29
Antipsychotic Agents & Lithium
CASE STUDY
A 17-year-old male high school student is referred to the psychiatry clinic for evaluation of suspected schizophrenia. After a diagnosis is made, haloperidol is prescribed at a gradually increasing dose on an outpatient basis. The drug improves the patient’s positive symptoms but ultimately causes intolerable adverse effects. Although more costly, risperidone is then prescribed, which, over the course of several weeks of treatment, improves his symptoms and is tolerated by the patient. What signs and symptoms would support an initial diagnosis of schizophrenia? In the treatment of schizophrenia, what benefits do the atypical antipsychotic drugs offer over the traditional agents such as haloperidol? In addition to the management of schizophrenia, what other clinical indications warrant consideration of the use of drugs nominally classified as antipsychotics?
 ANTIPSYCHOTIC AGENTS
ANTIPSYCHOTIC AGENTS
Antipsychotic drugs are able to reduce psychotic symptoms in a wide variety of conditions, including schizophrenia, bipolar disorder, psychotic depression, senile psychoses, various organic psychoses, and drug-induced psychoses. They are also able to improve mood and reduce anxiety and sleep disturbances, but they are not the treatment of choice when these symptoms are the primary disturbance in nonpsychotic patients. A neuroleptic is a subtype of antipsychotic drug that produces a high incidence of extrapyramidal side effects (EPS) at clinically effective doses, or catalepsy in laboratory animals. The “atypical” antipsychotic drugs are now the most widely used type of antipsychotic drug.
History
Reserpine and chlorpromazine were the first drugs found to be useful to reduce psychotic symptoms in schizophrenia. Reserpine was used only briefly for this purpose and is no longer of interest as an antipsychotic agent. Chlorpromazine is a neuroleptic agent; that is, it produces catalepsy in rodents and EPS in humans. The discovery that its antipsychotic action was related to dopamine (D or DA)-receptor blockade led to the identification of other compounds as antipsychotics between the 1950s and 1970s. The discovery of clozapine in 1959 led to the realization that antipsychotic drugs need not cause EPS in humans at clinically effective doses. Clozapine was called an atypical antipsychotic drug because of this dissociation; it produces fewer EPS at equivalent antipsychotic doses in man and laboratory animals. As a result, there has been a major shift in clinical practice away from typical antipsychotic drugs towards the use of an ever increasing number of atypical drugs, which have other advantages as well. The introduction of antipsychotic drugs led to massive changes in disease management, including brief instead of life-long hospitalizations. These drugs have also proved to be of great value in studying the pathophysiology of schizophrenia and other psychoses. It should be noted that schizophrenia and bipolar disorder are no longer believed by many to be separate disorders but rather to be part of a continuum of brain disorders with psychotic features.
Nature of Psychosis & Schizophrenia
The term “psychosis” denotes a variety of mental disorders: the presence of delusions (false beliefs), various types of hallucinations, usually auditory or visual, but sometimes tactile or olfactory, and grossly disorganized thinking in a clear sensorium. Schizophrenia is a particular kind of psychosis characterized mainly by a clear sensorium but a marked thinking disturbance. Psychosis is not unique to schizophrenia and is not present in all patients with schizophrenia at all times.
Schizophrenia is considered to be a neurodevelopmental disorder. This implies that structural and functional changes in the brain are present even in utero in some patients, or that they develop during childhood and adolescence, or both. Twin, adoption, and family studies have established that schizophrenia is a genetic disorder with high heritability. No single gene is involved. Current theories involve multiple genes with common and rare mutations, including large deletions and insertions (copy number variations), combining to produce a very variegated clinical presentation and course.
THE SEROTONIN HYPOTHESIS OF SCHIZOPHRENIA
The discovery that indole hallucinogens such as LSD (lysergic acid diethylamide) and mescaline are serotonin (5-HT) agonists led to the search for endogenous hallucinogens in the urine, blood, and brains of patients with schizophrenia. This proved fruitless, but the identification of many 5-HT-receptor subtypes led to the pivotal discovery that 5-HT2A-receptor and possibly 5-HT2C stimulation was the basis for the hallucinatory effects of these agents.
It has been found that 5-HT2A-receptor blockade is a key factor in the mechanism of action of the main class of atypical antipsychotic drugs, of which clozapine is the prototype, and includes, in order of their introduction around the world, melperone, risperidone, zotepine, blonanserin, olanzapine, quetiapine, ziprasidone, aripiprazole, sertindole, paliperidone, iloperidone, asenapine, and lurasidone. These drugs are inverse agonists of the 5-HT2A receptor; that is, they block the constitutive activity of these receptors. These receptors modulate the release of dopamine, norepinephrine, glutamate, GABA, and acetylcholine, among other neurotransmiters in the cortex, limbic region, and striatum. Stimulation of 5-HT2A receptors leads to depolarization of glutamate neurons, but also stabilization of N-methyl-D-aspartate (NMDA) receptors on postsynaptic neurons. Recently, it has been found that hallucinogens can modulate the stability of a complex consisting of 5-HT2A and NMDA receptors.
