Chapter 46 Bacterial Cell Wall Synthesis Inhibitors
| Abbreviations | |
|---|---|
| CSF | Cerebrospinal fluid |
| ESBLs | Extended-spectrum β-lactamases |
| GI | Gastrointestinal |
| IM | Intramuscular |
| IV | Intravenous |
| MIC | Minimal inhibitory concentration |
| MRSA | Methicillin-resistant Staphylococcus aureus |
| NAG | N-acetylglucosamine |
| NAM | N-acetylmuramate |
| PBPs | Penicillin-binding proteins |
Therapeutic Overview
Since its introduction, however, many strains of bacteria, particularly Staphylococcus aureus, have become resistant to penicillin through several mechanisms including the production of metabolic enzymes called β-lactamases, altered penicillin binding proteins (PBPs), and reduced drug permeability by resistant bacteria (see Table 45-6). Resistance to penicillin by the induction of β-lactamases led to the development of semisynthetic penicillins resistant to hydrolysis by these enzymes as well as numerous compounds with greater activity against gram-negative organisms. The development of resistance to β-lactams is an ongoing clinical problem and is increasing at a dramatic rate.
β-lactams. Resistance to vancomycin, however, is an increasing problem in enterococcus species. Even more alarming has been the discovery of clinical isolates of S. aureus with reduced vancomycin susceptibility, or even full resistance mediated by transfer of resistance genes from enterococcus species.
| Therapeutic Overview |
|---|
| β-Lactams |
| Penicillins, Cephalosporins, Carbapenems, and Monobactams |
| Bactericidal: inhibit many gram-positive and gram-negative organisms |
| Agents differ by: |
| Organism inhibited |
| Pharmacokinetics |
| Bacterial resistance |
| Glycopeptides and Polypeptides |
| Vancomycin—Bactericidal; inhibits many methicillin-resistant staphylococci |
| Bacitracin—Bactericidal; topical use only for gram-positive bacteria |
Mechanisms of Action
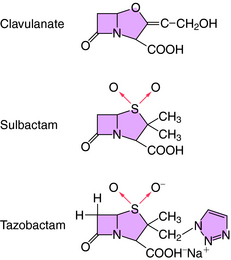
All β-lactam antibiotics have a four-membered ring structure containing a cyclic amide (the lactam); the β indicates that the amine is located on the second carbon relative to the carbonyl group (Fig. 46-1, A). This small ring is structurally strained with low inherent stability, which explains why some penicillins are not effective when administered orally, because they readily undergo hydrolysis, particularly in the presence of high stomach acidity. In addition, this ring is subject to hydrolysis by the β-lactamases, representing a major mechanism of resistance to these compounds. This is why some penicillin derivatives are marketed in combination with β-lactamase inhibitors such as clavulanate, sulbactam, and tazobactam, all of which contain the β-lactam ring structure (Fig. 46-2).
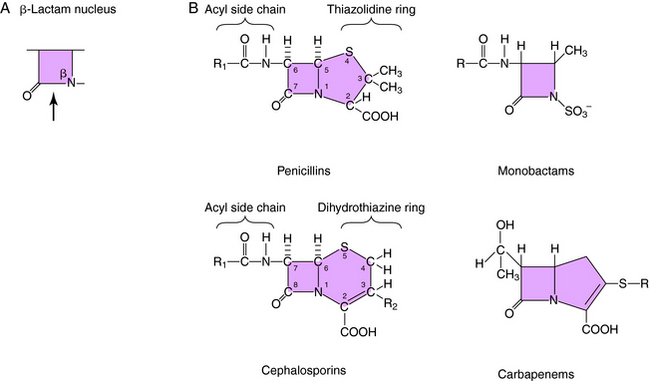
All of the β-lactam antibiotics, except the monobactams, have a second ring fused to the β-lactam ring (see Fig. 46-1, B). For penicillins the second ring is a thiazolidine, whereas for cephalosporins it is a dihydrothiazine. Carbapenems have an unsaturated ring with an external sulfur. Different structural groups positioned at the side chains (R) give rise to compounds with differing antibiotic properties.
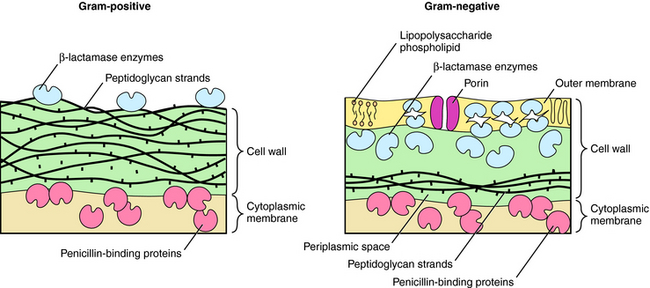
β-lactams interfere with bacterial cell wall synthesis. Although the outer cellular coverings of gram-positive and negative bacteria differ, both have a rigid cell wall composed of a highly cross-linked peptidoglycan matrix (Fig. 46-3). Gram-negative bacteria contain an outer lipopolysaccharide membrane exterior to several peptidoglycan layers. Gram-positive bacteria lack the lipopolysaccharide layer but contain many more (15 to 30) layers of peptidoglycan. The cell wall is assembled in a series of steps, originating within the cytoplasm of the bacteria and terminating outside the cytoplasmic membrane.
