1 Introduction
It is both a cliché and a truism to state that antibiotic resistance has been around for as long as antibiotics have been used to treat infection. Indeed, the origin of antibiotic resistance extends much further back in evolutionary terms and reflects the attack and counter-attack of complex microbial flora in order to establish ecological niches and survive. It is true to say that early treatment failures with antibiotics did not represent a significant clinical problem because other classes of agents, with different cellular targets, were available. It is the emergence of multiple resistance, i.e. resistance to several types of antibiotic agent, that is causing major problems in the clinic today. Several factors drove this situation in the 1970s and 1980s, including the introduction of extendedspectrum agents and advances in medical techniques, such as organ transplantation and cancer chemotherapy. The net result has been a huge selective pressure in favour of multiply resistant species. Coupled with this, there has been a sharp decline in the introduction of agents acting on new cellular targets over the last 30 years compared with the 20-year period following the Second World War. There are a number of resistant organisms causing concern at present. Notable Gram-positive organisms include meticillin-resistant Staphylococcus aureus (MRSA) and coagulase-negative staphylococci, glycopeptide-intermediate sensitivity Staphylococcus aureus (GISA), vancomycin-resistant Enterococcus (VRE) species and penicillin-resistant Streptococcus pneu-moniae. Concerns among the Gram-negative organisms include multidrug-resistant Pseudomonas aeruginosa, Stenotrophomonas maltophilia and Acinetobacter bau-mannii and members of the Enterobacteriaceae with extended-spectrum β-lactamases.Multidrug resistance in the acid-fast bacilli Mycobacterium tuberculosis and M. avium complex pose major health threats worldwide.
Some bacteria are said to have innate resistance against antibiotics and this typically reflects variations in the structure of their cell envelope. These will be identified in subsequent sections on resistance mechanisms. Resistance or reduced susceptibility may also be phenotypic, resulting from adaptation to growth within a specific environment. A characteristic of such phenotypic resistance is reversion to antibiotic susceptibility upon subculture in conventional laboratory media and failure to isolate genotypic resistant mutants (section 17). The origins of antibiotic resistance genes are unclear; however, studies using clinical isolates collected before the introduction of antibiotics demonstrated susceptibility, although conjugative plasmids (section 16.1) were present. Resistance can be achieved by horizontal acquisition of resistance genes, mobilized via insertion sequences, transposons and conjugative plasmids, by recombination of foreign DNA into the chromosome, or by mutations in different chromosomal loci. Given that it is only 60 years since the introduction of antibiotics, mutation of common ancestral genes could not be the only resistance mechanism. Many resistance genes will have derived from the diverse gene pool present in environmental microorganisms, most likely produced as protective mechanisms by antibiotic-producing organisms. Genetic exchange is likely to arise in soil and the general environment as well as in the gut of humans and animals. Rapid mutation can occur and there is clearly a heavy selective pressure resulting from the overuse of antibiotics in medical practice. Agricultural and veterinary use of antibiotics also makes an important and unhelpful contribution. The mutation process is not a static event and a complex network of factors influences the rate and type of mutants that can be selected under antibiotic selective pressure. Antibiotic concentration, physiological conditions such as nutrient availability and stress can each regulate mutation rates. The structure of a gene is relevant to mutability. Size is not the main factor, as not every mutation in a gene that encodes an antibiotic target leads to resistance. Resistance only occurs by mutations which are both permissive (i.e. not lethal or leading to an unacceptable reduction in ‘fitness ’ or ability to cause infection) and able to produce a resistance phenotype. The probability that such a mutation arises will be proportional to the number of target sites within the gene. In Escherichia coli, mutations in the gyrA gene, encoding the GyrA subunit of topoisomerase II and leading to fluoroquinolone resistance (section 8) have been identified in at least seven locations, whereas mutational changes in only three positions in the,parC gene, encoding a subunit of topoisomerase IV, have been observed. As a consequence, the prediction that the mutation rate would be higher in,gyrA than parC is correct. Such observations and predictions cannot be extrapolated to other organisms. Indeed, the opposite is true for fluoroquinolone resistance in Strep pneumoniae.
