Fig. 13.1
Gross image of AT/RT. Tumor (pink to tan mass) with areas of hemorrhage and necrosis located at the cerebellopontine angle
Histopathology
AT/RTs can be quite heterogeneous and are sometimes difficult to recognize solely on the basis of histopathology. The most striking feature in many cases is the presence of neoplastic cells with rhabdoid features: large cytologically atypical cells with irregular and well-defined cell borders eccentrically placed large nuclei with vesicular chromatin and prominent eosinophilic nucleoli, and abundant pink cytoplasm sometimes containing an eosinophilic cytoplasmic inclusion (Fig. 13.2). In practice, the appearance of these cells often falls along a spectrum, ranging from cells with classic rhabdoid features, to cells with epithelioid features (less striking nuclear atypia and large amounts of pale eosinophilic cytoplasm). Frequently, these cells can exhibit prominent cytoplasmic vacuolar degeneration (Fig. 13.2b). In whatever form they appear, these large cells are rarely the sole or even predominant histopathological feature in AT/RT. Typically, these cells are encountered in small collections or are interspersed among the more numerous primitive neuroectodermal tumor cells (Fig. 13.3). However, it is important to recognize that a relatively diverse range of histopathological patterns may be encountered in AT/RT. Mesenchymal differentiation in these tumors most commonly appears as areas with prominent spindle cell features (Fig. 13.4a) and accumulation of extracellular mucopolysaccharide (Fig. 13.4b). Variable degrees of epithelial differentiation may also be encountered in AT/RT (Fig. 13.5). This is the least common histopathological pattern seen in AT/RT and can manifest as poorly differentiated glandular structures, papillary structures, or poorly differentiated ribbons and cords of cells with epithelial features. Occasionally in the latter case, abundant mucopolysaccharide-rich material separates the nests and cords of tumors in a pattern reminiscent of chordoma. Irrespective of the histopathological pattern, mitotic figures are generally abundant in AT/RT. Both karyorrhexis and areas of geographic necrosis are commonly encountered [8, 9, 15, 16].
