Chapter 26 Antithrombotic Drugs
| Abbreviations | |
|---|---|
| APTT | Activated partial thromboplastin time |
| cAMP | Cyclic adenosine monophosphate |
| INR | International normalized ratio |
| IV | Intravenous |
| PT | Prothrombin time |
| t-PA | Tissue plasminogen activator |
| TXA2 | Thromboxane A2 |
| t-PA | Tissue plasminogen activator |
| u-PA | Urokinase plasminogen activator |
| vWF | Von Willebrand factor |
Therapeutic Overview
| Therapeutic Overview | |
|---|---|
| Anticoagulation | |
| Heparin and heparin derivatives, coumarins, directly acting thrombin inhibitors | Arterial thrombosis, atrial fibrillation, cardiomyopathy, cerebral emboli, hip surgery, vascular prostheses, heart valve disease, venous thromboembolism |
| Fibrinolysis | |
| Streptokinase, urokinase, tissue plasminogen activator and its derivatives | Acute myocardial infarction, deep venous thrombosis Pulmonary embolism |
| Platelet Aggregation Inhibition | |
| Aspirin | Cerebrovascular accident, stroke, after coronary artery bypass surgery, coronary angioplasty/stenting or thrombolysis, myocardial infarction, transient ischemic attack |
| Clopidogrel | Coronary artery disease, cerebrovascular accident, stroke, peripheral arterial disease |
| Glycoprotein IIb/IIIa inhibitors | Acute coronary syndromes, after coronary artery stenting |
contact with foreign materials, initiate coagulation and thrombus formation. In these settings prophylactic administration of anticoagulants diminishes unwanted thrombus formation. In situations where a thrombus has already formed, such as deep vein thrombosis, acute myocardial infarction, and pulmonary embolism, rapid activation of the fibrinolytic system to lyse the thrombus and initiation of anticoagulation therapy to minimize further clot formation are effective. In cardiovascular disease and stroke, clinical evidence supports the use of drugs that inhibit platelet function.
Mechanisms of Action
The interactions of the coagulation, fibrinolytic, and platelet systems are summarized in Figure 26-1. Endothelial cells in the blood vessel lumen normally present a nonthrombogenic surface. If the endothelium is damaged, blood comes into contact with thrombogenic substances within the subendothelium, such as collagen, which activates platelets, and tissue factor, which initiates blood coagulation. Foreign surfaces, such as prosthetic vascular grafts or mechanical cardiac valves, can also trigger clotting. Removal of thrombi by the fibrinolytic system depends on generation of plasmin from plasminogen by plasminogen activators.
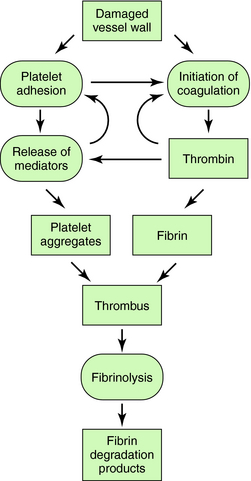
Blood coagulation occurs by sequential conversion of a series of inactive proteins into catalytically active proteases (Fig. 26-2). When the endothelium is damaged, blood comes into contact with cells that express tissue factor, a membrane-bound glycoprotein (the extrinsic pathway). A catalytically active complex of tissue factor and plasma factor VII is produced, which converts factor X to its enzymatically active form (Xa). In turn, factor Xa, in the presence of factor Va and a phospholipid surface (usually that of activated platelets), converts prothrombin to thrombin. Thrombin removes small peptides from fibrinogen (Fig. 26-3), converting it to fibrin monomer, which spontaneously polymerizes to form a clot. Fibrin is stabilized by factor XIIIa (transglutaminase), which introduces covalent bonds between fibrin molecules (see Fig. 26-3).
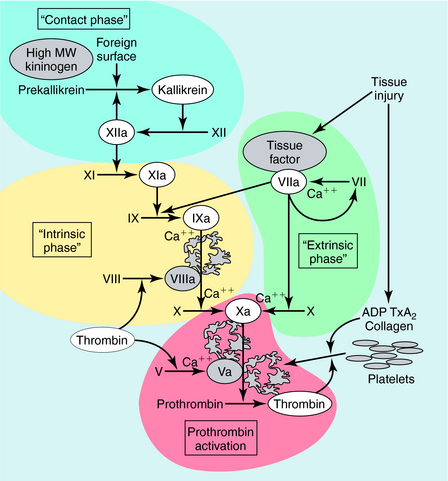
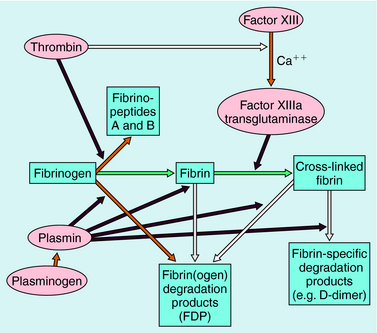
Most enzymes involved in coagulation are trypsin-like serine proteases with considerable homology. Plasma contains many inhibitors that regulate the coagulation cascade (Table 26-1). These proteins prevent inappropriate clotting and prevent appropriate, localized activation of the coagulation cascade from progressing to systemic coagulation.
