Anti-Arrhythmic Drugs
Cardiac cells undergo depolarization and repolarization to form cardiac action potentials ~60 times/min. The shape and duration of each action potential are determined by the activity of ion channel protein complexes in the membranes of individual cells, and the genes encoding most of these proteins now have been identified.
Arrhythmias can range from incidental, asymptomatic clinical findings to life-threatening abnormalities. In some human arrhythmias, precise mechanisms are known, and treatment can be targeted specifically against those mechanisms. In other cases, mechanisms can be only inferred, and the choice of drugs is based largely on the results of prior experience. Anti-arrhythmic drug therapy can have 2 goals: termination of an ongoing arrhythmia or prevention of an arrhythmia. Unfortunately, anti-arrhythmic drugs not only help to control arrhythmias but also can cause them, especially during long-term therapy. Thus, prescribing anti-arrhythmic drugs requires that precipitating factors be excluded or minimized, that a precise diagnosis of the type of arrhythmia be made, and that the risks of drug therapy can be minimized.
PRINCIPLES OF CARDIAC ELECTROPHYSIOLOGY
The flow of ions across cell membranes generates the currents that make up cardiac action potentials. Most anti-arrhythmic drugs affect more than one ion current, and many exert ancillary effects such as modification of cardiac contractility or autonomic nervous system function. Thus, anti-arrhythmic drugs usually exert multiple actions and can be beneficial or harmful in individual patients.
THE CARDIAC CELL AT REST: A K+-PERMEABLE MEMBRANE
The normal cardiac cell at rest maintains a transmembrane potential ~80-90 mV negative to the exterior; this gradient is established by pumps, especially the Na+, K+-ATPase, and fixed anionic charges within cells. There are both an electrical and a concentration gradient that would move Na+ ions into resting cells (Figure 29–1). However, Na+ channels, which allow Na+ to move along this gradient, are closed at negative transmembrane potentials, so Na+ does not enter normal resting cardiac cells. In contrast, a specific type of K+ channel protein (the inward rectifier channel) is in an open conformation at negative potentials. Hence, K+ can move through these channels across the cell membrane at negative potentials in response to either electrical or concentration gradients.
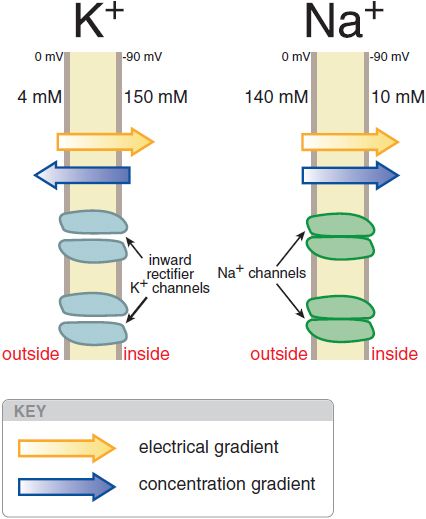
Figure 29–1 Electrical and chemical gradients for K+ and Na+in a resting cardiac cell. Inward rectifier K+ channels are open (left), allowing K+ ions to move across the membrane and the transmembrane potential to approach EK. In contrast, Na+ does not enter the cell despite a large net driving force because Na+ channel proteins are in the closed conformation (right) in resting cells.
For each individual ion, there is an equilibrium potential Ex at which there is no net driving force for the ion to move across the membrane. Ex can be calculated using the Nernst equation:
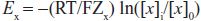
where Zx is the valence of the ion, T is the absolute temperature, R is the gas constant, F is Faraday’s constant, [x]o is the extracellular concentration of the ion, and [x]i is the intracellular concentration. For K+, [K]o = 4 mM and [K]i = 140 mM, the calculated K+ equilibrium potential EK is –94 mV. There is thus no net force driving K+ ions into or out of a cell when the transmembrane potential is –94 mV, close to the resting potential. Thus, at rest, the normal cardiac cell is permeable to K+ (because inward rectifier channels are open) and [K]o is the major determinant of resting potential. If [K]o is elevated to 10 mM, as might occur in diseases such as renal failure or myocardial ischemia, the calculated EK rises to –70 mV.
THE CARDIAC ACTION POTENTIAL
Na+ channels have a life cycle of openings and closings that help to regulate membrane excitability (Figure 29–2). To initiate an action potential, a cardiac myocyte at rest is depolarized above a threshold potential, usually via gap junctions by a neighboring myocyte. Upon membrane depolarization, Na+ channel proteins change from the “closed” (resting) state to the “open” (conducting) state, allowing up to 107 Na+ ions/sec to enter each cell and moving the transmembrane potential toward ENa (+65 mV). This surge of Na+ ions lasts only about a millisecond, after which the Na+ channel protein rapidly moves to an “inactivated,” nonconducting state. The maximum upstroke slope of phase 0 (dV/dtmax, or Vmax) of the action potential (Figure 29–3) is largely governed by Na+ current and plays a role in the conduction velocity of a propagating action potential. Under normal conditions, Na+ channels, once inactivated, cannot reopen until they reassume the closed conformation.