5-HT2C-receptor stimulation provides a further means of modulating cortical and limbic dopaminergic activity. Stimulation of 5-HT2C receptors leads to inhibition of cortical and limbic dopamine release. Many atypical antipsychotic drugs, eg, clozapine, asenapine, olanzapine, are 5-HT2C inverse agonists. 5-HT2C agonists are currently being studied as antipsychotic agents.
THE DOPAMINE HYPOTHESIS OF SCHIZOPHRENIA
The dopamine hypothesis for schizophrenia was the second neurotransmitter-based concept to be developed but is no longer considered adequate to explain all aspects of schizophrenia, especially the cognitive impairment. Nevertheless, it is still highly relevant to understanding the major dimensions of schizophrenia, such as positive and negative symptoms (emotional blunting, social withdrawal, lack of motivation), cognitive impairment, and possibly depression. It is also essential to understanding the mechanisms of action of most and probably all antipsychotic drugs.
Several lines of evidence suggest that excessive limbic dopaminergic activity plays a role in psychosis. (1) Many antipsychotic drugs strongly block postsynaptic D2 receptors in the central nervous system, especially in the mesolimbic and striatal-frontal system; this includes partial dopamine agonists, such as aripiprazole and bifeprunox. (2) Drugs that increase dopaminergic activity, such as levodopa, amphetamines, and bromocriptine and apomorphine, either aggravate schizophrenia psychosis or produce psychosis de novo in some patients. (3) Dopamine-receptor density has been found postmortem to be increased in the brains of schizophrenics who have not been treated with antipsychotic drugs. (4) Some but not all postmortem studies of schizophrenic subjects have reported increased dopamine levels and D2-receptor density in the nucleus accumbens, caudate, and putamen. (5) Imaging studies have shown increased amphetamine-induced striatal dopamine release, increased baseline occupancy of striatal D2 receptors by extracellular dopamine, and other measures consistent with increased striatal dopamine synthesis and release.
However, the dopamine hypothesis is far from a complete explanation of all aspects of schizophrenia. Diminished cortical or hippocampal dopaminergic activity has been suggested to underlie the cognitive impairment and negative symptoms of schizophrenia. Postmortem and in vivo imaging studies of cortical, limbic, nigral, and striatal dopaminergic neurotransmission in schizophrenic subjects have reported findings consistent with diminished dopaminergic activity in these regions. Decreased dopaminergic innervation in medial temporal cortex, dorsolateral prefrontal cortex, and hippocampus, and decreased levels of DOPAC, a metabolite of dopamine, in the anterior cingulate have been reported in postmortem studies. Imaging studies have found increased prefrontal D1-receptor levels that correlated with working memory impairments.
The fact that several of the atypical antipsychotic drugs have much less effect on D2 receptors and yet are effective in schizophrenia has redirected attention to the role of other dopamine receptors and to nondopamine receptors. Serotonin receptors—particularly the 5-HT2A-receptor subtype—may mediate synergistic effects or protect against the extrapyramidal consequences of D2 antagonism. As a result of these considerations, the direction of research has changed to a greater focus on compounds that may act on several transmitter-receptor systems, eg, serotonin and glutamate. The atypical antipsychotic drugs share the property of weak D2-receptor antagonism and more potent 5-HT2A-receptor blockade.
THE GLUTAMATE HYPOTHESIS OF SCHIZOPHRENIA
Glutamate is the major excitatory neurotransmitter in the brain (see Chapter 21). Phencyclidine (PCP) and ketamine are noncompetitive inhibitors of the NMDA receptor that exacerbate both cognitive impairment and psychosis in patients with schizophrenia. PCP and a related drug, MK-801, increase locomotor activity and, acutely or chronically, a variety of cognitive impairments in rodents and primates. These effects are widely employed as a means to develop novel antipsychotic and cognitive-enhancing drugs. Selective 5-HT2A antagonists, as well as atypical antipsychotic drugs, are much more potent than D2 antagonists in blocking these effects of PCP and MK-801. This was the starting point for the hypothesis that hypofunction of NMDA receptors, located on GABAergic interneurons, leading to diminished inhibitory influences on neuronal function, contributed to schizophrenia. The diminished GABAergic activity can induce disinhibition of downstream glutamatergic activity, which can lead to hyperstimulation of cortical neurons through non-NMDA receptors. Preliminary evidence suggests that LY2140023, a drug that acts as an agonist of the metabotropic 2/3 glutamate receptor (mGLuR2/3), may be effective in schizophrenia.