Resistance to antimicrobial agents typically occurs by one or more of the following mechanisms:
• Inactivation of the drug
• Alteration of the target
• Reduced cellular uptake
• Increased efflux
In this chapter, resistance will be examined by agent, but attention will be drawn to mechanisms which can permit resistance to multiple, chemically different agents. This is most commonly associated with efflux and this will be described in section 16.4.
4 Resistance to β-lactam antibiotics
β-Lactam antibiotics act by inhibiting the carboxy/ transpeptidase or penicillin-binding proteins (PBPs) involved in the late stages of peptidoglycan biosynthesis. Although introduced nearly 60 years ago, β-lactam antibiotics still represent the most widely used class of agents in the clinic today. Resistance to many β-lactam agents is common and is most often caused by β-lactamases or by mutation in the PBPs resulting in reduced affinity. Reduced uptake and efflux are also seen, but they are less significant.
4.1 β-Lactamases
A number of different β-lactamases have been described, but all share the feature of catalysing the ring-opening of the β-lactam moiety (Figure 13.1). Thus, the structural homology with the terminal D-Ala-d-Ala of maturing peptidoglycan, shared by all β-lactam antibiotics, is lost.
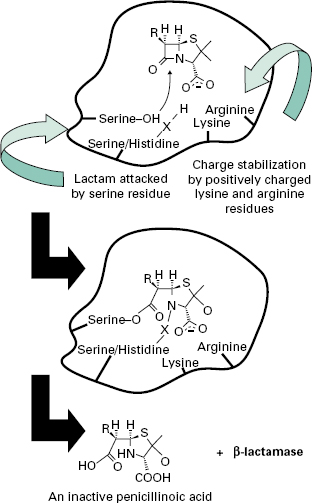
β-Lactamases may be chromosomal or plasmid-borne, inducible or constitutive, and for this reason their terminology can be confusing. A number of classification systems have been proposed, including classes A-D based on peptide sequence. Classes A, C and D have a serine at the active site, whereas class B enzymes have four zinc atoms at their active site and these are also called metallo-β-lactamases. Class A enzymes are highly active against benzylpenicillin; class B β-lactamases are effective against penicillins and cephalosporins. Class C enzymes are usually inducible, but mutation can lead to overexpression. Class D consists of the OXA-type enzymes, which can hydrolyse oxacillin. Increasing resistance to β-lactam agents, mainly by β-lactamase, prompted the discovery and introduction of agents with greater β-lactam stability such as cephalosporins, carbapenems and monobactams. Resistance first appeared in organisms such as Enterobacter cloacae and Pseudomonas aeruginosa, due to mutations causing overproduction of the class C chromosomal AmpC β-lactamase. Subsequently, in the late 1980s, resistance occurred in organisms such as Klebsiella pneumoniae and E. coli that lack an inducible AmpC enzyme. Resistance was found to be mediated by plasmids encoding extended-spectrum β-lactamases (ESBLs). These arose from mutational development of more limitedspectrum β-lactamases such as TEM and SHV that either increased the size of the active-site pocket or altered its binding characteristics to allow the larger cephalosporins to enter and be broken down. TEM derivatives predominate, possibly favoured by the use of ceftazidime and other slowly penetrating cephalosporins. These mutations also increase the binding of clavulanic acid and so these ESBLs remain susceptible to inhibition by this and other β-lactamase inhibitors such as sulbactam and tazobactam, which are generally ineffective against class C β-lactamases.
