Chapter 51 Antiviral Agents
| Abbreviations | |
|---|---|
| AIDS | Acquired immunodeficiency syndrome |
| AZT | Zidovudine (formerly known as azidothymidine) |
| CMV | Cytomegalovirus |
| CNS | Central nervous system |
| CSF | Cerebrospinal fluid |
| DNA | Deoxyribonucleic acid |
| GI | Gastrointestinal |
| HAART | Highly active antiretroviral therapy |
| HIV | Human immunodeficiency virus |
| HPV | Human papilloma virus |
| HSV | Herpes simplex virus |
| IM | Intramuscular |
| IV | Intravenous |
| NNRTIs | Non-nucleoside reverse transcriptase inhibitors |
| NRTIs | Nucleoside reverse transcriptase inhibitors |
| RNA | Ribonucleic acid |
| RSV | Respiratory syncytial virus |
| SC | Subcutaneous |
Therapeutic Overview
Viruses are responsible for significant morbidity and mortality in populations worldwide. These infectious agents consist of a core genome of nucleic acid (nucleoid) contained in a protein shell (capsid), which is sometimes surrounded by a lipoprotein membrane (envelope) (Fig. 51-1). Viruses cannot replicate independently. Rather, they must enter cells and use the energy-generating, deoxyribonucleic acid (DNA)-or ribonucleic acid (RNA)-replicating, and protein-synthesizing pathways of the host cell to replicate. Some viruses can integrate a copy of their genetic material into host chromosomes, achieving viral latency, in which clinical illness can recur without reexposure to the virus.
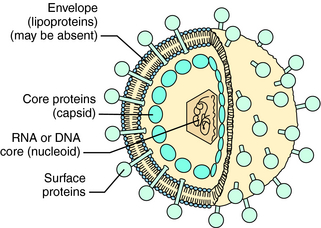
Some genera of viruses that cause human infections are listed in Table 51-1. Also listed is information about which genomic material—RNA or DNA—is present and examples of clinically important diseases attributed to each virus.
TABLE 51–1 Virus Groups of Clinical Importance
| Virus Genera or Groupings | Nucleic Acid | Clinical Examples of Illnesses |
|---|---|---|
| Adenovirus | DNA | Upper respiratory tract and eye infections |
| Hepadnaviridae | DNA | Hepatitis B, cancer |
| Herpesvirus | DNA | Genital herpes, varicella, meningoencephalitis, mononucleosis, retinitis |
| Papillomavirus | DNA | Papillomas (warts), cancer |
| Parvovirus | DNA | Erythema infectiosum |
| Arenavirus | RNA | Lymphocytic choriomeningitis |
| Bunyavirus | RNA | Encephalitis |
| Coronavirus | RNA | Upper respiratory tract infections |
| Influenzavirus | RNA | Influenza |
| Paramyxovirus | RNA | Measles, upper respiratory tract infections |
| Picornavirus | RNA | Poliomyelitis, diarrhea, upper respiratory tract infections |
| Retrovirus | RNA | Leukemia, AIDS |
| Rhabdovirus | RNA | Rabies |
| Togavirus | RNA | Rubella, yellow fever |
to allow detection of individual virus mutations, allowing physicians to predict viral susceptibility to many antiviral agents.
| Therapeutic Overview |
|---|
| Approaches to treatment of viral infections include: |
| Block viral attachment to cells |
| Block uncoating of virus |
| Inhibit viral DNA/RNA synthesis |
| Inhibit viral protein synthesis |
| Inhibit specific viral enzymes |
| Inhibit viral assembly |
| Inhibit viral release |
| Stimulate host immune system |
latency. Strains of viruses resistant to specific drugs can also develop.
Mechanisms of Action
Understanding the steps involved in virus infection and replication has led to the development of drugs that interfere with this process at various sites. This replication cycle and sites of action for the major classes of antiviral drugs are illustrated for the human immunodeficiency virus (HIV) in Figure 51-2.
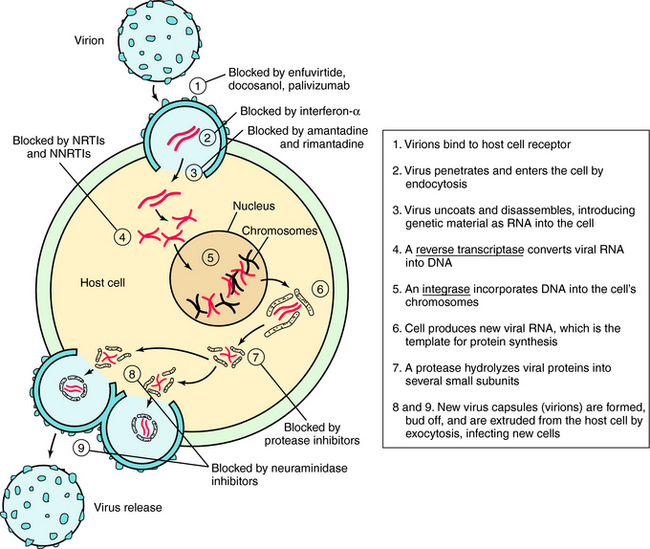
Inhibitors of Cell Penetration
HIV entry into cells is accomplished by a complex series of virus host interactions. Initially, virus approximates the CD4 cell by interactions of HIV surface protein and the host CD4 receptor. After approximation, host coreceptors interact with HIV surface proteins, resulting in folding of the HIV protein gp41. This folding results in fusion of the HIV membrane with the host cell membrane and insertion of the HIV nucleoid into the cell. Enfuvirtide prevents entry of HIV into cells by lying along the gp41 coils, causing steric hindrance of protein folding. Resistance occurs when mutations of gp41 occur that alter conformation and folding.