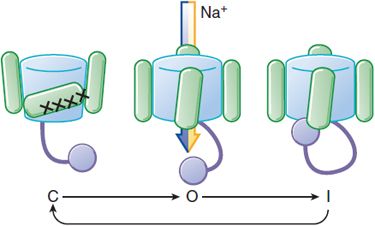
Figure 29–2 Life cycle of a voltage-sensitive channel. Voltage-dependent conformational changes determine current flow through Na+ channels. At hyperpolarized potentials, the channel is in a closed conformation (C) and no current flows. As depolarization begins, the pore opens (O), allowing conduction. As depolarization is maintained, a nearby region of a channel subunit moves to block current flow, putting the channel into an inactivated, nonconducting state (I). Restoration of the normal resting Em restores the conformation to the closed state (C). See Figure 20–2 for structural details.
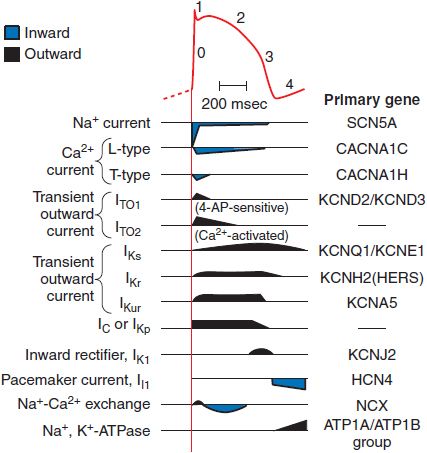
Figure 29–3 The relationship between an action potential from the conducting system and the currents that generate it. The current magnitudes are not to scale; the Na+ current is ordinarily 50 times larger than any other current, although the portion that persists into the plateau (phase 2) is small. Multiple types of Ca2+ current, transient outward current (IT0), and delayed rectifier (IK) have been identified. Each represents a different channel protein, usually associated with ancillary (function-modifying) subunits. 4-AP (4-aminopyridine) is a widely used in vitro blocker of K+ channels. ITO2 may be a Cl– current in some species. Components of IK have been separated on the basis of how rapidly they activate: slowly (IKs), rapidly (IKr), or ultra-rapidly (IKur). The voltage-activated, time-independent current may be carried by Cl– (ICl) or K+ (IKp, p for plateau). The genes encoding the major pore-forming proteins have been cloned for most of the channels shown here and are included in the right-hand column. The right-hand column lists the primary genes coding for the various ion channels and transporters.
A small population of Na+ channels may continue to open during the action potential plateau in some cells, providing further inward current. Certain mutations in the cardiac isoform of the Na+ channel can further increase the number of channels that do not properly inactivate, thereby prolonging the action potential, and cause a form of the congenital long QT syndrome. In general, however, as the cell membrane repolarizes, the negative membrane potential moves Na+ channel proteins from inactivated to “closed” conformations. The relationship between Na+ channel availability and transmembrane potential is an important determinant of conduction and refractoriness in many cells.
The changes in transmembrane potential generated by the inward Na+ current produce a series of openings (and in some cases subsequent inactivation) of other channels (see Figure 29–3). For example, when a cell is depolarized by the Na+ current, “transient outward” K+ channels quickly enter an open, or conducting, state; because the transmembrane potential at the end of phase 0 is positive to EK, the opening of transient outward channels results in an outward, or repolarizing, K+ current (termed IT0), which contributes to the phase 1 “notch” seen in action potentials from these tissues. Transient outward K+ channels, like Na+ channels, inactivate rapidly. During the phase 2 plateau of a normal cardiac action potential, inward, depolarizing currents, primarily through Ca2+ channels, are balanced by outward, repolarizing currents primarily through K+ (“delayed-rectifier”) channels. Delayed-rectifier currents (collectively termed IK) increase with time, whereas Ca2+ currents inactivate (and so decrease with time); as a result, cardiac cells repolarize (phase 3) several hundred milliseconds after the initial Na+ channel opening. Mutations in the genes encoding repolarizing K+ channels are responsible for the most common forms of the congenital long QT syndrome.