Continuing use of the third-generation cephalosporins Serine-OH X Lysine Serine/Histidine Charge stabilization Lactam attacked by positively charged by serine residue lysine and arginine residues RH H S N O and the introduction of β-lactamase inhibitor combinations (clavulanate with amoxycillin or ticarcillin, sulbactam with ampicillin, and tazobactam with piperacillin; see section 4.2) resulted in the appearance of plasmids encoding class C β-lactamases.After several unconfirmed reports, the first proof that a class C β-lactamase had been captured on a plasmid came in 1990 when transmis-sible resistance to α-methoxy and oxyimino-β-lactams was shown to be mediated by an enzyme whose gene was 90% identical to the ampC gene of Ent. cloacae. They have subsequently been found worldwide. Strains with Serine-O O X O Serine/Histidine O plasmid-mediated AmpC enzymes are typically resistant Arginine to aminopenicillins (ampicillin or amoxycillin), carboxLysine RH H S HN + -lactamase HO O O COOH An inactive penicillinoic acid ypenicillins (carbenicillin or ticarcillin) and ureidopenicillins (piperacillin). The enzymes also provide resistance to the oxyimino cephalosporins (ceftazidime, cefotaxime, ceftriaxone) and the 7-α-methoxy group (cefoxitin, cefmetazole and moxalactam) as well as the monobactam aztreonam.
In December 2009, the first report of a carbapenemase β-lactamase, referred to as New Delhi metallo-β-lactamase (NDM-1), was recorded. It was discovered in a carbapenem-resistant K. pneumoniae strain isolated in Sweden from a Swedish national who acquired the infection in India. The enzyme is one of the class of B metallo-β-lactamases and is conferred by the gene blaNDM-1. This is considered a serious threat to the carbapenem family of antibiotics.
4.2 β-Lactamase inhibitors
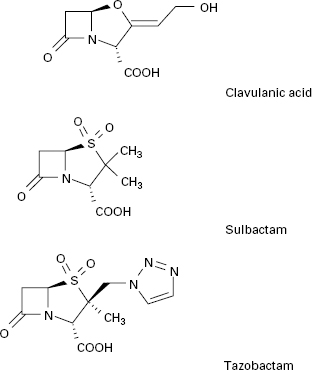
In addition to introducing agents with increased stability to β-lactamase inhibition, β-lactamase inhibitors including clavulanic acid, sulbactam and tazobactam have been developed (Figure 13.2). Clavulanic acid is produced by a streptomyces and is a suicide inhibitor of β-lactamases from a number of Gram-negative and Gram-positive organisms. These β-lactamase inhibitors do not have any significant antimicrobial activity against bacterial transpeptidases, but their combination with a β-lactam antibiotic (see above) has extended the clinical usefulness of the latter.
4.3 Altered penicillin-binding proteins and meticillin-resistant Staphylococcus aureus
Altered PBPs are responsible for reduced sensitivity to β-lactam agents by Strep. pneumoniae (PBP1a, PBP2b and PBP2x) and Haemophilus influenzae (PBP3a and PBP3b), but by far the most clinically significant exampleis MRSA. By the early 1950s, the acquisition and spread of plasmid-encoded β-lactamases had blunted the effectiveness of penicillin for treating Staph. aureus infections such as boils, carbuncles, pneumonia, endocarditis and osteomyelitis. The β-lactamase-stable agent meticillin was introduced in 1959, but by 1960, meticillin-resistant strains were identified. This was the result of Staph. aureus acquiring the mecA gene, which encodes an altered PBP gene, PBP2a. The mecA gene is chromosomal and expression is either constitutive or inducible, but not by meticillin. PBP2a has low affinity for most β-lactam antibiotics.
