diagnosis is made only after following the patient for several months and eliminating all other possible causes. Most patients with RA have a chronic course but may require red cell transfusion support. A small minority evolve to a more aggressive MDS and bone marrow failure or AML.
Refractory Anemia with Ringed Sideroblasts. The clinical and morphologic features of RA with ringed sideroblasts (RARS) are similar to those of RA (91,94). However, in RARS, 15% or more of the bone marrow erythroblasts are ringed sideroblasts. A dimorphic anemia with normal erythrocytes and microcytic or hypochromic poikilocytes is commonly found. Coarse basophilic stippling or Pappenheimer bodies are observed in some of the erythrocytes. Neutropenia and thrombocytopenia are not present or are minimal, and there are no dysplastic changes in either lineage. Myeloblasts are not present in the blood and monocytes are less than 1.0 × 109 per liter.
The bone marrow is hypercellular or normocellular with erythroid hyperplasia and markedly increased iron stores. The numerous ringed sideroblasts are the most prominent feature. Mild to moderate dyserythropoiesis may be observed but dysplastic changes in granulocytes and megakaryocytes are not present. Myeloblasts are rarely increased and never exceed 4%.
Ringed sideroblasts may be observed in any of the other categories of MDS, occasionally exceeding 15% of the erythroblasts. In some cases of refractory cytopenia with multilineage dysplasia, there are 15% or more ringed sideroblasts. This group is distinguished from RARS by the presence of bicytopenia or pancytopenia and dysplastic changes in more than one lineage. It is important to distinguish these cases from RARS because of their more aggressive clinical course.
Refractory Cytopenia with Multilineage Dysplasia. In refractory cytopenia with multilineage dysplasia (RCMD), there are one or more blood cytopenias and dysplastic changes in 10% or more of the cells in two or more myeloid lineages (91,230) (Fig. 16.17). Bone marrow and blood myeloblasts are less than 5% and less than 1%, respectively. RCMD is distinguished from RA by the significant changes in the granulocyte and platelet/megakaryocytic lineages in addition to the erythrocytes and differs from RA with excess blasts by having fewer than 5% myeloblasts. In some instances, dysgranulopoiesis or dysmegakaryopoiesis are the major abnormalities. RCMD usually has a more aggressive course than RA and a greater propensity for evolution to AML. The clinical course is often similar to that of refractory anemia with excess blasts (RAEB). In many respects, this category bridges RA and RAEB.
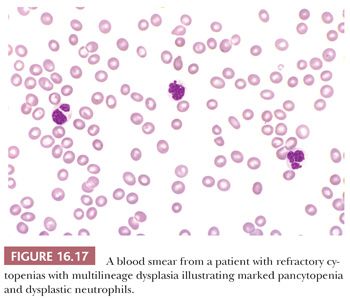
Refractory Anemia with Excess Blasts. Pancytopenia or bicytopenia are characteristic of RAEB (91,94). Dysplastic changes are commonly observed in erythrocytes, granulocytes, and platelets. Nucleated red blood cells and immature granulocytes, including myeloblasts and, occasionally, micromegakaryocytes, may be found in the blood smears. Myeloblasts may constitute up to 19% of the leukocytes in the blood.
The bone marrow is normocellular or hypercellular, and granulocytic or erythroid hyperplasia are present. Myeloblasts are increased to at least 5% but less than 20%. Auer rods may be present. Dyserythropoiesis is more severe than in RA or RARS. Dysgranulopoiesis is often prominent (Fig. 16.18). Megakaryocytic hyperplasia with dysmegakaryopoiesis and megakaryocytic clusters on biopsy sections may be observed. The features that distinguish RAEB from RA and RARS include the severity of the pancytopenia or bicytopenia, dysplastic changes in more than one cell lineage, and the presence of 5% or more myeloblasts in the blood or bone marrow.
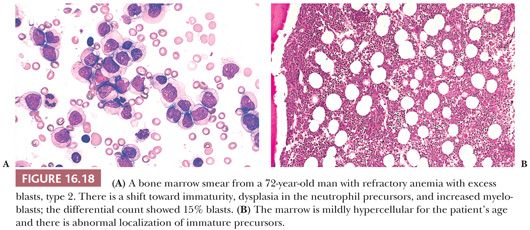
RAEB is distinguished from RCMD by the myeloblast percentage. The WHO classification separates RAEB into two types based on blast percentage (91). Type 1 RAEB is characterized by 2–4% blasts in the blood or 5–9% in the bone marrow. Type 2 RAEB has 5% to 19% blasts in the blood or 10–19% in the bone marrow (Fig. 16.18). The basis for this separation into two types stems from the finding that patients with RAEB with 10% or more myeloblasts often have a more aggressive course and a greater propensity for transformation to AML. The presence of Auer rods in a case of MDS, regardless of the blast count, elevates the diagnosis to RAEB type 2.