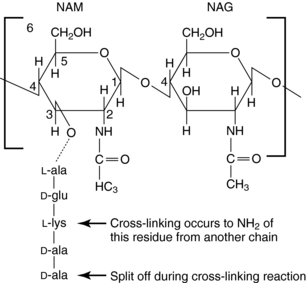
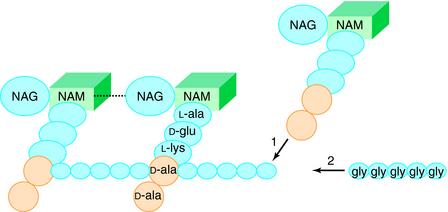
The glycan part of the peptidoglycan is composed of repeating disaccharide units of N-acetylmuramate (NAM) attached to a pentapeptide and N-acetylglucosamine (NAG), connected through β-1,4-linkages (Fig. 46-4). This initial stage of peptidoglycan synthesis occurs in the cytoplasm. The NAG-NAM-pentapeptide is then transferred by a carrier across the cytoplasmic membrane, and the saccharide units are linked in sequence via the pentapeptides to form long chains of alternating disaccharides. This final stage involves a cross-linking reaction to form continuous two-dimensional sheets, a process that occurs outside the cytoplasmic membrane in the periplasm but is catalyzed by membrane-bound transpeptidase enzymes (Fig. 46-5). During the cross-linking reaction, the D-ala-D-ala terminus of the pentapeptide reacts with the transpeptidase to displace the final D-ala, forming an acylenzyme intermediate. This intermediate is reactive and readily couples to the free amino group of the third residue (L-lys) of the pentapeptide of an adjacent chain, thus completing the cross-linking and regenerating the enzyme. It is at the final cross-linking step during synthesis of the rigid peptidoglycan matrix that β-lactam and glycopeptide antibiotics exert their actions, albeit by different mechanisms.
Mechanisms of Resistance to β-Lactams
As discussed in Chapter 45, bacterial resistance to antibiotics is a major therapeutic concern. Resistance to the β-lactams is common and occurs by three major mechanisms:
These mechanisms may coexist, and in some cases it takes a combination of mechanisms, such as decreased permeability and poor binding within a single organism, to confer resistance.
In gram-negative bacteria, β-lactams must pass an outer lipid membrane to reach the PBPs on the cytoplasmic membrane (see Fig. 46-3). Channels in the outer membrane, referred to as porins, allow β-lactams to pass through. Alterations in porin proteins that reduce the amount of drug reaching the PBPs have been observed. For example, P. aeruginosa can delete the porin protein through which imipenem passes and develop resistance. Some β-lactams are extremely resistant to β-lactamases but do not readily pass through porins of gram-negative outer membranes and thus fail to inhibit these bacteria.
However, more than 25% of nosocomial enterococcal isolates, primarily E. faecium, are now vancomycin resistant. Resistance is caused by production of a new pentapeptide ending in a terminal D-ala-D-lac instead of D-ala-D-ala, which does not bind vancomycin. There are also other types of resistance to vancomycin (Table 46-1); the vanA cluster of genes, transferred by a transposable genetic element, is the best characterized. This is an elegant resistance mechanism that contains at least eight genes. One gene product, vanS, functions like a transmembrane receptor, senses the presence of vancomycin, and activates the gene vanR to up regulate expression of three additional genes. The products of these three genes cleave the terminal D-ala-D-ala and insert D-ala-D-lac. Similar resistance genes are present in streptomyces that produce glycopeptide antibiotics and probably evolved as a self-preservation strategy for the organism. Vancomycin-resistant strains of S. aureus are emerging, mediated by increased numbers of vancomycin-binding sites in the cell wall. In 2002, the first two clinical strains of S. aureus isolates with high levels of vancomycin resistance appeared. These strains acquired the vanA gene cluster from vancomycin-resistant enterococci. Bacitracin is a polypeptide bactericidal antibiotic. It inhibits bacterial cell wall synthesis by interfering with dephosphorylation of the lipid carrier that moves the early cell wall components through the membrane.

Pharmacokinetics
All β-lactam agents exhibit time-dependent killing of bacteria and have little postantibiotic effect, unlike fluoroquinolones and aminoglycosides, which function by concentration-dependent mechanisms. Consequently, efficacy of the β-lactams depends on the amount of time the concentration of drug is above the minimal inhibitory concentration (MIC) for the organism at the site of infection. The pharmacokinetic parameters for penicillins of clinical interest are summarized in Table 46-2.
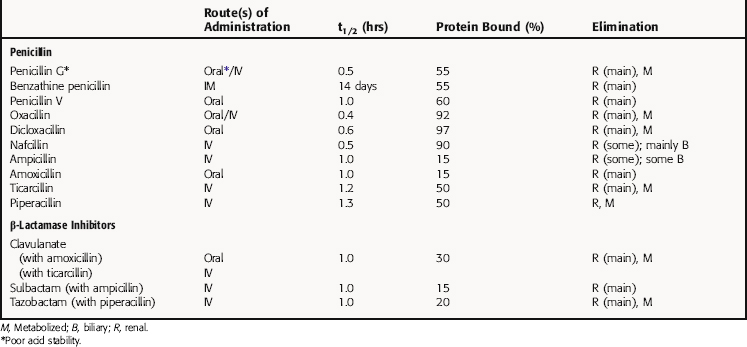
β-Lactamase-Resistant Penicillins
The β-lactamase-resistant penicillins oxacillin, cloxacillin, and dicloxacillin are acid stable and orally absorbed, but absorption is decreased in the presence of food. Peak plasma concentrations are achieved approximately 1 hour after ingestion, and they are all highly protein bound. Elimination is primarily via the kidney, with some biliary excretion and some liver metabolism. These drugs are minimally removed from the body by hemodialysis. Oxacillin is less effective orally; however, adequate plasma and CSF concentrations are achieved when it is administered by the intravenous (IV) route. Nafcillin is erratically absorbed when ingested orally, and the preferred route is IV. Elimination is primarily by biliary excretion. It enters the CSF in concentrations adequate to treat staphylococcal meningitis or brain abscesses.