5 Resistance to glycopeptide antibiotics
Vancomycin and teicoplanin are the two glycopeptides used clinically. They bind the terminal D-alanyl-D-alanine side chains of peptidoglycan and prevent cross-linking in a number of Gram-positive organisms. They are not active against Gram-negative organisms because of the presence of the outer membrane. Vancomycin use increased dramatically in response to the increasing incidence of MRSA and resistance was first reported in the enterococci in 1988. VRE now account for more than 20% of all enterococcal infections. Resistance is greatest amongst Ent. faecium strains, but significant numbers of the more clinically significant Ent. faecalis are also resist-ant. Five types of resistance, VanA-VanE, have now been reported. Phenotypic VanA resistance is the most common and confers high-level resistance to vancomycin and teicoplanin. VanA resistance is mediated by a sevengene cluster on the transposable genetic element Tn 1546 (Figure 13.3).
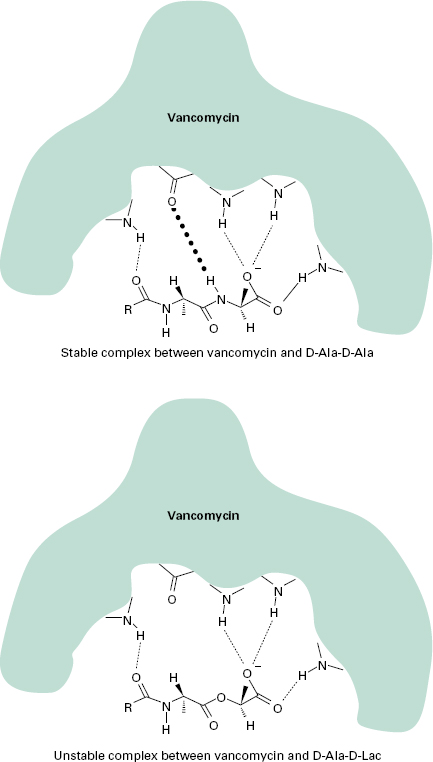
Resistance to vancomycin is via a sensor histidine kinase (VanS) and a response regulator (VanR). VanH encodes a D-lactate dehydrogenase/α-keto acid reductase and generates D-lactate, which is the substrate for VanA, a D-Ala-D-Lac ligase. The result is cell wall precursors terminating in D-Ala-D-Lac to which vancomycin binds with very low affinity. This change in affinity is mediated by one hydrogen bond. The complex formed between vancomycin and D-Ala-D-Ala is stabilized by five hydrogen bonds, whereas only four hydrogen bonds can form between vancomycin and D-Ala-D-Lac and the complex is unstable (Figure 13.4). Further, VanX encodes a D-Ala-D-Ala dipeptidase which can modify endogenous D-Ala-D-Ala precursors. Recent genetic analysis has identified close homology between this cluster and genes present in the vancomycin-producing organism Amycolatopsis orientalis, suggesting that selective pressure has forced genes originally present to protect antibioticproducing organisms to jump to other species. VanB resistance is also acquired and the peptidoglycan precursor is again D-Ala-D-Lac, but isolates often remain susceptible to teicoplanin. VanC resistance is intrinsic and chromosomally encoded in some enterococcal species such as Ent. gallinarum and the peptidoglycan precursor is D-Ala-D-Ser. Less is known of VanD and VanE resistance, but both are acquired. VanD uses D-Ala-D-Lac and VanE uses D-Ala-D-Ser.