Myelodysplastic Syndrome Unclassifiable. MDS-U has features warranting a diagnosis of MDS but lacks the typical findings that define RA, RARS, RCMD, and RAEB (230). Cases are generally placed in this category for one of three reasons: (a) cases that exhibit features of RCUD but with 1% or more blasts in the blood, (b) cases otherwise classifiable as RCUD but with pancytopenia, (c) cases with cytopenias and cytogenetic abnormalities typical of MDS but lacking evidence of dysplasia in any lineage.
Myelodysplastic Syndrome with Isolated del(5q). MDS with isolated del(5q)chromosome abnormality (5q- syndrome) is characterized by macrocytic anemia, often thrombocytosis (approximately 50%), erythroblastopenia, megakaryocyte hyperplasia with nuclear hypolobation, and an isolated interstitial deletion of chromosome 5 (Fig. 16.19). The 5q- syndrome is found predominantly in older women (242). Most patients have a stable clinical course but are often transfusion dependent.
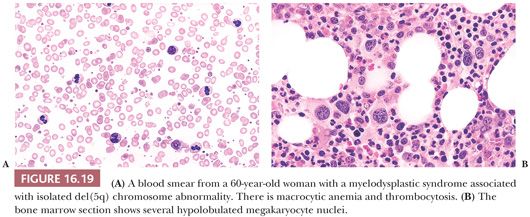
Childhood Myelodysplastic Syndrome. MDS is uncommon in childhood and about one-third of cases are associated with a congenital predisposing condition; these usually manifest in young children (249–251). Any of the standard categories of MDS that are seen in adults may be seen in children, but RARS and MDS with isolated del(5q) are extremely rare (91). The most common MDS in children is refractory cytopenia of childhood (RCC), which accounts for about half of all cases. RCC is characterized by persistent cytopenia, less than 5% blasts in the bone marrow and less than 2% in the blood (252). Dysplastic changes are present in two different lineages or in more than 10% of cells of a single lineage. In about 75% of cases, the bone marrow is hypocellular, often making RCC difficult to distinguish from other congenital or acquired causes of bone marrow failure (91). Most patients with RCC have normal cytogenetics; in those with a cytogenetic abnormality, monosomy 7 is most common (253). Patients with normal cytogenetics may experience a long indolent course of their disease. Patients with a monosomy 7 are the most likely to progress to a more aggressive MDS (253).
MYELOPROLIFERATIVE NEOPLASMS
The myeloproliferative neoplasms (MPNs) arise from clonal hematopoietic stem cells. They are characterized by effective autonomous proliferation of one or more myeloid lineages resulting in increased numbers of leukocytes, erythrocytes, and/or platelets in the blood. The elevation in blood counts is often marked. In the early phase of the disorders, the morphology of the expanded cell population(s) is/are usually normal. The WHO classification of the chronic MPNs is shown in Table 16.12 (254,255).

CHRONIC MYELOGENOUS LEUKEMIA
The diagnosis of chronic myelogenous leukemia (CML) is usually made from examination of a blood smear (256–258) (Fig. 16.20A). There is marked leukocytosis, with the entire neutrophil series represented from myeloblasts to segmented neutrophils; neutrophil myelocytes and segmented neutrophils are most numerous. Myeloblasts generally account for less than 5% of the leukocytes. All patients have basophilia, and the platelet count is elevated in more than half.
The bone marrow in CML is markedly hypercellular, often with a complete absence of adipose tissue. There is marked granulocytic hyperplasia, primarily of the neutrophil lineage, but an increase in basophils, eosinophils, and sometimes monocytes may be observed (Fig. 16.20B). Megakaryocytes are increased in most cases and may be markedly increased and clustered in biopsy sections; they are typically smaller than normal and hypolobated. This finding can be helpful in distinguishing CML from other chronic myeloproliferative disorders in which megakaryocytes are normal sized or larger than normal and frequently hyperlobated. In the absence of these megakaryocytic findings, the marrow smears and sections of some cases are not readily distinguishable from a profound leukemoid reaction (259).
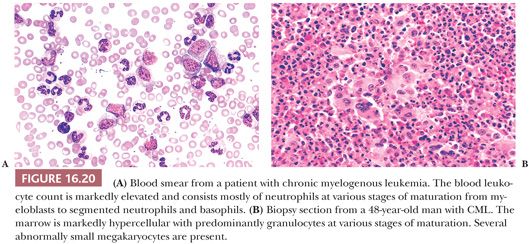
A mild increase in reticulin fibers may be found in marrow biopsy sections in the majority of patients, but prominent myelofibrosis is uncommon at diagnosis (260). Extensive myelofibrosis evolves during the course of the disease in a minority of patients. In these cases, the histopathologic features may be indistinguishable from those of primary myelofibrosis or advanced polycythemia vera (postpolycythemic myelofibrosis). Bone marrows from patients with CML have demonstrated decreased reticulin fibrosis by quantitative measures in the short term following initiation of imatinib (tyrosine kinase inhibitor), although imatinib did not eliminate the overall progression of fibrosis in these patients (261). The evolution to myelofibrosis in CML is reported to be a poor prognostic factor.