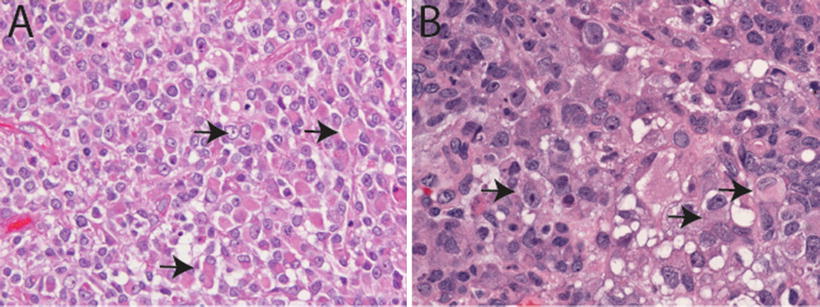
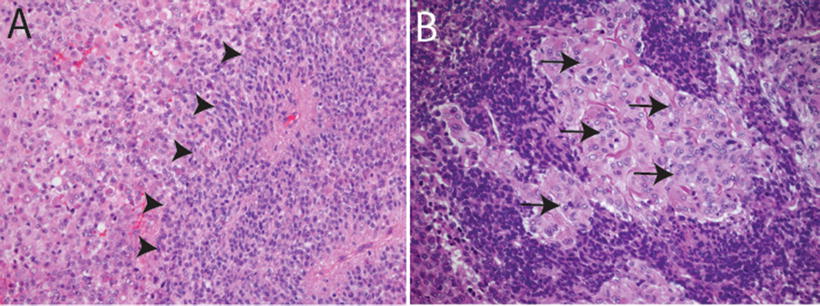
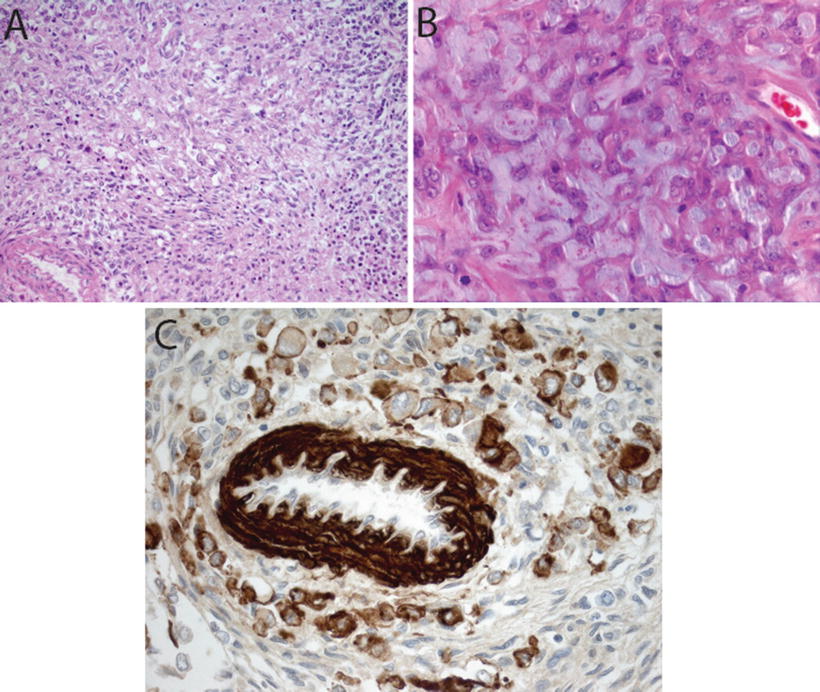
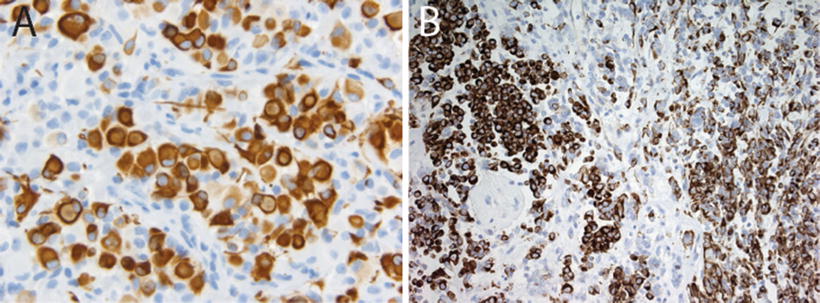
Fig. 13.2
Classic rhabdoid morphology in AT/RT. H&E sections (20×, a, and 40×, b) illustrating classic rhabdoid tumor cells in AT/RT characterized by large cells with eccentrically placed nuclei with vesicular chromatin and abundant pink cytoplasm sometimes containing an eosinophilic cytoplasmic inclusion (arrows). Vacuolar cytoplasmic degeneration typical of ATR/RT in (b)
Fig. 13.3
Primitive neuroectodermal features in AT/RT. H&E sections showing primitive neuroectodermal component (arrowheads in a, 20×) with adjacent rhabdoid areas (arrows in b, 40×)
Fig. 13.4
Mesenchymal differentiation in AT/RT. H&E sections showing spindle cell component (20×, a) and extracellular mucopolysaccharide accumulation (40×, b) in AT/RT. Immunostain for smooth muscle antigen (SMA, 40×) showing staining in vessel wall and tumor cells
Fig. 13.5
Vimentin and epithelial membrane antigen expression in AT/RT. Immunostains for vimentin (a, 20×) and epithelial membrane antigen (EMA, 40×, showing membranous staining, (b) in AT/RT
Differential Diagnosis
The differential diagnosis of AT/RT includes MB, CNS PNET, pineoblastoma, anaplastic ependymoma, choroid plexus carcinoma, and germ cell tumors [9, 15, 17–19]. Particular care must be taken to avoid confusing anaplastic/large cell MB for AT/RT and vice versa. Misdiagnosis can generally be avoided with attention to a few critical details.
First, at the cytologic level, anaplastic/large cell MB show a range of nuclear features that while overlapping with AT/RT in some regards, generally fail to recapitulate the vesicular chromatin-staining pattern typical of these tumors. While they are variably prominent, at least some proportion of rhabdoid tumor cells demonstrates prominent eosinophilic nucleoli. These are not encountered in anaplastic/large cell MB except in the rare case with predominant or exclusively large cell features; these tumors lack the vesicular chromatin staining and variability seen in AT/RT.