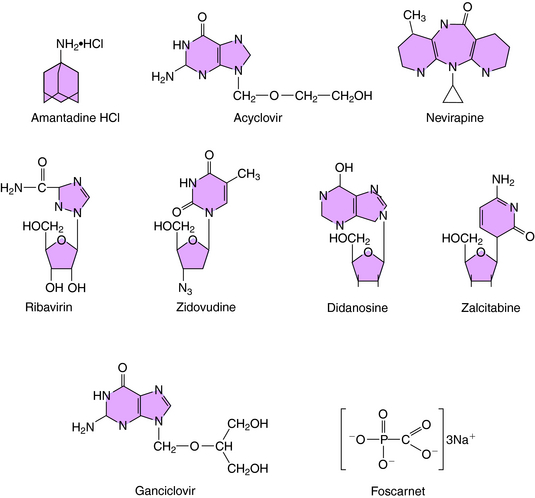
The structure of amantadine is shown in (Fig. 51-3). Its mechanism of action is not fully established but appears to involve blocking the ion channel activity of the M2 protein, thereby inhibiting late-stage uncoating of influenza A virions. This drug is not effective against influenza B, which lacks the M2 protein. A single amino acid change in the M2 protein results in amantadine resistance. Resistant virus is virulent and causes disease in exposed people. Rimantadine is a related compound with similar actions but an improved side effect profile.
Inhibitors of Viral DNA and RNA Synthesis
Acyclovir (see Fig. 51-3) is a synthetic guanosine analog and is the prototypical agent for this group of anti-HSV drugs. The group includes the related drugs valacyclovir and famciclovir, a prodrug for penciclovir. All of these drugs must be phosphorylated to be active and are initially monophosphorylated by viral thymidine kinase. Because the thymidine kinases of HSV types 1 and 2 are many times more active on acyclovir than host thymidine kinase, high concentrations of acyclovir monophosphate accumulate in infected cells. This is then further phosphorylated to the active compound acyclovir triphosphate. The triphosphate cannot cross cell membranes and accumulates further. The resulting concentration of acyclovir triphosphate is 50 to 100 times greater in infected cells than in uninfected cells.
The structure of ganciclovir is shown in Figure 51-3. It is also a synthetic guanosine analog active against many herpesviruses and must also be phosphorylated to be active. Infection-induced kinases, viral thymidine kinase, or deoxyguanosine kinase of various herpesviruses can catalyze this reaction. After monophosphorylation, cellular enzymes convert ganciclovir to the triphosphorylated form, and the triphosphate inhibits viral DNA polymerase rather than cellular DNA polymerase. Ganciclovir triphosphate competitively inhibits the incorporation of guanosine triphosphate into DNA. Because of its toxicity and the availability of acyclovir for treatment of many herpesvirus infections, its use is currently restricted to treatment of CMV retinitis.
Foscarnet (see Fig. 51-3) inhibits DNA polymerases, RNA polymerases, and reverse transcriptases. In vitro it is active against herpesviruses, influenza virus, and HIV. Foscarnet is used primarily in treatment of CMV retinitis. Viral resistance is attributable to structural alterations in CMV DNA polymerase. Foscarnet inhibits CMV herpesviruses that are resistant to acyclovir and ganciclovir.
Ribavirin is a synthetic purine nucleoside active against many viruses, including respiratory syncytial virus, Lassa fever virus, and influenza viruses (see Fig. 51-3). Ribavirin appears to be phosphorylated in host cells by host adenosine kinase. The 5’-monophosphate subsequently inhibits cellular inosine monophosphate formation, resulting in depletion of intracellular guanosine triphosphate. In some situations ribavirin triphosphate suppresses guanosine triphosphate-dependent capping of messenger RNA, thereby inhibiting viral protein synthesis. It also acts by suppressing the initiation or elongation of viral messenger RNA. Exogenous guanosine can reverse the antiviral effects of ribavirin with some viruses.
Nucleoside Reverse Transcriptase Inhibitors and Nucleotides
These drugs include zidovudine, didanosine, lamivudine, stavudine, and others (Box 51-1), and all work through a similar mechanism. Zidovudine (AZT) is the prototype for use in HIV infection. It is a thymidine analog that is phosphorylated to monophosphate, diphosphate, and triphosphate forms by cellular kinases in infected and uninfected cells. NRTIs have two primary methods of action. First, the triphosphate form acts as a competitive inhibitor of HIV reverse transcriptase. Second, after the nucleoside is incorporated into the elongating DNA chain, the forming sugar phosphate backbone of the DNA is blocked from further elongation by substitution at the 3 position. This results in chain termination. In the case of zidovudine, this substitution is an azido (N3) group. Zidovudine inhibits HIV reverse transcriptase at much lower concentrations than those needed to inhibit cellular DNA polymerases, leading to a more targeted effect against HIV.
BOX 51–1 Mechanism of Action of Antiviral Agents