CML is defined by the presence of a specific chromosome rearrangement, the t(9;22) (q34;q11.2), also known as the Philadelphia chromosome. This translocation is identified by conventional cytogenetics in 95% of cases, with the remaining cases representing cryptic translocations. The breakpoints of the translocation involve the ABL gene on chromosome 9 and the BCR gene on chromosome 22. The chimeric BCR/ABL gene can be detected by PCR or FISH in all cases (262,263). Its presence distinguishes CML from other myelodysplastic/myeloproliferative neoplasms that may share some of the morphologic features of CML (e.g., CMML, atypical CML) (256).
The natural course of untreated CML is to evolve to an accelerated or blast phase in an average of approximately 3 years (264–268). Features of accelerated phase may include any of the following, singly or in various combinations: blast count of 10% to 19% in the blood or marrow; blood basophils of greater than 20%; persistent thrombocytopenia of less than 100 × 109 per liter; persistent thrombocytosis of greater than 1000 × l09 per liter, increasing spleen size, and/or leukocyte count (>10 × 109 per liter) while on adequate therapy; and chromosomal evolution (254). In blast phase, the blast count is 20% or more in the blood or marrow, thus meeting the acute leukemia definition with either myeloid or lymphoid differentiation (265). In some instances, there is an extramedullary blast proliferation prior to obvious bone marrow blast phase, or the marrow blast proliferation is focal in biopsy sections. The blast phase is generally poorly responsive to therapy and portends a short survival. In both accelerated and blast phases, granulocytic dysplasia may develop (269).
CHRONIC NEUTROPHILIC LEUKEMIA
Chronic neutrophilic leukemia (CNL) is a rare MPN characterized by a sustained increase (>25 × 109 per liter) in mature blood neutrophils without an identifiable cause (24,270–272). The marrow is hypercellular with granulocytic hyperplasia; erythroid and megakaryocyte hyperplasia may occur in some cases. There is minimal or no myelodysplasia, and myeloblasts are not increased. Splenomegaly is frequently present resulting from infiltration by mature neutrophils. Cytogenetics are normal in approximately 90% of cases. In cases with cytogenetic abnormalities, changes common to other myeloid neoplasms are the usual finding, for example, +8, del(20q), etc. Other MPNs must be effectively ruled out, including rare cases of CML with a variant BCR/ABL fusion gene, p230, which exhibits predominantly mature neutrophils in the blood and should not be mistaken for CNL (273).
Several cases of CNL have been reported in association with other neoplasms, most commonly plasma cell myeloma (274,275). None of these cases has had documented clonal cytogenetic changes in the granulocytes, and they may represent secondary proliferations due to abnormal cytokine release by the neoplastic cells.
CNL has a chronic course in most instances. Cases undergoing transformation to MDS or AML have been reported (271,272).
CHRONIC EOSINOPHILIC LEUKEMIA AND THE HYPEREOSINOPHILIC SYNDROME
In chronic eosinophilic leukemia (CEL), there is a sustained eosinophilia of greater than 1.5 × 109 per liter, increased eosinophils in the marrow, and less than 20% myeloblasts (254,276). Causes of reactive eosinophilia must be excluded by clonality assessment (cytogenetics or molecular abnormality), or an increase in myeloblasts (>2% in blood; >5% in marrow) should be demonstrated. Other myeloid disorders that may be associated with eosinophilia, such as CML and other MPNs, CMML, AML, and MDS, must be excluded. Disorders with eosinophilia and PDGFRA, PDGFRB, or FGFR1 abnormalities are also excluded (see “Myeloid and Lymphoid Neoplasms with Eosinophilia and Abnormalities of PDGFRA, PDGFRB, and FGFR1” section.) In cases where clonality of the eosinophils cannot be demonstrated and there is no increase in myeloblasts despite persistent eosinophilia and eosinophilia-associated organ dysfunction, the designation “idiopathic hypereosinophilic syndrome” is preferred.
The degree of eosinophilia is variable from the minimum necessary for a diagnosis to greater than 100 × 109 per liter. Most of the eosinophils are mature but scattered, and sometimes numerous immature eosinophils may be present. The eosinophils may display abnormal features, but these changes are often similar to findings observed in marked reactive eosinophilia. Neutrophilia is present in some cases. The marrow is hypercellular with a marked increase in eosinophil precursors. Usually, neutrophil, erythroid, and megakaryocyte maturation are normal. Myeloblasts may be increased but, in many cases, are normal in number at initial diagnosis. Dysplastic changes vary from no obvious abnormalities to dysplastic changes in both eosinophils and other lineages.
There are no specific cytogenetic abnormalities in CEL and idiopathic hypereosinophilic syndrome, but changes common to other myeloid neoplasms such as +8, i(17q) are found in some cases. Chromosome 8p11 rearrangements have been reported in several cases of CEL.
Most patients suffer multiorgan disease due to tissue infiltration by eosinophils and release of cytokines and other factors in the eosinophil granules. The most commonly affected organs are the heart, lungs, CNS, gastrointestinal (GI) tract, and skin. Survival is variable, from relatively short to more than 5 years. Massive splenomegaly, increased blasts, dysplasia, cytogenetic abnormalities, and severe clinical symptoms with organ damage are associated with more aggressive disease (276,277).