Second, evaluation of the cytoplasmic features is extremely helpful. The presence of tumor cells with rhabdoid cytoplasmic inclusions is reassuring. However, the prominence of such cells within any given case can be extremely variable. This may be a function of biology, sampling, or both. In cases where no classic rhabdoid cells are encountered, it is critical to recognize the presence of poorly preserved rhabdoid tumor cells. The presence of scattered cells with large poorly preserved nuclei, vacuolar cytoplasmic degeneration, and prominent well-defined cell borders may be the only histologic evidence of an AT/RT. In other cases, it is the presence of cells with epithelioid or vaguely epithelioid features that alerts the pathologist to the presence of an AT/RT.
Finally, it is important to evaluate the overall histologic pattern for clues to the presence of an AT/RT. In many cases the overall growth pattern may appear essentially indistinguishable from a classic MB or CNS PNET. However, in some cases it is possible to recognize features suggestive of epithelial differentiation in what is otherwise an unremarkable MB or CNS PNET. These features include evidence of epithelial (the presence of poorly differentiated glandular and epithelial structures or the growth of tumor cells in small nests and cords) and/or mesenchymal structures [most often a spindle cell growth pattern (Fig. 13.4a), but occasionally tumors show evidence of more advanced mesenchymal differentiation including the presence of bone and cartilage]. Finally, the accumulation of extracellular myxohyaline material (Fig. 13.4b) in a CNS embryonal tumor is a histologic feature that should raise the differential diagnosis of AT/RT.
Among the non-CNS embryonal neoplasms, there are several features that deserve consideration in the differential diagnosis of AT/RT. Anaplastic ependymomas in the posterior fossa can resemble AT/RT due to their high cellularity and necrosis. It is not uncommon to encounter areas where AT/RTs undermine and appear to grow into the choroid plexus. Combined with occasional papillary features or cells arranged in cord-like structures resembling poorly differentiated epithelial structures, choroid plexus carcinomas should be considered in the differential diagnosis. Prominent spindle cell pattern may resemble a sarcoma and glandular or epithelial differentiations may suggest a teratoma or metastatic carcinoma. In the pineal and other midline locations, germ cell tumors should also enter into the differential diagnosis. Immunohistochemical studies and molecular testing can play a pivotal role in helping the pathologist refine the diagnosis in such cases.
Immunohistochemistry
Immunohistochemical staining for the SMARCB1 protein has been shown to be a highly sensitive and specific tool for detecting the presence of alterations in the SMARCB1 gene on chromosome 22q11.2 (Judkins et al., 2004). In addition to SMARCB1, immunohistochemical expression of several other markers may play an important role in the diagnosis of AT/RT. The heterogeneity in appearance of AT/RT and suggestion of differentiation along multiple cell lineages is reflected in the polyphenotypic immunostaining profile characteristic of these tumors. AT/RTs demonstrate almost universal expression of smooth muscle antigen (SMA, Fig. 13.4c), epithelial membrane antigen (EMA, Fig. 13.5b) and vimentin (Figs. 13.5a). While the latter stain is of limited specificity, it can occasionally prove a useful tool for demonstrating the presence of hard to detect rhabdoid cells. The combined expression of mesenchymal (SMA, Fig. 13.4c) and epithelial markers (EMA, Fig. 13.5b) is unique for AT/RT among other CNS tumors. Expression of glial fibrillary acid protein (GFAP, Fig. 13.6a) and neuronal markers including neurofilament protein (NFP), synaptophysin (SYN, Fig. 13.6b, and NeuN is typically seen in up to about 75 % of AT/RT. Cytokeratin markers such as AE1.3 are also positive in some AT/RT, though typically they are not as frequently expressed as the other markers discussed above [8, 9]. Classical germ cell markers such as placental alkaline phosphatase (PLAP), β–human chorionic gonadotropin (β–HCG) and octomer-binding transcription factor (OCT-4) may be negative. However, other germ cell markers such as Sal-like protein-4 (SALL4), sex determining region Y-box 2 (SOX2) and Nanog may be positive suggesting the potential for pluripotency in these tumors [20]. The expression of these various markers is highly variable from case to case and may reflect the complex biology of these tumors and their ability to differentiate along multiple lineages.