The NMDA receptor, an ion channel, requires glycine for full activation. It has been suggested that in patients with schizophrenia, the glycine site of the NMDA receptor is not fully saturated. There have been several trials of high doses of glycine to promote glutamatergic activity, but the results are far from convincing. Currently, glycine transport inhibitors are in development as possible antipsychotic agents.
Ampakines are drugs that potentiate currents mediated by AMPA-type glutamate receptors. In behavioral tests, ampakines are effective in correcting behaviors in various animal models of schizophrenia and depression. They protect neurons against neurotoxic insults, in part by mobilizing growth factors such as brain-derived neurotrophic factor (BDNF, see also Chapter 30).
BASIC PHARMACOLOGY OF ANTIPSYCHOTIC AGENTS
Chemical Types
A number of chemical structures have been associated with antipsychotic properties. The drugs can be classified into several groups as shown in Figures 29–1 and 29–2.
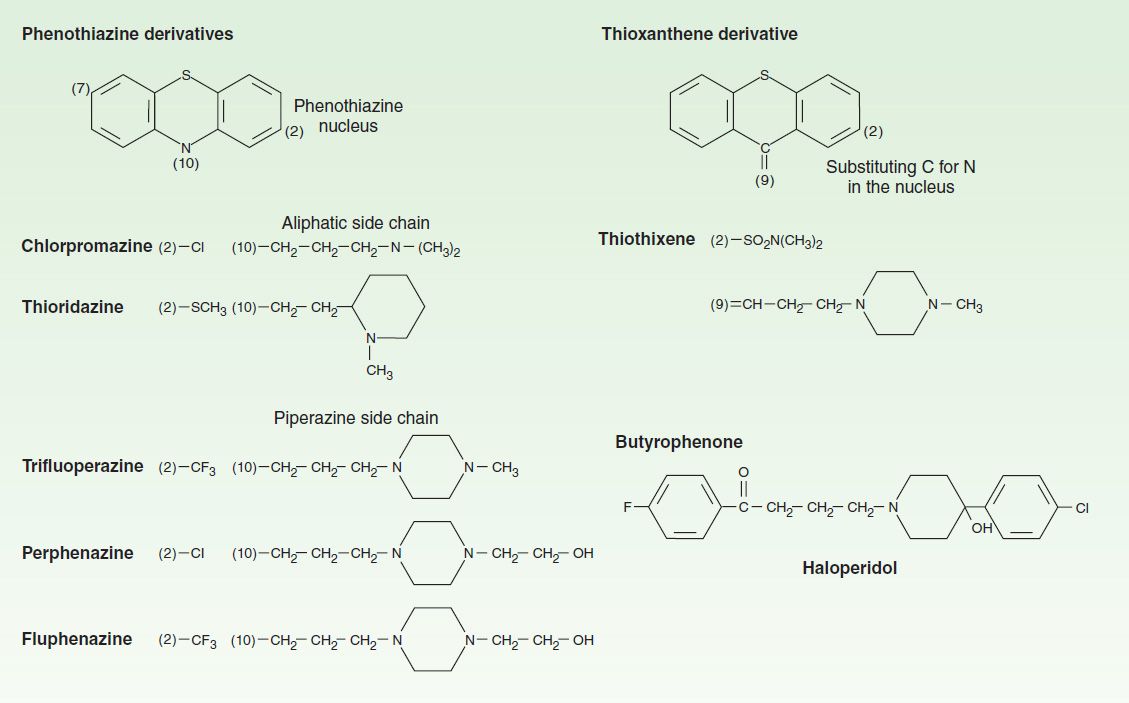
FIGURE 29–1 Structural formulas of some older antipsychotic drugs: phenothiazines, thioxanthenes, and butyrophenones. Only representative members of each type are shown.