POLYCYTHEMIA VERA
Polycythemia vera (PV) is a myeloproliferative disorder characterized by panhyperplasia of the bone marrow with erythrocytosis, leukocytosis, and thrombocytosis in the blood (255,278–282). Erythroid and megakaryocytic hyperplasia are the most profound. The hemoglobin, hematocrit, and platelet count are often markedly increased. Diagnostic criteria in the WHO classification include increased red cell volume, presence of the JAK2 V617F or JAK2 exon 12 mutation, hypercellular marrow with panmyelosis, and low serum erythropoietin level (254). Markedly decreased erythropoietin levels usually distinguish PV from secondary causes of erythrocytosis. Trephine biopsy sections are usually moderately to markedly hypercellular, with striking erythroid hyperplasia and increased megakaryocytes, many of which are larger than usual and often polymorphic (283–286) (Fig. 16.21). The marrow may show increased vascularity and depleted storage iron. The prominent erythroid hyperplasia distinguishes PV from other MPNs. Not all PV cases require marrow biopsy for disease confirmation, as many can be diagnosed based on molecular and complete blood count studies.
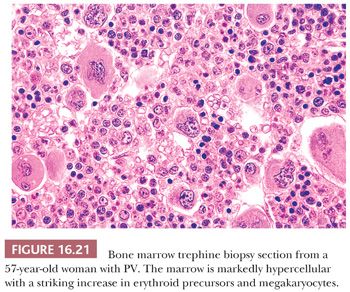
Reticulin may be increased in PV, particularly in areas where megakaryocytes are concentrated. There may be a gradual increase in reticulin fibrosis, culminating in advanced myelofibrosis and osteosclerosis at the terminal stage of disease (postpolycythemic myelofibrosis), at which time the changes in trephine biopsy sections are indistinguishable from primary myelofibrosis. Given the extensive myelofibrosis at this stage, the marrow may be hypocellular for age. The incidence of transformation of PV to AML is low and appears to be related to the type of chemotherapy used to control the polycythemia (286,287).
The histopathologic changes in PV must be distinguished from those in cases of secondary erythrocytosis. In secondary erythroid hyperplasia, megakaryocytes are not affected, the cellularity may be less than in PV, there is no increase in reticulin, and iron stores are less likely to be depleted. In most instances, PV and secondary erythrocytosis are readily distinguished by the erythropoietin levels and the WHO classification criteria (254).
ESSENTIAL THROMBOCYTHEMIA
Essential thrombocythemia is an MPN in which the most striking feature is a marked increase in megakaryocytes and florid thrombocytosis (282,288–293). The WHO classification criteria for diagnosis include a sustained platelet count of greater than 450 × 109 per liter, a marrow biopsy showing proliferation mainly of the megakaryocyte lineage with increased numbers of enlarged mature megakaryocytes, and exclusion of other causes of thrombocytosis (254) (Fig. 16.22). The morphology of the platelets on blood smears may vary from normal to large, atypical forms. The bone marrow is normocellular or hypercellular (292). Megakaryocytes show considerable variation in size, similar to those in PV, and may be present scattered or in loose clusters. The characteristic megakaryocytes are termed staghorn-like and have large size and deeply hyperlobated nuclei. Mildly increased reticulin is demonstrable in some cases, but prominent fibrosis is exclusionary.
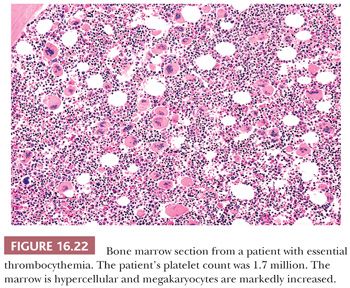
Essential thrombocythemia must be distinguished from secondary thrombocytosis and the other chronic MPNs, especially primary myelofibrosis and PV. The platelet count is generally not as elevated in secondary thrombocytosis, but the distinction is best made by the clinical findings. Panhyperplasia is present in some cases of essential thrombocythemia, but erythroid precursors are less strikingly increased than in PV and the red cell mass is normal (292,294). Primary myelofibrosis tends to have tight clusters of large, atypical megakaryocytes and a background of granulocyte hyperplasia, in contrast to the loose clustering and sole megakaryocyte hyperplasia seen in essential thrombocythemia (295).
There is no consistent cytogenetic abnormality identified in essential thrombocythemia. Approximately 50% of cases will have a JAK2 V617F mutation; identification of this abnormality allows for distinction from nonclonal reactive thrombocytoses.