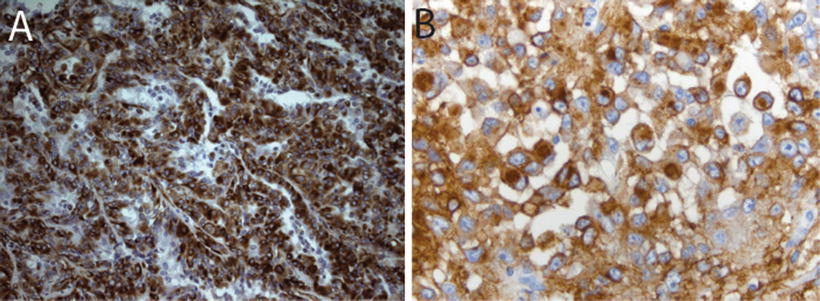
Fig. 13.6
Neuroglial markers in AT/RT. Immunostains for glia fibrillary acidic protein (GFAP, 40×, a) and synaptophysin (SYN, 20×, b) in AT/RT
Loss of SMARCB1 expression in tumor cells enables differentiation of AT/RT from other CNS tumors, which demonstrate retained expression of SMARCB1 (Fig. 13.7). Correct interpretation of SMARCB1 immunohistochemical staining is aided by the fact that SMARCB1 is a ubiquitously expressed nuclear protein and therefore it demonstrates positive immunostaining in endothelial cells and infiltrating tumor lymphocytes (Fig. 13.7). The loss of expression of SMARCB1 in these cells, as well as the failure of the stain to be expressed in any adjacent normal tissues, should signal to the pathologist the need to repeat the SMARCB1 immunohistochemical staining.
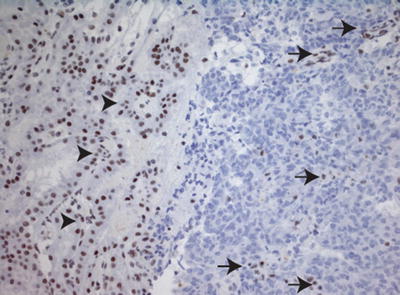
Fig. 13.7
Loss of SMARCB1 staining in AT/RT. Immunostain for SMARCB1 (20×), showing loss of SMARCB1 expression in tumor cells. As an internal control, there is preserved SMARCB1 staining in adjacent choroid plexus (arrow heads) and intra-tumoral endothelial cells and infiltrating lymphocytes (arrows)
As a result of the utility of SMARCB1, combined with the histopathological diversity of AT/RT, it has become the standard of care to stain all CNS embryonal neoplasms with SMARCB1. By so doing, cases of previously unrecognized AT/RT have been reported in some institutions [21]. Routine application of SMARCB1 immunostaining has led to the recognition of loss of expression in other tumor types including CNS low-grade tumors undergoing malignant transformation (ganglioglioma, pleomorphic xanthoastrocytoma) [22, 23] as well as extra-CNS tumors including epithelioid sarcoma, extraskeletal myxoid chondrosarcoma, renal medullary carcinoma, and epithelioid malignant peripheral nerve sheath tumor [24–26]. The pathogenic role, if any, of loss of SMARCB1 in these non-rhabdoid tumors remains to be determined. It is particularly critical that the neuropathologist who makes the diagnosis of AT/RT recognizes that at least a third of all newly diagnosis AT/RTs are attributable to a germline mutation [12] (see below). It is therefore essential that a referral for genetic counseling and testing be made for the patient and their family.
Cytogenetics and Molecular Genetics
Molecular Genetics
SMARCB1 is a tumor suppressor gene biallelically mutated in >95 % of all AT/RTs [27–30]. SMARCB1 is also referred to as: (1) INI1 (Integrase Interactor 1) as the mammalian homologue was first discovered as an HIV-1 integrase-binding protein, (2) BAF47 (BRG1-associated factor 47), and (3) hSNF5 (human sucrose non-fermenting) [31]. SMARCB1 is a component of the mammalian chromatin remodeling SWI/SNF (Switch/Sucrose Non-Fermentable) complex, which reorganizes and/or repositions nucleosomes in an ATP-dependent manner [32]. This complex is believed to be a critical epigenetic regulator for normal development and maintenance of tissue-specific gene expression [33].