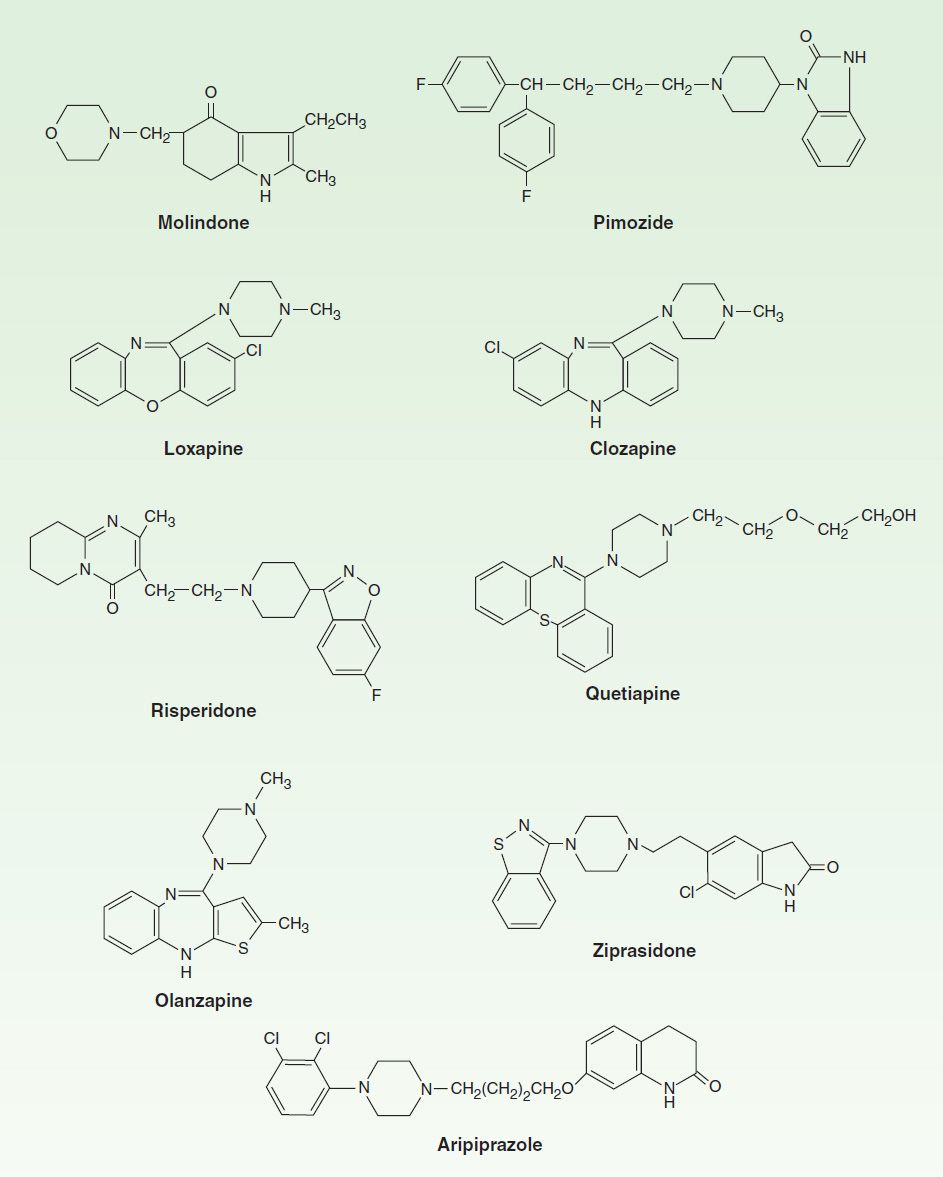
FIGURE 29–2 Structural formulas of some newer antipsychotic drugs.
A. Phenothiazine Derivatives
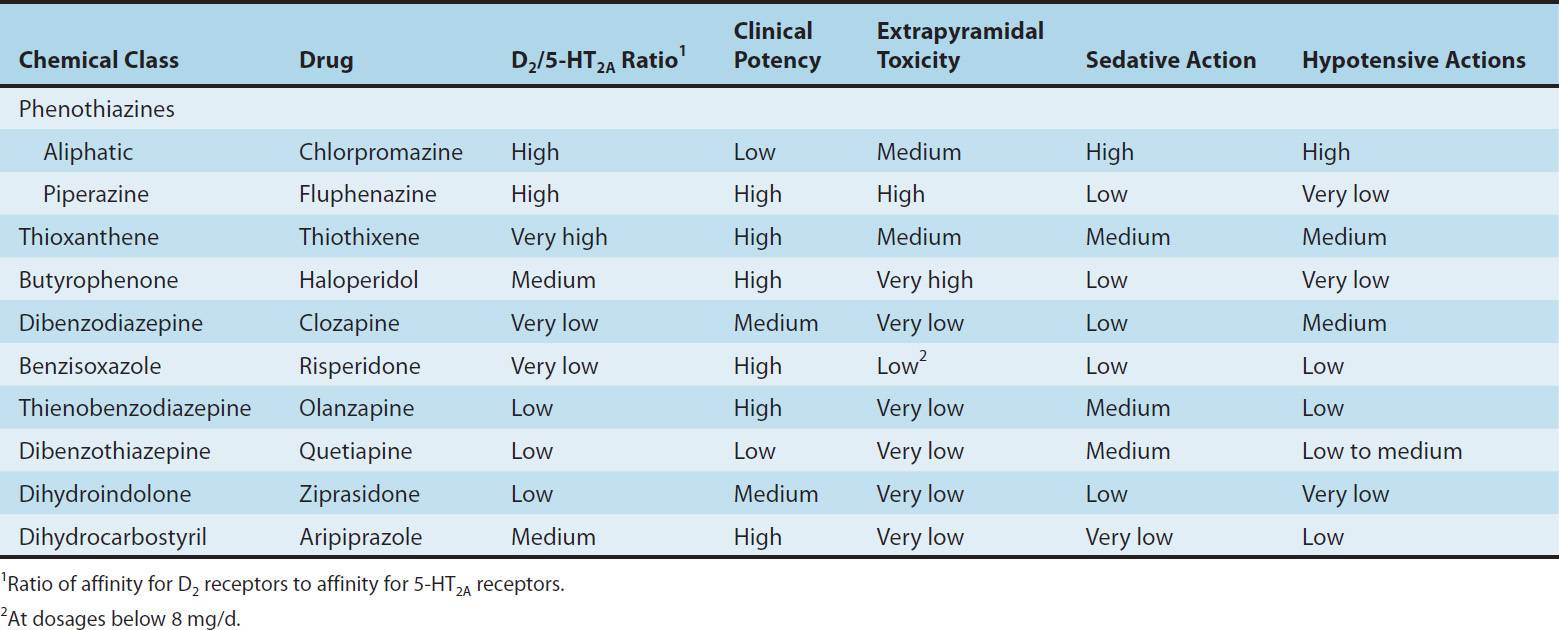
Three subfamilies of phenothiazines, based primarily on the side chain of the molecule, were once the most widely used of the antipsychotic agents. Aliphatic derivatives (eg, chlorpromazine) and piperidine derivatives (eg, thioridazine) are the least potent. These drugs produce more sedation and weight gain. Piperazine derivatives are more potent (effective in lower doses) but not necessarily more efficacious. The piperazine derivatives are also more selective in their pharmacologic effects (Table 29–1).
TABLE 29–1 Antipsychotic drugs: Relation of chemical structure to potency and toxicities.
The National Institute of Mental Health (NIMH)-funded Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE) reported that perphenazine, a piperazine derivative, was as effective as atypical antipsychotic drugs, with the modest exception of olanzapine, and concluded that typical antipsychotic drugs are the treatment of choice for schizophrenia based on their lower cost. However, there were numerous flaws in the design, execution and analysis of this study, leading to it having only modest impact on clinical practice. In particular, it failed to consider issues such as dosage of olanzapine, inclusion of treatment resistant patients, encouragement of patients to switch medications inherent in the design, risk for tardive dyskinesia following long-term use of even low dose typical antipsychotics, and the necessity of large sample sizes in equivalency studies.
B. Thioxanthene Derivatives
This group of drugs is exemplified primarily by thiothixene.
C. Butyrophenone Derivatives
This group, of which haloperidol is the most widely used, has a very different structure from those of the two preceding groups. Haloperidol, a butyrophenone, is the most widely used typical antipsychotic drug, despite its high level of EPS relative to typical antipsychotic drugs. Diphenylbutylpiperidines are closely related compounds. The butyrophenones and congeners tend to be more potent and to have fewer autonomic effects but greater extrapyramidal effects than phenothiazines (Table 29–1).