Figure 13.4 Mechanism of high-level vancomycin resistance. Hydrogen bonds are denoted by dotted lines. The key hydrogen bond present in the stable complex with d-Ala-d-Ala, but missing in the unstable complex with d-Ala-d-Lac, is shown in bold type
5.1 MRSA and reduced glycopeptide susceptibility
There is major concern that high-level, VanA-type resistance could transfer to staphylococci, particularly MRSA. Experimental transfer of the enterococcal VanA system to Staph. aureus on the skin of mice has been reported, but other mechanisms resulting in intermediate-level resistance occur in clinical isolates. In the 1960s and 1970s MRSA was not feared because several other treatment options existed, including use of tetracyclines, macrolides and aminoglycosides. But multiple resistance was accumulating and by the 1980s empirical therapy of staphylococcal infections, particularly nosocomial sepsis, was changed to the glycopeptide antibiotic vancomycin. MRSA levels were rising and the early 1990s saw a major increase in vancomycin use. The inevitable consequence of the selective pressure was the isolation in 1997 of the first Staph. aureus strain with reduced susceptibility to vancomycin and teicoplanin (vancomycin MIC = 8μg/ ml). At the beginning of the 21st century, MRSA is responsible for up to 25% of nosocomial infections in the USA and reports of community-acquired MRSA infections are increasing. While reports of ‘superbugs ’ resistant to all known antibiotics abound, it is important to distinguish between reduced susceptibility and resistance, recognizing that there are conflicting definitions of resistance and resistance breakpoints. Strains with MIC values <4μg/ml are considered sensitive, 8-16 μ g/ml intermedi-ate and >32μg/ml resistant. Thus the acronyms VISA (vancomycin-intermediate Staph. aureus) and GISA (glycopeptide-insensitive Staph. aureus) are used to denote strains with vancomycin or teicoplanin MICs of 8μg/ml, whereas VRSA (vancomyin-resistant Staph. aureus) is reserved for strains with MIC values >32μg/ml. The mechanism of glycopeptide resistance is poorly understood, but strains show longer doubling times and decreased susceptibility to lysostaphin. Increased quantities of PBP2 and PBP2′ and cell wall precursors are presumed to trap vancomycin, while amidation of glutamine residues in cell wall muropeptides reduces the cross-linking and consequently the number of vancomycin target molecules.
6 Resistance to aminoglycoside antibiotics.
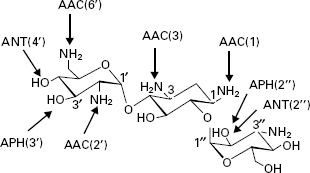
The aminoglycosides are hydrophilic sugars possessing a number of amino and hydroxy substituents. The amine groups are protonated at biological pH and it is the polycationic nature of the molecules that affords them their affinity for nucleic acids, particularly the acceptor (A) site of 16S ribosomal RNA. Aminoglycoside binding to the A site interferes with the accurate recognition of cognate tRNA by rRNA during translation and may also perturb translocation of the tRNA from the A site to the peptidyl-tRNA site (P site). While high-level resistance in aminoglycoside-producing microorganisms is by methylation of the rRNA, this is not the mechanism of resistance in previously susceptible strains. The most common mechanism for clinical aminoglycoside resistance is their structural modification by enzymes expressed in resistant organisms, which compromises their ability to interact with rRNA. There are three classes of these enzymes: aminoglycoside phosphatases (APHs), aminoglycoside nucleotidyltransferases (ANTs) and aminoglycoside acetyltransferases (AACs). Within each class, there are enzymes with differing specificities around the sugars. There are four ANTs (ANT(6), ANT(4′), ANT(3″) and ANT(2″)), seven APHs (APH(3′), APH(2″), APH(3″), APH(6), APH(9), APH(4) and APH(7 ″)) and four AACs (AAC(2′), AAC(6′), AAC(1) and AAC(3)). There is also a bifunctional enzyme, AAC(6′)-AAC(2″). Aminoglycosides are typically susceptible to attack by multiple enzymes (Figure 13.5). Attempts to circumvent these modifying enzymes have centred on structural modifications. Examples include tobramycin which lacks the 3′-hydroxyl group and is thus not a substrate for APH(3 ′), and amikacin which has an acylated N-1 group and is not a substrate for several modifying enzymes. Other strategies are exemplified by experimental compounds such as 3′-oxo-kanamycin. This molecule is a substrate for APH(3′), but the phosphorylation product is unstable and regenerates the original antibiotic.
Figure 13.5 Structure of kanamycin B showing system of ring numbering and sites of action of some aminoglycoside-modifying enzymes. AAC, aminoglycoside acetyltransferase; ANT, aminoglycoside nucleotidyltransferase; APH, aminoglycoside phosphotransferase.