ACTION POTENTIAL HETEROGENEITY IN THE HEART. The general description of the action potential and the currents that underlie it must be modified for certain cell types, primarily due to variability in the expression of ion channels and electrogenic ion transport pumps. In the ventricle, action potential duration (APD) and shape vary across the wall of each chamber, as well as apico-basally (Figure 29–4). Atrial cells have short action potentials, probably because ITO is larger, and an additional repolarizing K+ current, activated by the neurotransmitter acetylcholine, is present. As a result, vagal stimulation further shortens atrial action potentials. Cells of the sinus and atrioventricular (AV) nodes lack substantial Na+ currents. In addition, these cells, as well as cells from the conducting system, normally display the phenomenon of spontaneous diastolic, or phase 4 depolarization and thus spontaneously reach threshold for regeneration of action potentials. The rate of spontaneous firing usually is fastest in sinus node cells, which therefore serve as the natural pacemaker of the heart.
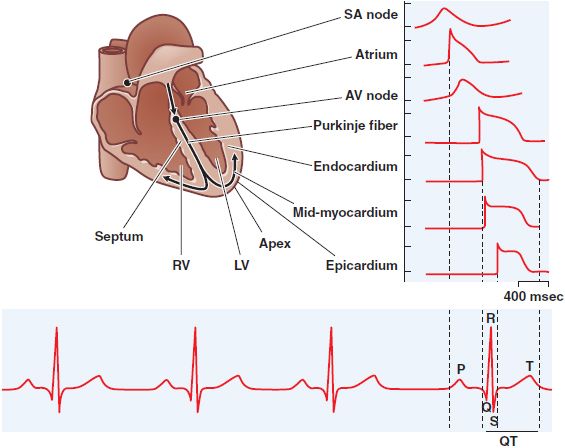
Figure 29–4 Normal impulse propagation. A schematic of the human heart with example action potentials from different regions of the heart (top) for a normal beat and their corresponding contributions to the macroscopic ECG (bottom). AV, atrioventricular; LV, left ventricle; RV, right ventricle; SA, sinoatrial. (Used with permission from The Am Physiol Soc. Nerbonne JM, Kass RS. Physiol Rev, 2005;85:1205-1253.)
One of the pacemaking currents responsible for this automaticity is generated via specialized K+ channels, the hyperpolarization-activated, cyclic nucleotide-gated (HCN) channels that are permeable to both potassium and sodium. Another mechanism responsible for automaticity is the repetitive spontaneous Ca2+ release from the sarcoplasmic reticulum (SR). The rise in cytosolic Ca2+ causes membrane depolarizations when Ca2+ is extruded from the cell via the electrogenic Na-Ca exchanger (NCX). In addition, sinus node cells lack inward rectifier K+ currents that are primarily responsible for protecting working myocardium against spontaneous membrane depolarizations.
MAINTENANCE OF INTRACELLULAR ION HOMEOSTASIS
With each action potential, the cell interior gains Na+ ions and loses K+ ions. The Na+, K+-ATPase (Na+ pump) is activated in most cells to maintain intracellular homeostasis, extruding 3 Na+ ions for every 2 K+ ions shuttled from the exterior of the cell to the interior; as a result, the act of pumping itself generates a net outward (repolarizing) current.
Normally, basal intracellular Ca2+ is maintained at very low levels (<100 nM). In cardiac myocytes, the entry of Ca2+ during each action potential through L-type Ca2+ channels is a signal to the SR to release its Ca2+ stores. The efflux of Ca2+ from the SR occurs through ryanodine-receptor (RyR2) Ca2+ release channels, and the resulting increase in intracellular Ca2+ subsequently triggers Ca2+-dependent contractile processes (= excitation-contraction coupling). Removal of intracellular Ca2+ occurs by both Ca2+-ATPase (which moves Ca2+ ions back into the SR) and NCX, which exchanges 3 Na+ ions from the exterior for each Ca2+ ion extruded (see Figure 28–5). Abnormal regulation of intracellular Ca2+ is increasingly well described in heart failure and contributes to arrhythmias in this setting. Furthermore, mutations that disrupt the normal activity of the RyR2 channels and the cardiac isoform of calsequestrin have been linked to catecholaminergic polymorphic ventricular tachycardia (CPVT), thereby demonstrating a direct link between spontaneous SR Ca2+ release and cardiac arrhythmias.