D. Miscellaneous Structures
Pimozide and molindone are typical antipsychotic drugs. There is no significant difference in efficacy between these newer typical and the older typical antipsychotic drugs.
E. Atypical Antipsychotic Drugs
Clozapine, asenapine, olanzapine, quetiapine, paliperidone, risperidone, sertindole, ziprasidone, zotepine, and aripiprazole are atypical antipsychotic drugs (some of which are shown in Figure 29–2). Clozapine is the prototype. Paliperidone is 9-hydroxyrisperidone, the active metabolite of risperidone. Risperidone is rapidly converted to 9-hydroxyrisperidone in vivo in most patients, except for about 10% of patients who are poor metabolizers. Sertindole is approved in some European countries but not in the USA.
These drugs have complex pharmacology but they share a greater ability to alter 5-HT2A-receptor activity than to interfere with D2-receptor action. In most cases, they act as partial agonists at the 5-HT1A receptor, which produces synergistic effects with 5-HT2A receptor antagonism. Most are either 5-HT6 or 5-HT7 receptor antagonists.
Sulpride and sulpiride constitute another class of atypical agents. They have equivalent potency for D2 and D3 receptors, but they are also 5-HT7 antagonists. They dissociate EPS and antipsychotic efficacy. However, they also produce marked increases in serum prolactin levels and are not as free of the risk of tardive dyskinesia as are drugs such as clozapine and quetiapine. They are not approved in the USA.
Cariprazine represents another class of atypical agents. In addition to D2/5-HT2 antagonism, cariprazine is also a D3 partial agonist with selectivity for the D3 receptor. Cariprazine’s selectivity for the D3 receptor may be associated with greater effects on the negative symptoms of schizophrenia. This drug is currently under review for possible approval in 2014.