The subunits of the SWI/SNF complex are grouped into two major subfamilies: (1) BAF (BRG1 or hBRM-associated factor) complex and (2) PBAF (Polybromo-associated BAF) [34–41]. Among the subunits, there are four core components that are present in all versions of the SWI/SNF complex including: (1) the ATPase enzymatic subunits SMARCA2 (hBRM, hBrahma) or SMARCA4 (BRG1, or Brahma-related gene 1), (2) SMARCB1 (INI1/SNF5/BAF47), (3) SMARCC1 (BAF155), and (4) SMARCC2 (BAF170).
Mutations in almost all subunits of the SWI/SNF complex have been identified in many human cancers [42–44]. SMARCB1 is biallelically inactivated in >95 % of MRT but is variably mutated in other cancer types (discussed below), suggesting that the function of SMARCB1 through SWI/SNF is essential for protecting a specific cell type from becoming cancerous and preventing the genesis of rhabdoid tumors including AT/RT [45].
Mutational Spectrum of SMARCB1 Found in AT/RT
Notwithstanding their histological and immunohistochemical diversity, nearly all AT/RTs involve mutation, deletion, or loss of expression of the SMARCB1 gene. Approximately 70 % MRT arise due to biallelic loss of the SMARCB1 tumor suppressor, and an additional 20–25 % exhibit loss of SMARCB1 function due to reduced RNA or protein expression [12].
Several studies have defined the spectrum of SMARCB1 mutations within AT/RT. These studies have shown that approximately 40 % of SMARCB1 mutations are homozygous deletions, frequently associated with chromosomal rearrangements of 22q11 [3, 46, 47]. In addition to homozygous deletion of SMARCB1, 96 coding-sequence mutations were identified in nine exons of the SMARCB1 gene among 119 ATRT tumor samples analyzed [12].
These mutations occurred with the highest frequency in exons 5 and 9 in AT/RT, while mutations within exon 8 have yet to be detected, and mutations in exons 1 and 3 are largely underrepresented [12, 48]. The majority of coding-sequence mutations (48/96) were single base-pair point mutations, 47 of which were nonsense mutations predicting premature truncation of the protein, and one of which was a missense mutation in exon 9 [12]. The second-most frequent (31/96) type of mutation was deletion and appeared to be localized to a few spots within SMARCB1; 20 were found within exon 9 and 14 involved deletion of one of four cytosines in bases 1,145–1,148, and six involved deletion of one of two guanines at position 1,143 or 1,144 [12]. The remaining mutations identified were duplications of 4–19 bases (7/96), insertions (6/96), and intragenic deletions of one or two exons (4/96) [12]. In addition to coding-sequence mutations, a mutation affecting splicing of the SMARCB1 transcript has also been identified: an A to G mutation in intron 5, which disrupts the splice acceptor site [49].
Most mutations discussed were likely somatic mutations, however; a study on 49 tumor and matched blood DNA samples showed that 33 % (16/49) mutations were germline [12]. These germline mutations were coding-sequence mutations resulting in introduction of a premature stop codon found within exons 2, 4, 5, 6, and 7 [12].
Some of the mutations identified occur repeatedly in different MRT samples indicating possible hot-spots for mutation [12, 48]. Examples include the cytosine deletion at the 3′ end of the SMARCB1 coding sequence (detected in ten different AT/RTs) and C601T (p. Arg201x, found in 12 RTs), and C472T (p. Arg158x, found in seven different tumors). Interestingly, the C601T and C472T mutations have also been identified as germline mutations in families with rhabdoid tumor predisposition syndrome (RTPS) [47]. Other germline mutations identified in RTPS included deletions and insertions causing frame shifts, nonsense mutations, missense mutations, and mutations affecting splicing of the SMARCB1 transcript [11, 47, 50–54]. Notably, many of the SMARCB1 mutations associated with RTPS are de novo germline mutations [11, 47]. LOH is common in RTPS, causing loss of the second allele of SMARCB1 [11, 47, 50].
Exome analysis has revealed that the genomes of these cancers are remarkably simple, showing extremely low rate of mutations with loss of SMARCB1 being the primary recurrent event [55]. This observation is consistent with the data that mouse models with heterozygous deletions of SMARCB1 gene develop a high frequency of tumors with early onset resembling AT/RT with loss of heterozygosity (LOH) at the SMARCB1 locus [56].