IMPULSE PROPAGATION AND THE ELECTROCARDIOGRAM
Normal cardiac impulses originate in the sinus node. Once impulses leave the sinus node, they propagate rapidly throughout the atria, resulting in atrial systole and the P wave of the surface electrocardiogram (ECG; see Figure 29–4). Propagation slows markedly through the AV node, where the inward current (through Ca2+ channels) is much smaller than the Na+ current in atria, ventricles, or the subendocardial conducting system. This conduction delay allows the atrial contraction to propel blood into the ventricle, thereby optimizing cardiac output. Once impulses exit the AV node, they enter the conducting system, where Na+ currents are larger than elsewhere and propagation is correspondingly faster, up to 0.75 m/s longitudinally. Activation spreads from the His–Purkinje system on the endocardium of the ventricles throughout the rest of the ventricles, stimulating coordinated ventricular contraction. This electrical activation manifests itself as the QRS complex on the ECG. The T wave of the ECG represents ventricular repolarization.
The ECG can be used as a rough guide to some cellular properties of cardiac tissue:
• Heart rate reflects sinus node automaticity.
• PR-interval duration reflects AV nodal conduction time.
• QRS duration reflects conduction time in the ventricle.
• The QT interval is a measure of ventricular APD.
REFRACTORINESS AND CONDUCTION FAILURE
If a single action potential is restimulated very early during the plateau, no Na+ channels are available to open, no inward current results, and no action potential is generated: the cell is termed refractory (Figure 29–5). Conversely, if a stimulus occurs after the cell has repolarized completely, Na+ channels have recovered, and a normal Na+ channel–dependent upstroke results (Figure 29–5A). When a stimulus occurs during phase 3 of the action potential, the magnitude of the resultant Na+ current depends on the number of Na+ channels that have recovered (Figure 29–5B). The recovery from inactivation is faster at more hyperpolarized membrane potentials. Thus, refractoriness is determined by the voltage-dependent recovery of Na+ channels from inactivation. The effective refractory period (ERP) is the longest interval at which a premature stimulus fails to generate a propagated response.
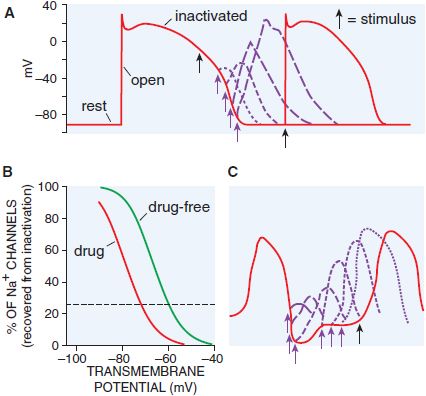
Figure 29–5 Qualitative differences in responses of nodal and conducting tissues to premature stimuli. A. With a very early premature stimulus (black arrow) in ventricular myocardium, all Na+ channels still are in the inactivated state, and no upstroke results. As the action potential repolarizes, Na+ channels recover from the inactivated to the resting state, from which opening can occur. The phase 0 upstroke slope of the premature action potentials (purple) are greater with later stimuli because recovery from inactivation is voltage dependent. B. The relationship between transmembrane potential and degree of recovery of Na+ channels from inactivation. The dotted line indicates 25% recovery. Most Na+ channel–blocking drugs shift this relationship to the left. C. In nodal tissues, premature stimuli delivered even after full repolarization of the action potential are depressed; recovery from inactivation is time dependent.
The situation is different in tissue whose depolarization is largely controlled by Ca2+ channel current such as the AV node. As Ca2+ channels have a slower recovery from inactivation, these tissues often are referred to as slow response, in contrast to fast response in the remaining cardiac tissues (Figure 29–5C). Even after a Ca2+ channel–dependent action potential has repolarized to its initial resting potential, not all Ca2+ channels are available for re-excitation. Therefore, an extra stimulus applied shortly after repolarization is complete and generates a reduced Ca2+ current, which may propagate slowly to adjacent cells prior to extinction. An extra stimulus applied later will result in a larger Ca2+ current and faster propagation. Thus, in Ca2+ channel–dependent tissues, which include not only the AV node but also tissues whose underlying characteristics have been altered by factors such as myocardial ischemia, refractoriness is prolonged and propagation occurs slowly. Slow conduction in the heart, a critical factor in the genesis of reentrant arrhythmias (see next section), also can occur when Na+ currents are depressed by disease or membrane depolarization (e.g., elevated [K]o), resulting in decreased steady-state Na+ channel availability (see Figure 29–5B).
MECHANISMS OF CARDIAC ARRHYTHMIAS
When the normal sequence of impulse initiation and propagation is perturbed, an arrhythmia occurs. Failure of impulse initiation, in the sinus node, may result in slow heart rates (bradyarrhythmias), whereas failure in the normal propagation of action potentials from atrium to ventricle results in dropped beats (commonly referred to as heart block) that usually reflect an abnormality in either the AV node or the His–Purkinje system. These abnormalities may be caused by drugs (Table 29–1) or by structural heart disease; in the latter case, permanent cardiac pacing may be required.