F. Glutamatergic Antipsychotics
No glutamate-specific agents are currently approved for the treatment of schizophrenia. However, several agents are in late clinical testing. Among these is bitopertin, a glycine transporter 1 receptor inhibitor (GlyT1). Glycine is a required co-agonist with glutamate at NMDA receptors. Phase 2 studies indicated that bitopertin used adjunctively with standard antipsychotics significantly improved negative symptoms of schizophrenia. Sarcoserine (N-methylglycine), another GlyT1 inhibitor, in combination with a standard antipsychotic has also shown benefit in improving both negative and positive symptoms of schizophrenia in acutely ill as well as in more chronic patients with schizophrenia.
Another class of investigational antipsychotic agents includes the metabotropic glutamate receptor agonists. Eight metabotropic glutamate receptors are divided into three groups: group I (mGluR1,5), group II (mGluR2,3), and group III (mGluR4,6,7,8). mGluR2,3 inhibits glutamate release presynaptically. Several mGluR2,3 agents are being investigated in the treatment of schizophrenia. One agent, pomaglumetad methionil, showed antipsychotic efficacy in early phase 2 trials, but subsequent trials failed to show benefit in either positive or negative symptoms of schizophrenia. Other metabotropic glutamate receptor agonists are being explored for the treatment of negative and cognitive symptoms of schizophrenia.
Pharmacokinetics
A. Absorption and Distribution
Most antipsychotic drugs are readily but incompletely absorbed. Furthermore, many undergo significant first-pass metabolism. Thus, oral doses of chlorpromazine and thioridazine have systemic availability of 25–35%, whereas haloperidol, which has less first-pass metabolism, has an average systemic availability of about 65%.
Most antipsychotic drugs are highly lipid soluble and protein bound (92–99%). They tend to have large volumes of distribution (usually more than 7 L/kg). They generally have a much longer clinical duration of action than would be estimated from their plasma half-lives. This is paralleled by prolonged occupancy of D2 dopamine receptors in the brain by the typical antipsychotic drugs.
Metabolites of chlorpromazine may be excreted in the urine weeks after the last dose of chronically administered drug. Long-acting injectable formulations may cause some blockade of D2 receptors 3–6 months after the last injection. Time to recurrence of psychotic symptoms is highly variable after discontinuation of antipsychotic drugs. The average time for relapse in stable patients with schizophrenia who discontinue their medication is 6 months. Clozapine is an exception in that relapse after discontinuation is usually rapid and severe. Thus, clozapine should never be discontinued abruptly unless clinically needed because of adverse effects such as myocarditis or agranulocytosis, which are true medical emergencies.
B. Metabolism
Most antipsychotic drugs are almost completely metabolized by oxidation or demethylation, catalyzed by liver microsomal cytochrome P450 enzymes. CYP2D6, CYP1A2, and CYP3A4 are the major isoforms involved (see Chapter 4). Drug-drug interactions should be considered when combining antipsychotic drugs with various other psychotropic drugs or drugs—such as ketoconazole—that inhibit various cytochrome P450 enzymes. At the typical clinical doses, antipsychotic drugs do not usually interfere with the metabolism of other drugs.
Pharmacodynamics
The first phenothiazine antipsychotic drugs, with chlorpromazine as the prototype, proved to have a wide variety of central nervous system, autonomic, and endocrine effects. Although efficacy of these drugs is primarily driven by D2-receptor blockade, their adverse actions were traced to blocking effects at a wide range of receptors including α adrenoceptors and muscarinic, H1 histaminic, and 5-HT2 receptors.
A. Dopaminergic Systems
Five dopaminergic systems or pathways are important for understanding schizophrenia and the mechanism of action of antipsychotic drugs. The first pathway—the one most closely related to behavior and psychosis—is the mesolimbic-mesocortical pathway, which projects from cell bodies in the ventral tegmentum in separate bundles of axons to the limbic system and neocortex. The second system—the nigrostriatal pathway—consists of neurons that project from the substantia nigra to the dorsal striatum, which includes the caudate and putamen; it is involved in the coordination of voluntary movement. Blockade of the D2 receptors in the nigrostriatal pathway is responsible for EPS. The third pathway—the tuberoinfundibular system—arises in the arcuate nuclei and periventricular neurons and releases dopamine into the pituitary portal circulation. Dopamine released by these neurons physiologically inhibits prolactin secretion from the anterior pituitary. The fourth dopaminergic system—the medullary-periventricular pathway—consists of neurons in the motor nucleus of the vagus whose projections are not well defined. This system may be involved in eating behavior. The fifth pathway—the incertohypothalamic pathway—forms connections from the medial zona incerta to the hypothalamus and the amygdala. It appears to regulate the anticipatory motivational phase of copulatory behavior in rats.
After dopamine was identified as a neurotransmitter in 1959, it was shown that its effects on electrical activity in central synapses and on production of the second messenger cAMP synthesized by adenylyl cyclase could be blocked by antipsychotic drugs such as chlorpromazine, haloperidol, and thiothixene. This evidence led to the conclusion in the early 1960s that these drugs should be considered dopamine-receptor antagonists and was a key factor in the development of the dopamine hypothesis of schizophrenia described earlier in this chapter. The antipsychotic action is now thought to be produced (at least in part) by their ability to block the effect of dopamine to inhibit the activity of adenylyl cyclase in the mesolimbic system.