TABLE 26–1 Plasma Protease Inhibitors That Regulate Blood Coagulation and Fibrinolysis
| Name | Principal Target |
|---|---|
| α1-Protease inhibitor | Elastase |
| α1-Antichymotrypsin | Cathepsin G1 |
| Antithrombin | Thrombin, Xa, IXa |
| α2-Macroglobulin | Plasmin, kallikrein, and other proteases |
| C1 inhibitor | Complement, XIIa |
| α2-Antiplasmin | Plasmin |
| Heparin cofactor II | Thrombin, Xa |
| Plasminogen activator inhibitor-1 (PAI-1) | t-PA, u-PA |
Parenteral Anticoagulant Drugs
Heparin is a linear polysaccharide with alternating residues of glucosamine and either glucuronic or iduronic acid (Fig. 26-4) derived from animal sources. The amino group of glucosamine is either acetylated or sulfated, and there is a variable degree of sulfation (≤40%) on the hydroxyl groups, rendering heparin a heterogeneous compound. Heparin acts by increasing the activity of antithrombin, a plasma glycoprotein that inhibits serine protease clotting enzymes. Heparin binds to antithrombin, causing a conformational change that renders the reactive site on antithrombin more accessible to serine proteases, inactivating thrombin and factors IXa and Xa; low molecular weight heparin inhibits mainly factor Xa. After the binding of antithrombin to thrombin, the heparin molecule is released and can bind to another antithrombin molecule. Although low doses of heparin act primarily by neutralizing factor Xa, at high doses it acts by preventing thrombin-induced platelet activation and prolongs bleeding time. Although the heparin-antithrombin complex is a very efficient inhibitor of free thrombin, clot-bound thrombin is resistant to inhibition.
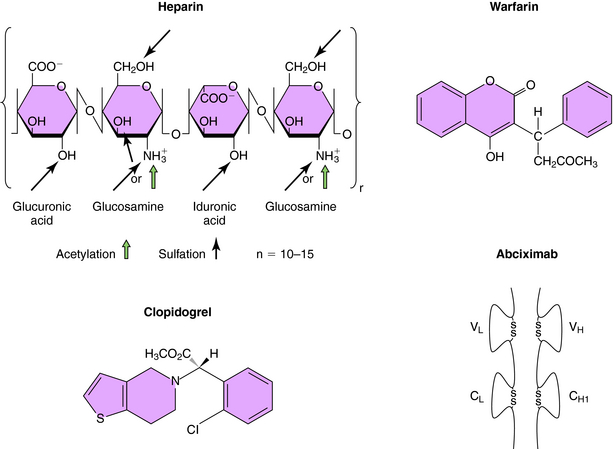
The oral anticoagulants, typified by warfarin and the coumarins (Fig. 26-4), represent a very important class of agents whose action involves their ability to inhibit vitamin K. A subset of blood coagulation factors (II, VII, IX, X) and anticoagulant proteins C and S are activated via the γ-carboxylation of several glutamic acid residues, which mediate their Ca++-dependent binding to phospholipid surfaces, critical for assembly of complexes necessary to generate thrombin. This activation requires vitamin K as a cofactor, and carboxylation of these vitamin K-dependent coagulation factors leads to the concomitant oxidation of vitamin K to its corresponding epoxide. The regeneration of vitamin K necessary to sustain the carboxylation reaction is mediated by vitamin K epoxide reductase, an enzyme inhibited by warfarin and the coumarins. Thus these compounds block recycling of the oxidized form of vitamin K to the reduced form required for cofactor function. Because these compounds inhibit the synthesis of clotting factors but have no direct effect on previously synthesized factors, plasma levels of preexisting vitamin K-dependent factors must decline before the anticoagulant effect of these agents becomes apparent, which requires several days. The first to decline is factor VII, followed by other factors with longer half-lives (see Table 26-2). The full anticoagulant effect of warfarin is typically reached within 4 to 7 days. Because of genetic variations in metabolism, drug interactions, and differences in vitamin K intake, significant variations between individuals exist in the time required for a maximal effect and in doses required for maintenance. Consequently, careful monitoring of prothrombin time (PT), a standard laboratory measure, is necessary.
TABLE 26–2 Rates of Disappearance (Half-Lives) of Vitamin K-dependent Proteins from Blood
| Protein | Time |
|---|---|
| Coagulation Factors | |
| Factor VII | 5 hours |
| Factor IX | |