Table 29–1
Drug-Induced Cardiac Arrhythmias
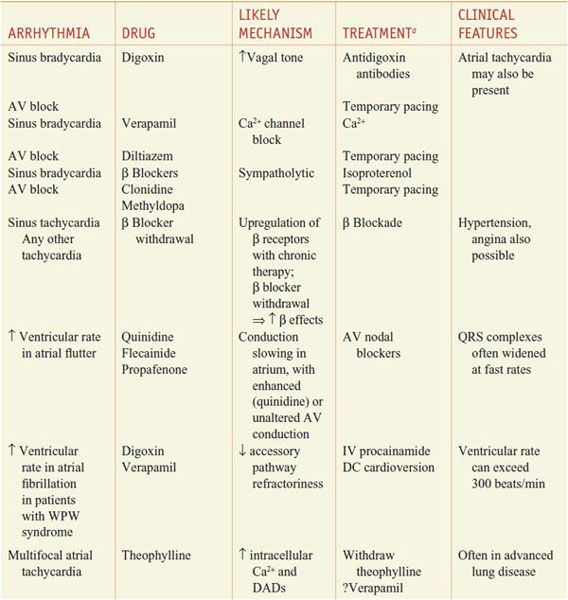
Abnormally rapid heart rhythms (tachyarrhythmias) are common clinical problems that may be treated with anti-arrhythmic drugs. Three major underlying mechanisms have been identified: enhanced automaticity, triggered automaticity, and reentry. These often are interrelated mechanisms, as the first 2 often serve to initiate reentry.
ENHANCED AUTOMATICITY. Enhanced automaticity may occur in cells that normally display spontaneous diastolic depolarization—the sinus and AV nodes and the His–Purkinje system. β Adrenergic stimulation, hypokalemia, and mechanical stretch of cardiac muscle cells increase phase 4 slope and accelerate pacemaker rate; acetylcholine reduces pacemaker rate both by decreasing phase 4 slope and through hyperpolarization (making the maximum diastolic potential more negative). Automatic behavior may also occur in sites that ordinarily lack spontaneous pacemaker activity; e.g., depolarization of ventricular cells (e.g., by ischemia) may produce “abnormal” automaticity. When impulses propagate from a region of enhanced normal or abnormal automaticity to excite the rest of the heart, more complex arrhythmias may result from the induction of functional reentry.
AFTERDEPOLARIZATIONS AND TRIGGERED AUTOMATICITY. Under some pathophysiologic conditions, a normal cardiac action potential may be interrupted or followed by an abnormal depolarization (Figure 29–6). If this abnormal depolarization reaches threshold, it may give rise to secondary upstrokes that can propagate and create abnormal rhythms. These abnormal secondary upstrokes occur only after an initial normal, or “triggering,” upstroke and are termed triggered rhythms.
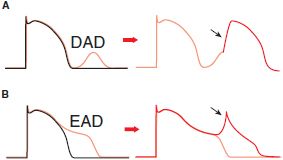
Figure 29–6 Afterdepolarizations and triggered activity. A. Delayed afterdepolarization (DAD) arising after full repolarization. DADs are typically caused by spontaneous Ca2+ release from the sarcoplasmic reticulum under conditions of Ca2+ overload. The extra cytosolic Ca2+ is removed from the cytosol by the electrogenic Na-Ca exchanger (NCX), which produces Na+ influx and causes a cell membrane depolarization in the form of a DAD. A DAD that reaches threshold results in a triggered upstroke (black arrow, right). B. Early afterdepolarization (EAD) interrupting phase 3 repolarization. Multiple ion channels and transporters can contribute to EADs (e.g., Na+ channel, L-type Ca2+ channel, NCX). Under some conditions, triggered beat(s) can arise from an EAD (black arrow, right).
In the first form of triggered rhythm, under conditions of intracellular or SR Ca2+ overload (e.g., myocardial ischemia, adrenergic stress, digitalis intoxication, heart failure), a normal action potential may be followed by a delayed afterdepolarization (DAD; Figure 29–6A). If this afterdepolarization reaches threshold, a secondary triggered beat or beats may occur. In the second type of triggered activity, the key abnormality is marked prolongation of the cardiac action potential. When this occurs, phase 3 repolarization may be interrupted by an early afterdepolarization (EAD; Figure 29–6B). EAD-mediated triggering in vitro and clinical arrhythmias are most common when the underlying heart rate is slow, extracellular K+ is low, and certain drugs that prolong APD are present. EAD-related triggered upstrokes probably reflect inward current through Na+ or Ca2+ channels. EADs are induced much more readily in Purkinje cells than in epicardial or endocardial cells. When cardiac repolarization is markedly prolonged, polymorphic ventricular tachycardia with a long QT interval, known as the torsade de pointes syndrome, may occur. This arrhythmia is thought to be caused by EADs, which trigger functional reentry owing to heterogeneity of APDs across the ventricular wall. Congenital long QT syndrome, a disease in which torsade de pointes is common, can most often be caused by mutations in the genes encoding the Na+ channels or the channels underlying the repolarizing currents IKr and IKs.