PRIMARY MYELOFIBROSIS
Primary myelofibrosis (PM) is a chronic MPN characterized by marrow fibrosis, extramedullary hematopoiesis, and moderate to marked splenomegaly (255,282,296–304) (Fig. 16.23). In the natural course of the disease, the bone marrow becomes increasingly fibrotic and in some cases osteosclerotic; normal hematopoietic tissue is gradually reduced. The myelofibrosis appears to be caused by factors produced and released by the abnormal megakaryocytes (305,306). The disease is characterized by a chronic course, often of several years’ duration. The designation “malignant myelosclerosis” has been used to describe cases with an unusually rapid clinical course (307).
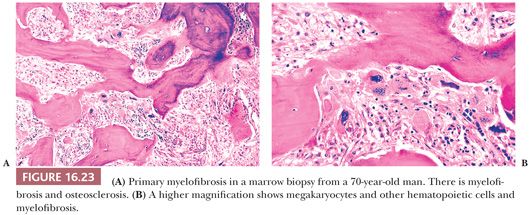
The histopathology of the marrow reflects the progression of the disease and can be divided into three phases (308). A hypercellular (prefibrotic) phase is characterized by an increase in hematopoietic elements, primarily megakaryocytes and granulocytes. Increased fibrous connective tissue is not always apparent in the hypercellular phase, but increased reticulin can usually be demonstrated around clusters of megakaryocytes. A patchy phase features alternating areas of hematopoiesis and fibrosis. The phase of obliterative myelosclerosis (fibrosis) is characterized by extensive marrow replacement with fibrous connective tissue. The remaining scattered clusters of hematopoietic cells have the appearance of being entrapped and compressed in the fibrotic marrow; megakaryocytes may be markedly distorted. Osteosclerosis is often prominent in this terminal phase of the disease. To standardize interpretations, scoring systems exist for the semiquantitative assessment of marrow fibrosis (309). Although the marrow histopathologic features may vary among the three phases in biopsies from different anatomic sites (308), megakaryocytic atypia is a feature common to all three phases (295,309). Megakaryocytes show sizes ranging from small to large, frequent arrangement in tight clusters, and morphology characterized by “cloud-like” bulbous nuclear lobation and hyperchromatic nuclei. Of all the MPNs, PM has the most megakaryocytic atypia—a feature which can be used to discriminate the different diseases (254).
In the hypercellular phase, blood counts are normal or increased; immature granulocytes and atypical enlarged/giant platelets are commonly observed. A leukoerythroblastic reaction may be observed, including occasional circulating myeloblasts. As the marrow becomes increasingly fibrotic, the blood counts drop and more abnormal cells appear in blood smears. Red cell changes are particularly striking, with abundant teardrop cells present. Extramedullary hematopoiesis and spleen size increase as the marrow becomes progressively sclerotic.
There is no consistent cytogenetic abnormality identified in PM. Approximately 50% of cases will have a JAK2 V617F mutation; the presence of this abnormality does not distinguish PV from PM.
MASTOCYTOSIS
Mastocytosis encompasses several clinical syndromes that result from an abnormal, clonal proliferation of mast cells in one or more organs. The diseases range from solitary organ involvement to widespread systemic disease and have clinical behaviors ranging from benign lesions to aggressive leukemia. The WHO classification of mastocytosis is shown in Table 16.13 (254,310). Only those disorders involving the bone marrow are discussed.

According to the WHO, in order to diagnose systemic mastocytosis, cases must fulfill the major criterion plus one minor criterion or three minor criteria. The major criterion is the presence of multifocal, dense, or compact infiltrates of 15 or more mast cells in marrow biopsies or other extracutaneous organs. These should be confirmed by tryptase or other special stains. The minor criteria include:
• More than 25% of the mast cells in the infiltrate on a biopsy section are spindle shaped or have atypical morphology, or more than 25% of the mast cells in a marrow aspirate are immature or atypical
• Detection of a KIT point mutation
• Mast cells abnormally expressing CD2 and/or CD25
• Serum tryptase persistently greater than 20 ng/mL (unless there is an associated clonal myeloid disorder)
Bone marrow is the tissue most commonly biopsied to establish the diagnosis of systemic mastocytosis and is involved in most cases (310–317). There is a wide variation in degree and pattern of involvement from case to case. In more than 80% of patients, the pattern of involvement is focal and diffuse in less than 20% (313,315,318). Focal lesions may be paratrabecular, perivascular, or randomly distributed; all three types may be observed in the same biopsy specimen (313,316). The paratrabecular lesions are associated with fibrosis and expansion of the trabeculae (Fig. 16.24). The perivascular lesions induce prominent medial and adventitial hypertrophy.