B. Dopamine Receptors and Their Effects
At present, five dopamine receptors have been described, consisting of two separate families, the D1-like and D2-like receptor groups. The D1 receptor is coded by a gene on chromosome 5, increases cAMP by Gs-coupled activation of adenylyl cyclase, and is located mainly in the putamen, nucleus accumbens, and olfactory tubercle and cortex. The other member of this family, D5, is coded by a gene on chromosome 4, also increases cAMP, and is found in the hippocampus and hypothalamus. The therapeutic potency of antipsychotic drugs does not correlate with their affinity for binding to the D1 receptor (Figure 29–3, top) nor did a selective D1 antagonist prove to be an effective antipsychotic in patients with schizophrenia. The D2 receptor is coded on chromosome 11, decreases cAMP (by Gi-coupled inhibition of adenylyl cyclase), and inhibits calcium channels but opens potassium channels. It is found both pre- and postsynaptically on neurons in the caudate-putamen, nucleus accumbens, and olfactory tubercle. A second member of this family, the D3 receptor, also coded by a gene on chromosome 11, is thought to also decrease cAMP and is located in the frontal cortex, medulla, and midbrain. D4 receptors also decrease cAMP and are concentrated in the cortex.
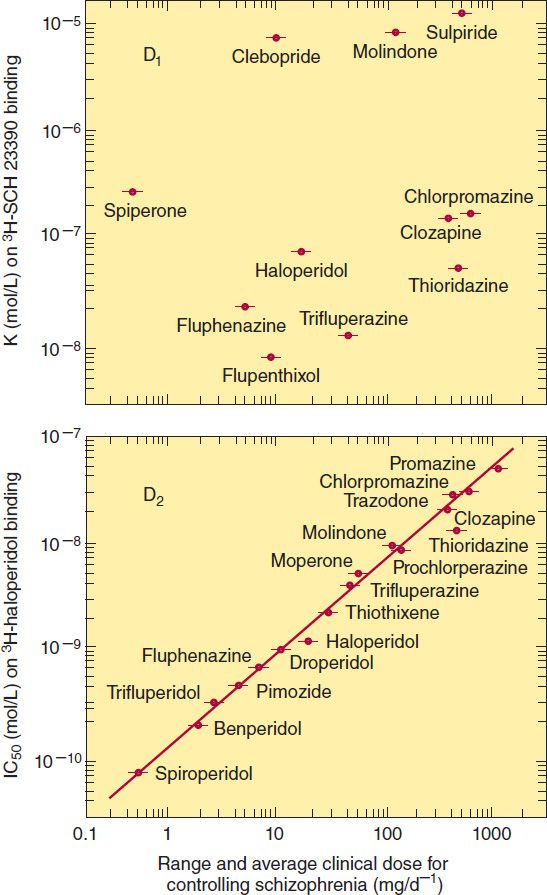
FIGURE 29–3 Correlations between the therapeutic potency of antipsychotic drugs and their affinity for binding to dopamine D1 (top) or D2 receptors (bottom). Potency is indicated on the horizontal axes; it decreases to the right. Binding affinity for D1 receptors was measured by displacing the selective D1 ligand SCH 23390; affinity for D2 receptors was similarly measured by displacing the selective D2 ligand haloperidol. Binding affinity decreases upward. (Reprinted, with permission, of Wiley-Liss, Inc., a subsidiary of John Wiley & Sons, Inc., from Seeman P: Dopamine receptors and the dopamine hypothesis of schizophrenia. Synapse 1987;1:133.)
The typical antipsychotic agents block D2 receptors stereoselectively for the most part, and their binding affinity is very strongly correlated with clinical antipsychotic and extrapyramidal potency (Figure 29–3, bottom). In vivo imaging studies of D2-receptor occupancy indicate that for antipsychotic efficacy, the typical antipsychotic drugs must be given in sufficient doses to achieve at least 60% occupancy of striatal D2 receptors. This is not required for the atypical antipsychotic drugs such as clozapine and olanzapine, which are effective at lower occupancy levels of 30–50%, most likely because of their concurrent high occupancy of 5-HT2A receptors. The typical antipsychotic drugs produce EPS when the occupancy of striatal D2 receptors reaches 80% or higher.
Positron emission tomography (PET) studies with aripiprazole show very high occupancy of D2 receptors, but this drug does not cause EPS because it is a partial D2-receptor agonist. Aripiprazole also gains therapeutic efficacy through its 5-HT2A antagonism and possibly 5-HT1A partial agonism.