REENTRY. Reentry occurs when a cardiac impulse travels in a path such as to return to its original site and reactivate the original site and self-perpetuate rapid activation independent of the normal sinus node conduction. This abnormal activation path (or reentrant circuit) requires an isotropic conduction slowing (or failure) due to either an anatomic or functional barrier.
ANATOMICALLY DEFINED REENTRY. Reentry can occur when impulses propagate by more than 1 pathway between 2 points in the heart, and those pathways have heterogeneous electrophysiologic properties. Patients with Wolff–Parkinson–White (WPW) syndrome have accessory connections between the atrium and ventricle (Figure 29–7). With each sinus node depolarization, impulses can excite the ventricle via the normal structures (AV node) or the accessory pathway. However, the electrophysiologic properties of the AV node and accessory pathways are different: Accessory pathways usually consist of nonnodal tissue and consequently vastly differ in refractoriness with the AV node. Thus, with a premature atrial beat, conduction may fail in the accessory pathway but continue, albeit slowly, in the AV node and then through the His–Purkinje system; there, the propagating impulse may encounter the ventricular end of the accessory pathway when it is no longer refractory. The likelihood that the accessory pathway is no longer refractory increases as AV nodal conduction slows. When the impulse reenters the atrium, it then can reenter the ventricle via the AV node, reenter the atrium via the accessory pathway, and so on. Reentry of this type, referred to as AV reentrant tachycardia, is determined by:
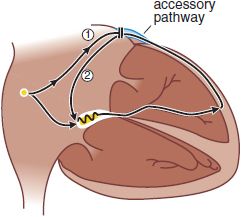
Figure 29–7 Atrioventricular reentrant tachycardia in the Wolff–Parkinson–White syndrome. In these patients, an accessory atrioventricular (AV) connection is present (light blue). A premature atrial impulse blocks in the accessory pathway (1) and propagates slowly through the AV node and conducting system. On reaching the accessory pathway (by now no longer refractory), the impulse reenters the atrium (2), where it then can reenter the ventricle via the AV node and become self-sustaining (see Figure 29–9C). AV nodal blocking drugs readily terminate this tachycardia. Recurrences can be prevented by drugs that prevent atrial premature beats, by drugs that alter the electrophysi-ologic characteristics of tissue in the circuit (e.g., they prolong AV nodal refractoriness), and by nonpharmacologic techniques that section the accessory pathway.
• The presence of an anatomically defined circuit
• Heterogeneity in refractoriness among regions in the circuit
• Slow conduction in one part of the circuit
Similar “anatomically defined” reentry commonly occurs in the region of the AV node (AV nodal reentrant tachycardia) and in the atrium (atrial flutter). The term paroxysmal supraventricular tachycardia (PSVT) includes both AV reentry and AV nodal reentry, which share many clinical features. It now is sometimes possible to identify and ablate critical segments of reentrant pathway (or automatic foci), thus curing the patient and obviating the need for long-term drug therapy.
FUNCTIONALLY DEFINED REENTRY. Reentry also may occur in the absence of a distinct, anatomically defined pathway (Figure 29–8). If ischemia or other electrophysiologic perturbations result in an area of sufficiently slow conduction in the ventricle, the impulses exiting from that area find the rest of the myocardium re-excitable and reentry may ensue. Atrial or ventricular fibrillation (VF) is an extreme example of “functionally defined” (or “leading circle”) reentry. Cells are re-excited as soon as they are repolarized sufficiently to allow enough Na+ channels to recover from inactivation. In this setting, neither organized activation patterns nor coordinated contractile activity is present.