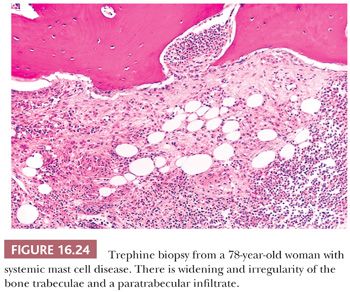
Focal lesions may be polycellular or relatively monocellular (313,316,317). Polycellular lesions are usually randomly distributed and characterized by a mixture of mast cells, lymphocytes, eosinophils, neutrophils, histiocytes, endothelial cells, and fibroblasts. The various cell types may be uniformly mixed, but often there is an element of compartmentalization of cell types with a central focus of lymphocytes encircled by mast cells (Fig. 16.25). The mast cells are usually round or oval with abundant eosinophilic cytoplasm. The monocellular focal lesions are composed primarily of mast cells with occasional lymphocytes and eosinophils. The mast cells are frequently spindle shaped with pale to lightly eosinophilic cytoplasm. The nuclei are round, oval, elongated, or monocytoid in configuration; these mast cells resemble histiocytes or fibroblasts. In all types of focal lesions, nucleoli are inconspicuous and mitotic figures absent. The uninvolved portion of the bone marrow may be normocellular or hypercellular with increased granulocytes (313,316).
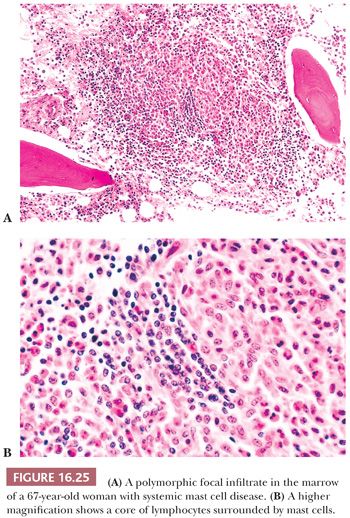
In diffuse lesions, the entire marrow space between trabeculae is replaced. The mast cells vary from round to elongated and may resemble fibroblasts. They are frequently mixed with neutrophils, eosinophils, and macrophages. Moderate to marked fibrosis is present, and normal hematopoietic cells are markedly reduced.
Bone changes are common in both focal and diffuse lesions. Widened, irregular trabeculae are often observed, particularly in areas near paratrabecular lesions. In some cases, osteoclasts are increased and are associated with thinning of trabeculae (313,319).
There is a relatively common association of systemic mastocytosis and myeloid neoplasms, including AML, MDS, MPN, or CMML (systemic mastocytosis with associated clonal hematologic non–mast cell lineage disease). In these cases, the bone marrow biopsy shows features of both disorders (316,320,321) (Fig. 16.26). Normally, bone marrow aspirate smears contain only a few scattered mast cells, with mast cells morphologically varying from normal appearing with round nuclei and densely packed granules to unusually large cells with abundant cytoplasm and scattered fine granules. When there is a concurrent myeloid neoplasm, atypical-appearing mast cells may be found in the marrow smears along with increased myeloblasts and dysplastic or proliferative myeloid compartments (318,320,321). Collections of atypical mast cells may be concentrated in bone marrow particles and thus difficult to identify.
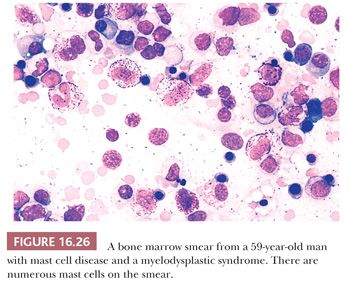
Recognition of mast cells in a lesion on H&E-stained sections of bone marrow may be problematic because of their morphologic similarity to histiocytes and fibroblasts. When histologic features suggest mastocytosis, the mast cell tryptase and CD117 (c-kit) immunocytochemical stains are the most useful for confirmation of the diagnosis (322). The tryptase stain is highly sensitive and the most specific for mast cells in the bone marrow (Fig. 16.27). CD117 is strongly expressed by mast cells but may also be positive, although weaker, in early myeloid cells; the strength of staining can help differentiate these cell populations in difficult cases. CD33, CD45, CD68, vimentin, lysozyme, α1-antitrypsin, and α1-antichymotrypsin are expressed in normal and most neoplastic mast cells. Mast cells lack myeloperoxidase, CD14, CD15, CD16, CD34, and most B- and T-cell–associated antigens (322–325). Antibodies for several of the chemical mediators produced by mast cells can also be applied by IHC methods (323). Mast cell granules exhibit prominent metachromasia with a Giemsa or toluidine blue stain in both marrow smears and sections (304). The chloroacetate esterase stain may be helpful in identifying mast cells in nondecalcified, formalin-fixed specimens (313,315).
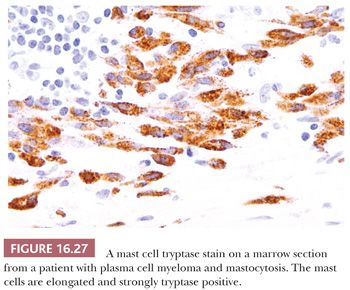
IHC staining or flow cytometry analysis for CD2 and CD25 expression can be particularly useful in distinguishing normal mast cells from mastocytosis. Neoplastic mast cells differ from their normal counterparts by the frequent aberrant expression of CD2 and/or CD25 (325–329). Recent studies have identified CD30 expression in neoplastic mast cells, preferentially in mastocytosis with aggressive clinical courses (330).