Rarely, other components of SWI/SNF may be implicated in the development of AT/RT. There are a few exceptions to the above observations. There are reports indicating that tumors that exhibit typical AT/RT features showed the presence of intact SMARCB1 gene and protein, but show nonsense mutation and inactivation of SMARCA4 [57]. Conversely, recently, there has been an explosion of data describing pathological SMARCB1 mutations/loss of expression in several other tumor types including schwannoma, epithelioid MPNST, epithelioid sarcoma, extraskeletal myxoid chondrosarcoma, synovial sarcoma, pediatric undifferentiated sarcoma, renal medullary carcinoma, small cell undifferentiated variant of hepatoblastoma and myoepithelial carcinoma, and schwannomatosis [26, 58–68]. The role of SMARCB1 mutation/loss in these tumors remains to be clearly established.
Molecular Diagnosis
The ubiquitous expression of SMARCB1 and its loss in nearly all AT/RTs makes it a powerful target as a diagnostic tool. Immunohistochemical analysis for loss of SMARCB1 expression was shown to effectively establish the diagnosis of AT/RT and distinguish MRTs from other pediatric soft tissue tumors [18, 19, 69]. Earlier studies demonstrated that AT/RTs could be distinguished from other histologically similar CNS tumors by cytogenetic studies using FISH of chromosome region 22q11.2 to visualize loss of SMARCB1 locus [70]. Additionally, quantitative real-time PCR (q-RT-PCR) can be performed when DNA of the tumor tissues are available to determine the presence of missense and non-synonymous mutations in the exons of SMARCB1 gene.
Prognostic Stratification and Treatments
Prognosis and Treatment
The treatment of children with AT/RT presents several challenges. These tumors are very aggressive and often resistant to even the most intensive therapies. In addition, because these tumors are more commonly found in very young children, treatment modalities such as craniospinal irradiation are not always an option.
Chemotherapy
One of the earliest regimens resulting in long-term survival even in young children with AT/RT incorporated the approach used by the rhabdomyosarcoma group for patients with parameningeal tumors known as Intergroup Rhabdomyosarcoma Study-III (IRS-III). Weinblatt and Kochen treated a patient with a CNS rhabdoid tumor who underwent gross total resection of the tumor followed by 4,140 cGy focal irradiation and intensive chemotherapy using the IRS-III regimen [71]. The patient was alive and without evidence of disease 4.5 years at the time of the report. Olson et al. published their results on three additional patients with AT/RT using a similar approach [72].
Chi et al. published one of the largest prospective studies to date incorporating the IRS III chemotherapy into an intensive multimodality treatment approach [73]. They reported the results of 20 children newly diagnosed with AT/RT treated between 2004 and 2006. The 2-year progression-free survival rate was 53 ± 13 % with an overall survival rate of 70 ± 10 %.
High Dose Chemotherapy
Finlay et al. have used a strategy known as Head Start therapy for very young children newly diagnosed with malignant brain tumors including AT/RT. This approach uses high dose chemotherapy with autologous stem cell reinfusion in order to avoid or at least postpone the use of irradiation in very young children. The initial report included patients treated on Head Start I and Head Start II [74]. Patients enrolled on Head Start I received 5 cycles of induction chemotherapy with cisplatin, vincristine (during the first 3 cycles), cyclophosphamide, and etoposide. This was followed by a single course of high dose chemotherapy including carboplatin, thiotepa, and etoposide with autologous stem cell reinfusion. Head Start II had identical therapy except for the addition of high dose methotrexate with each induction course. One patient received craniospinal irradiation following autologous stem cell reinfusion, but prior to relapse. At the time of the report, there were three patients treated on Head Start II who were event-free survivors 42+, 54+, and 67+ months following diagnosis without radiation therapy.
Lafay-Cousin et al. have recently reported the experience of the Canadian Brain Tumor Consortium. This was a retrospective review of children treated between 1995 and 2007 [75]. The majority of the patients were <36 months of age and over 1/3 had metastatic disease. . Of the 40 patients who were treated, 22 received standard dose chemotherapy and 18 received high dose chemotherapy with autologous stem cell reinfusion. Adjuvant radiation therapy was administered to 15 patients and nine received intrathecal chemotherapy. Children who received high dose chemotherapy had a 2-year overall survival of 60 ± 12.6 % compared to 21.7 ± 8.5 % for those treated with standard dose chemotherapy. Half of the survivors did not receive any irradiation.