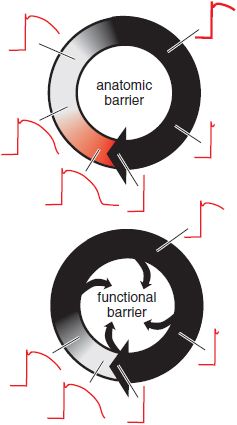
Figure 29–8 Two types of reentry. The border of a propagating wavefront is denoted by a heavy black arrowhead. In anatomically defined reentry (top), a fixed pathway is present (e.g., Figure 29–7). The black area denotes tissue in the reentrant circuit that is completely refractory because of the recent passage of the propagating wavefront; the gray area denotes tissue in which depressed upstrokes can be elicited (see Figure 29–5A), and the red area represents tissue in which restimulation would result in action potentials with normal upstrokes. The red area is termed an excitable gap. In functionally defined, or “leading circle,” reentry (bottom), there is no anatomic pathway and no excitable gap. Rather, the circulating wavefront creates an area of inexcitable tissue at its core. In this type of reentry, the circuit does not necessarily remain in the same anatomic position during consecutive beats, and multiple such “rotors” may be present.
COMMON ARRHYTHMIAS AND THEIR MECHANISMS
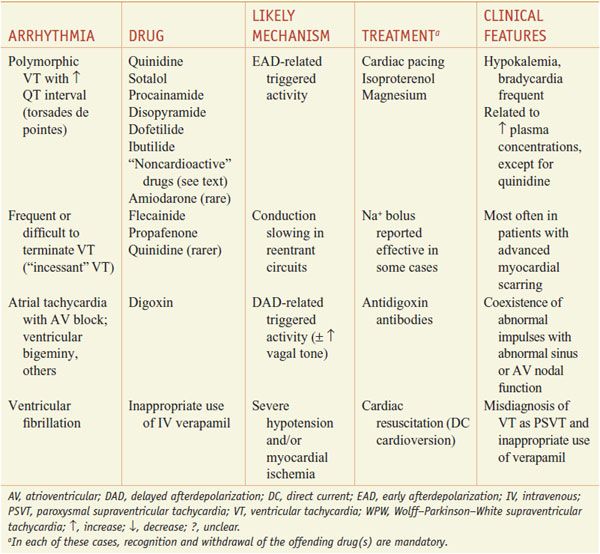
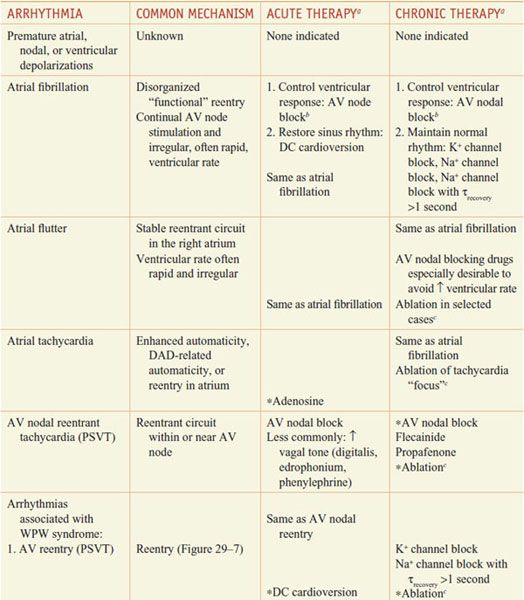
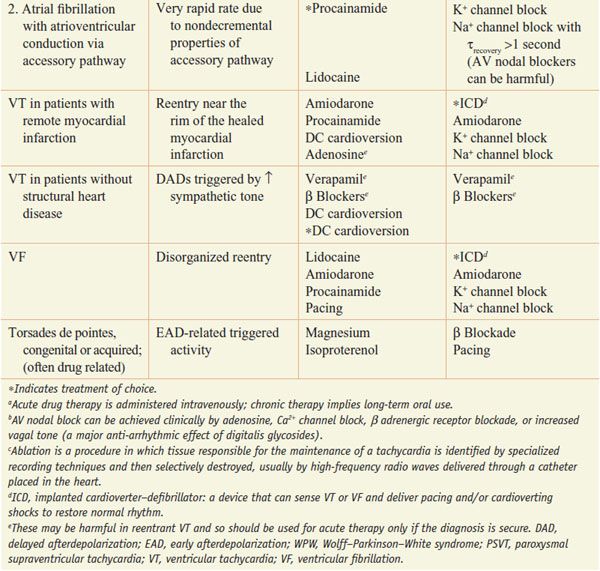
Table 29–2 lists common arrhythmias, their likely mechanisms, and approaches that should be considered for their acute termination and for long-term therapy to prevent recurrence.
Table 29–2
A Mechanistic Approach to Anti-Arrhythmic Therapy
MECHANISMS OF ANTI-ARRHYTHMIC DRUG ACTION
Anti-arrhythmic drugs almost invariably have multiple effects in patients, and their effects on arrhythmias can be complex. A drug can modulate additional targets in addition to its primary mode of action. A single arrhythmia may result from multiple underlying mechanisms. Drugs may be anti-arrhythmic by suppressing the initiating mechanism or by altering the reentrant circuit. In some cases, drugs may suppress the initiator but nonetheless promote reentry.