Conditions that merit consideration in the differential diagnosis of systemic mastocytosis in marrow sections include chronic idiopathic myelofibrosis, angioimmunoblastic T-cell lymphoma, hairy cell leukemia, Hodgkin lymphoma, and eosinophilic granuloma. IHC stains will provide the distinction from these disorders in nearly all cases.
An activating KIT point mutation is identified in 95% of systemic mastocytosis. Bone marrow aspirate specimens are among the specimens of choice for this molecular analysis (331). The identification of a KIT mutation is not only of diagnostic utility in mastocytosis but also confers resistance to tyrosine kinase inhibitor therapy.
In the rare cases of mast cell leukemia, the marrow is markedly hypercellular with replacement of normal hematopoietic cells by atypical mast cells that are frequently immature appearing with decreased granules and irregular-shaped nuclei or bilobed nuclei (313–315,320,332). Mast cells are rarely found in blood smears except in cases of mast cell leukemia, where they account for greater than 20% of the cellularity (315,321,332).
MYELODYSPLASTIC/MYELOPROLIFERATIVE DISEASES
Some cases of myeloid neoplasm overlap MDS and MPN in their morphologic and clinical features and do not clearly fit into the categories of either group. The WHO classification recognizes four such myeloid neoplasms; these are described in the following discussion (91) (Table 16.14).

CMML exhibits features that overlap MDS and MPNs. CMML is associated with a broad spectrum of clinical and hematologic presentations (94). Some patients exhibit the typical clinical and morphologic features of MDS, including blood cytopenias, ineffective hematopoiesis, dysplastic changes, and increased blasts. The morphologic features of CMML in these cases may be similar to those of RCUD, RCMD, or RAEB, with the addition of monocytosis of greater than 1.0 × 109 per liter. The monocytes may exhibit dysplastic features in blood smears in the form of hyperlobated nuclei, increased basophilia of the cytoplasm, and abnormal granulation. Blasts and promonocytes comprise fewer than 20% of the nucleated cells in the blood and marrow. Other cases present with leukocytosis, sometimes marked leukocytosis, with monocytosis, organomegaly, and minimal or no dysplasia or increase in blasts; there is effective hematopoiesis, and erythrocyte and platelet counts may be normal. Regardless of the presenting features in CMML, it is not always predictable whether a case will evolve clinically like MDS or MPN. There are two subcategories of CMML based on the blast count. In CMML type 1, there are fewer than 10% blasts in the blood and bone marrow; in CMML type 2, there are 10% to 19%.
Cytogenetic abnormalities are found in CMML in 20% to 40% of cases (247,91,333). There are no cytogenetic abnormalities that are specific to CMML, but the most common are +8, −7, del(7q), and abnormalities of 12p. Point mutations of RAS genes are found in up to 40% of cases at diagnosis or later in the course of disease (91,334).
The course of disease varies from aggressive to indolent with corresponding survivals. Up to 30% of patients experience eventual transformation to AML (91).
Atypical chronic myeloid leukemia (aCML) lacks a t(9;22)(q34;q11.2) BCR-ABL1 and is characterized by an increased leukocyte count composed predominantly of cells in the neutrophil lineage. Mature neutrophils predominate but immature granulocytes usually account for more than 10% of the blood leukocytes. Monocytosis may occur but is less than 10% of the leukocytes (256). The bone marrow is hypercellular and there is granulocytic hyperplasia with dysplastic changes. In some cases, there is dyserythropoiesis and dysmegakaryopoiesis. Several features of aCML are similar to CML but, unlike CML, basophilia is minimal or lacking; dysplasia is a prominent feature in the neutrophils; and anemia and thrombocytopenia are common (256). Cytogenetic abnormalities are found in up to 80% of patients; the most common abnormalities are +8 and del(20q) (91,334–336). The prognosis for aCML is similar to an aggressive MDS, with reported median survivals of less than 2 years. Patients may manifest terminal bone marrow failure or AML (336).
Juvenile myelomonocytic leukemia (JMML) was formerly referred to as juvenile chronic myeloid leukemia (JCML) (249,337). The designation JMML is more appropriate because its morphologic and clinical features more closely mimic CMML than CML. Approximately 60% of cases are diagnosed in patients less than 2 years of age. However, cases have been diagnosed in children from less than 1 month of age to early adolescence. JMML arises from a stem cell defect that leads to deranged hematopoiesis. The disorder is characterized by leukocytosis in the range of 20 to 30 × 109 per liter, composed of granulocytes and monocytes. Immature and dysplastic forms can be identified but dysplasia is usually not prominent. Blasts and promonocytes are fewer than 20% of the blood and bone marrow cells. The bone marrow is hypercellular with granulocytic hyperplasia. The degree of monocyte involvement is variable, from 5% to greater than 30% of the bone marrow cells. In vitro cell culture studies show spontaneous formation of high numbers of abnormal colony-forming units. Hypersensitivity of the myeloid progenitor cells in JMML to GM-CSF has also been repeatedly demonstrated and has become an important diagnostic feature of JMML (338).