Radiation Therapy
The group from St. Jude Children’s Research Hospital has investigated the role of radiation therapy in a retrospective review of patients with AT/RT treated at their institution between 1987 and 2007 [76]. The timing and field of irradiation were age- and risk dependent with the youngest patients receiving irradiation at least a month or more following resection and usually limited to the involved-field. Cox regression modeling revealed that overall survival in their cohort was adversely affected by disease progression prior to irradiation, time from diagnosis to start of irradiation and age at diagnosis. They concluded that early postoperative irradiation is important for local control particularly in patients without evidence of metastatic disease at diagnosis.
The Children’s Oncology Group has recently closed accrual to a study for children newly diagnosed with AT/RT. This is a single arm study which incorporated high dose methotrexate during induction and consolidation with triple high dose chemotherapy using carboplatin and thiotepa following the previous infant brain tumor study, CCG99703. Radiation therapy was administered between induction and consolidation to children >6 months of age with localized posterior fossa tumors and to those >12 months of age with supratentorial disease. The remaining patients received irradiation following consolidation. Although the study was closed for a year as a result of a toxic pulmonary death, accrual was recently completed and results should be available shortly.
Molecular Signaling Pathways
AT/RTs are relatively free of the genomic instability and widespread accumulation of mutations that are common in many cancers [55]. However, since SMARCB1 is a component of the SWI/SNF complex, its loss may result in modifications of the epigenome that result in large expression changes which may lead to the development of tumors. It has been demonstrated that SWI/SNF regulates transcription of approximately 2–10 % of cellular genes [77].
The SWI/SNF complex mediates both activation and repression of transcription by chromatin remodeling. The exact function of SMARCB1 within SWI/SNF complex is not understood. However, SMARCB1 is one of the subunits that may bridge the interaction of SWI/SNF with specific transcription factors. For example, SMARCB1 interacts with cMYC and the SWI/SNF complex is required for the transactivation functions of cMYC [78]. Another transcription factor that has been shown to bind to SMARCB1 is Hedgehog-Gli1 transcription factor. Inactivation of SMARCB1 may lead to disruption of specific nucleosome patterning and occupancy with accompanying gene expression changes [79].
Loss of SMARCB1 results in a widespread but specific deregulation of genes and pathways important for the cell cycle, differentiation, senescence, or apoptosis, all with tumorigenic potential. Gene expression profile studies carried out to define the changes in transcriptome that occur when SMARCB1 is reintroduced into SMARCB1-null MRT cells identified downstream effectors and several pathways and genes important for normal cell division and homeostasis [80–82]. Genes upregulated by SMARCB1 tend to be antiproliferative or involved in inducing senescence and differentiation, while genes repressed by SMARCB1 tend to be involved in cell cycle progression [82–84]. Several important cell cycle regulatory genes are over-expressed in AT/RT due to loss of SMARCB1 including Cyclin D1 and Aurora A [84–86]. Experimental studies in murine models and human RT cell lines have demonstrated that Cyclin D1 is a key regulator of AT/RT tumor cell growth and that abrogation of Cyclin D1 leads to complete loss of tumor formation in genetically engineered smarcb1± heterozygous mouse models [84, 85, 87]. Furthermore, reintroduction of SMARCB1 leads to induction of senescence [85, 88–90].
In mammalian cells SMARCB1 loss results in transcriptional activation of EZH2 gene, a polycomb gene (PcG) protein and a component of the mammalian polycomb complex, PRC2 as well as in repression and increased H3K27-trimethylation of polycomb targets [91]. It thus appears that SMARCB1 represses and EZH2 activates stem cell-associated programs [91]. This is consistent with the observation that there is frequent activation/upregulation of PcG proteins and frequent inactivation of SWI/SNF components in human cancers [91, 92]. Thus it appears that SWI/SNF modulates the expression and activities of PcG complex to maintain the epigenome and proper expression of cellular genes required for preventing tumor formation [91, 92].
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