Drugs may slow automatic rhythms by altering any of the 4 determinants of spontaneous pacemaker discharge (Figure 29–9):
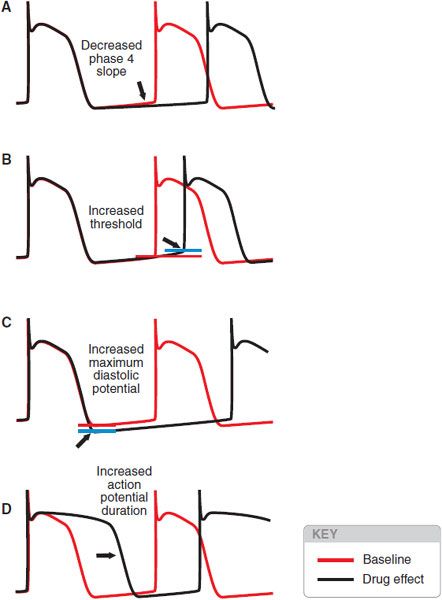
Figure 29–9 Four ways to reduce the rate of spontaneous discharge. The blue horizontal line represents threshold potential.
• Decrease phase 4 slope
• Increase threshold potential
• Increase maximum diastolic potential
• Increase APD
Adenosine and acetylcholine may increase maximum diastolic potential, and βreceptor antagonists may decrease phase 4 slope. Blockade of Na+ or Ca2+ channels usually results in altered threshold, and blockade of cardiac K+ channels prolongs the action potential.
Anti-arrhythmic drugs may block arrhythmias owing to DADs or EADs by 2 major mechanisms:
• Inhibition of the development of afterdepolarizations
• Interference with the inward current (usually through Na+ or Ca2+ channels), which is responsible for the upstroke
Thus, arrhythmias owing to digitalis-induced DADs may be inhibited by verapamil (which blocks the development of DAD by reducing Ca2+ influx and subsequent storage/release) or by quinidine (which blocks Na+ channels, thereby elevating the threshold required to produce the abnormal upstroke). Similarly, 2 approaches are used in arrhythmias related to EAD-triggered beats (see Tables 29–1 and 29–2). EADs can be inhibited by shortening APD; in practice, heart rate is accelerated by isoproterenol infusion or by pacing. Triggered beats arising from EADs can be inhibited by Mg2+, without normalizing repolarization in vitro or QT interval. In patients with a congenitally prolonged QT interval, torsade de pointes often occurs with adrenergic stress; therapy includes β adrenergic blockade (which does not shorten the QT interval) as well as pacing.
In anatomically determined reentry, drugs may terminate the arrhythmia by blocking propagation of the action potential. In the example of the WPW-related arrhythmia, drugs that prolong AV nodal refractoriness and slow AV nodal conduction, such as Ca2+ channel blockers, βadrenergic receptor antagonists, or digitalis glycosides, are likely to be effective. Conversely, slowing conduction in functionally determined reentrant circuits may change the pathway without extinguishing the circuit. Slow conduction generally promotes the development of reentrant arrhythmias, whereas the most likely approach for terminating functionally determined reentry is prolongation of refractoriness. In atrial and ventricular myocytes, refractoriness can be prolonged by delaying the recovery of Na+ channels from inactivation. Drugs blocking Na+ channels generally shift the voltage dependence of recovery from block (see Figure 29–5B) and so prolong refractoriness (Figure 29–10). Drugs that increase APD without direct action on Na+ channels (e.g., by blocking delayed-rectifier currents) also will prolong refractoriness. In sinoatrial or AV nodal tissues, Ca2+ channel blockade prolongs refractoriness.
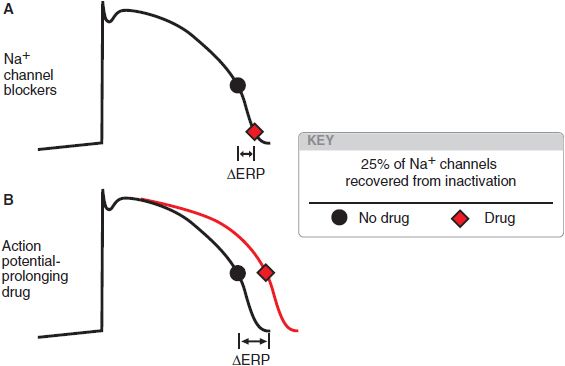
Figure 29–10 Two ways to increase refractoriness. The black dot indicates the point at which a sufficient number of Na+ channels (an arbitrary 25%; see Figure 29–5
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