Other features of JMML are thrombocytopenia, hepatosplenomegaly and lymphadenopathy, skin rash, and an elevated hemoglobin F level (91). The latter can be a helpful diagnostic clue in the early stages of the disease. Cytogenetic studies are often normal; the most commonly reported abnormality is a monosomy 7. In about 10% of patients, there is deletion of the NF1 tumor suppressor gene and an associated neurofibromatosis type 1 (337).
Unfavorable risk factors in JMML include age greater than 1 year, low platelet counts, elevated hemoglobin F levels, and abnormal cytogenetics (337,339). The disease course may wax and wane in some cases, but ultimately, most patients without hematopoietic stem cell transplant succumb to the disease; stem cell transplant is curative in about half of patients (340).
Cases of myelodysplastic/myeloproliferative neoplasm, unclassifiable (MDS/MPN-U) exhibit the features for inclusion in the MDS/MPN group but do not meet the specific criteria for CMML, aCML, or JMML (91). One provisional category of MDS/MPN, refractory anemia with ringed sideroblasts associated with marked thrombocytosis (RARS-T), is presently placed under the category of MDS/MPN-U (341). This myeloid neoplasm has the morphologic features of the MDS RARS but in addition has marked thrombocytosis and abnormal megakaryocytes of the type found in essential thrombocythemia or early PM. Criteria for a diagnosis of RARS-T include dysplastic ineffective erythropoiesis with a minimum of 15% ringed sideroblasts, less than 5% blasts, and thrombocytosis of more than 450 × 109 per liter with associated large atypical megakaryocytes (91). A majority of patients with RARS-T have a JAK2 mutation (341,342).
MYELOID AND LYMPHOID NEOPLASMS WITH EOSINOPHILIA AND ABNORMALITIES OF PDGFRA, PDGFRB, OR FGFR1
Myeloid and lymphoid neoplasms with eosinophilia and abnormalities of platelet-derived growth factor receptor alpha (PDGFRA), platelet-derived growth factor receptor beta (PDGFRB), and fibroblast growth factor receptor-1 (FGFR1) are recognized in the WHO classification as distinct clinicopathologic entities (254). The disorders are collectively characterized by persistent peripheral and marrow eosinophilia and cytogenetic translocations resulting in abnormal tyrosine kinase function. Reactive causes of eosinophilia are excluded, as the cytogenetic abnormalities represent evidence of clonality. Although one cytogenetic abnormality characterizes the majority of PDGFRA-rearranged diseases, multiple cytogenetic abnormalities have been described in PDGFRB and FGFR1 neoplasms.
MYELOID AND LYMPHOID NEOPLASMS WITH PDGFRA REARRANGEMENT
The majority of PDGFRA rearrangements present as CEL, although fewer cases of acute leukemia of myeloid and/or T lineage have been described. The peripheral blood reveals a leukocytosis with absolute eosinophilia. Eosinophils may have variable morphology, ranging from normal to abnormal, including abnormal localization of the eosinophilic granules within the cytoplasm with associated cleared zones of cytoplasm, immature granules, and abnormal segmentation (295). The bone marrow biopsy has myeloproliferative features, with hypercellularity, prominent marrow eosinophilia, and variable concomitant granulocytic hyperplasia. Blasts are not increased in number generally. Mast cells are hyperplastic in many cases and characteristically arranged in inconspicuous, loose, ill-defined aggregates, which are best identified with immunostaining for CD117 and/or mast cell tryptase (343). The loosely clustered mast cells in this disorder are in contrast to the tightly arranged mast cell aggregates of systemic mastocytosis. In a subset of cases, mast cells aberrantly express CD25 (343). Increased reticulin fibrosis may be present.
The FIP1L1-PDGFRA fusion product, which characterizes the majority of PDGFRA-rearranged neoplasms, is indirectly or directly identified by FISH analysis. Conventional karyotype analysis fails to identify this cryptic translocation in the majority of cases.
MYELOID NEOPLASMS WITH PDGFRB REARRANGEMENT
The PDGFRB-rearranged neoplasms represent myeloid disorders, most commonly CMML or CEL. The subset of CMMLs with eosinophilia characterized by the t(5;12) are currently classified in this new WHO diagnostic category. Other translocation variants have been identified.
The peripheral blood demonstrates leukocytosis with anemia and thrombocytopenia. There is variable eosinophilia, neutrophilia, monocytosis, and basophilia. The marrow is hypercellular with eosinophilia (Fig. 16.28). Many cases show features consistent with CMML (344). Rare cases morphologically resembling CML have been described (295).
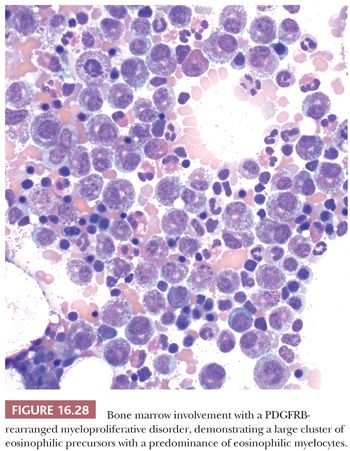
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