These findings have been incorporated into the dopamine hypothesis of schizophrenia. However, additional factors complicate interpretation of dopamine receptor data. For example, dopamine receptors exist in both high- and low-affinity forms, and it is not known whether schizophrenia or the antipsychotic drugs alter the proportions of receptors in these two forms.
It has not been convincingly demonstrated that antagonism of any dopamine receptor other than the D2 receptor plays a role in the action of antipsychotic drugs. Selective and relatively specific D1-, D3-, and D4-receptor antagonists have been tested repeatedly with no evidence of antipsychotic action. Most of the newer atypical antipsychotic agents and some of the traditional ones have a higher affinity for the 5-HT2A receptor than for the D2 receptor (Table 29–1), suggesting an important role for the serotonin 5-HT system in the etiology of schizophrenia and the action of these drugs.
C. Differences among Antipsychotic Drugs
Although all effective antipsychotic drugs block D2 receptors, the degree of this blockade in relation to other actions on receptors varies considerably among drugs. Vast numbers of ligand-receptor binding experiments have been performed in an effort to discover a single receptor action that would best predict antipsychotic efficacy. A summary of the relative receptor-binding affinities of several key agents in such comparisons illustrates the difficulty in drawing simple conclusions from such experiments:
Chlorpromazine: α1 = 5-HT2A > D2 > D1
Haloperidol: D2 > α1 > D4 > 5-HT2A > D1 > H1
Clozapine: D4 = α1 > 5-HT2A > D2 = D1
Olanzapine: 5-HT2A > H1 > D4 > D2 > α1 > D1
Aripiprazole: D2 = 5-HT2A > D4 > α1 = H1 >> D1
Quetiapine: H1 > α1 > M1,3 > D2 > 5-HT2A
Thus, most of the atypical and some typical antipsychotic agents are at least as potent in inhibiting 5-HT2 receptors as they are in inhibiting D2 receptors. The newest, aripiprazole, appears to be a partial agonist of D2 receptors. Varying degrees of antagonism of α2 adrenoceptors are also seen with risperidone, clozapine, olanzapine, quetiapine, and aripiprazole.
Current research is directed toward discovering atypical antipsychotic compounds that are either more selective for the mesolimbic system (to reduce their effects on the extrapyramidal system) or have effects on central neurotransmitter receptors—such as those for acetylcholine and excitatory amino acids—that have been proposed as new targets for antipsychotic action.
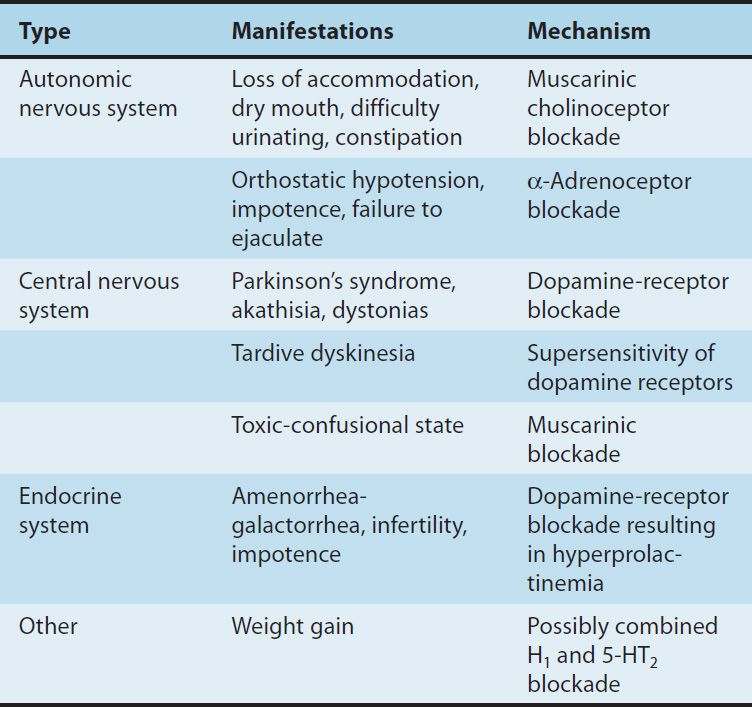
In contrast to the difficult search for receptors responsible for antipsychotic efficacy, the differences in receptor effects of various antipsychotics do explain many of their toxicities (Tables 29–1 and 29–2). In particular, extrapyramidal toxicity appears to be consistently associated with high D2 potency.
TABLE 29–2 Adverse pharmacologic effects of antipsychotic drugs.
D. Psychological Effects
Most antipsychotic drugs cause unpleasant subjective effects in nonpsychotic individuals. The mild to severe EPS, including akathisia, sleepiness, restlessness, and autonomic effects are unlike any associated with more familiar sedatives or hypnotics. Nevertheless, low doses of some of these drugs, particularly quetiapine, are used to promote sleep onset and maintenance, although there is no approved indication for such usage.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


