Chapter 45
Alterations of Musculoskeletal Function in Children
Kristen Lee Carroll and Lynn M. Kerr

Musculoskeletal Development in Children
Bone Formation
Bone formation, which begins at about the sixth week of gestation, involves two phases: (1) the delivery of bone cell precursors to sites of bone formation and (2) the aggregation of these cells at primary centers of ossification, where they mature and begin to secrete osteoid (see Chapter 43). Some of the bone cell precursors are present in fetal connective tissues, whereas others migrate in blood to sites of bone formation after blood vessels have grown into the tissue.
Endochondral formation of bone is the development of new bone from cartilage (Figure 45-1). First, mesenchymal tissue forms a cartilage anlage, which defines the shape of the bone. This is usually found by 6 weeks of gestation. Blood vessel invasion to inside the anlage brings osteoprogenitor cells leading to primary centers of calcification by 8 weeks. Endochondral bone formation begins in the outer layer of the cartilage model, which consists of a layer of dense connective tissue called perichondrium. The perichondrium contains cells that develop into osteoblasts, forming a collar of bone, termed the periosteal collar, around the cartilage model. Cartilage enclosed within the periosteal collar degenerates, and capillaries from outside the perichondrium invade the degenerating cartilage cells, carrying with them osteoblast precursors from the inner layer of the perichondrium and osteoclast precursors from the blood itself.
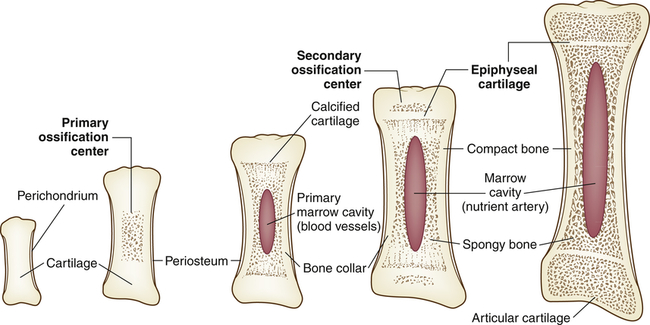
Two regions of cartilage remain at the ends of long bones: (1) articular cartilage over the free ends of the bone, and (2) the physeal plate, a layer of cartilage between the metaphysis and epiphysis. (These structures are described and illustrated in Chapter 43; see Figure 43-3.) The physeal plate retains the ability to form and calcify new cartilage and deposit bone until the skeleton matures approximately 1 year after sexual maturity (11 to 15 years of age in females, 15 to 18 in males).
Bone Growth
Even after physeal closure at skeletal maturity, bone is constantly being destroyed and re-formed (see Chapter 43). This is a rapid process in young children, allowing them to heal bone injury more quickly than adults. By adulthood, however, bone turnover, or remodeling, occurs at a relatively slow rate. Peak bone mass is achieved by the middle to late twenties and slowly decreases throughout life; therefore, ensuring appropriate levels of calcium and phosphorous intake, performing weightbearing lifting and exercise, and minimizing caffeine intake are especially important for a young female if she is to avoid osteoporosis in later life. Recently, the importance of vitamin D levels also has been emphasized. In one study, nearly 70% of American children had low levels of vitamin D.1
Skeletal Development
The axial skeleton changes shape with growth. (The axial skeleton and appendicular skeleton are described and illustrated in Chapter 43; see Figure 43-5). In a newborn the entire spine is concave anteriorly, or kyphosed. In the first 3 months of life, with the infant’s ability to control the head, the upper (cervical) spine begins to arch, or become lordotic. The normal lordotic curve of the lower (lumbar) spine begins to develop with sitting.
The appendicular skeleton (the extremities) grows faster during childhood than does the axial skeleton (see Figure 43-5). The newborn has a relatively large head and long spine with disproportionately shorter limbs than an adult. By 1 year of age, 50% of the total growth of the spine has occurred and is more than 70% complete by age 8.2 Therefore, failure of the spine to grow (e.g., spinal fusion) does not limit eventual height as much as the premature fusion of the growth plates of the lower extremities. In children with congenital curvature of the spine, growth tends to worsen the deformity rather than to increase the length of the spine.
Besides getting longer, growing bones of the extremities undergo changes in rotation and alignment. In the newborn the proximal femur is rotated forward up to 40 degrees and the tibia is rotated inward. With growth the femur assumes its normal alignment (by 12 years of age) and tibial rotation neutralizes at 8 years of age.3 Bowlegs and knock knees can be normal at certain stages of growth. At birth the newborn’s legs are bowed because of stresses in utero. Genu varum (bowleg) reaches a peak by 30 months of age, whereas genu valgum (knock knee) maximizes by 5 to 6 years of age. If genu varum or genu valgum persists past these ages, a pathologic process rather than a physiologic phase may be present. Pathologic causes of genu varum are Blount disease, rickets, skeletal dysplasias (such as achondroplastic dwarfism), and traumatic injury. Genu valgum may persist also as a result of skeletal dysplasia or genetic predisposition.
Muscle Growth
The composition and size of muscles vary with age. In the fetus, muscle tissue contains a large amount of water and much intercellular matrix. After birth, both are reduced considerably as the muscle fibers (cells) enlarge by accumulating cytoplasm. Little information is available about the numbers of fibers in a given muscle at various ages, but the total mass of muscle in the body can be estimated from the amount of creatinine excreted in the urine, because the conversion of creatine to creatinine takes place only in muscle (see Chapter 43). Between birth and maturity the number of muscle nuclei in the body increases 14 times in boys and 10 times in girls. Muscle fibers reach their maximal size in girls at approximately 10 years of age and in boys by 14 years. Growth in length occurs at the ends of muscles, and the increase in length is accompanied by an increase in number of nuclei in the fibers. Muscle fibers increase in diameter as the fibrils become more numerous. The fibrils themselves do not increase in diameter. Connective tissue components of muscle grow where the tendon and muscle meet.
Musculoskeletal Alterations in Children
Congenital Defects
Syndactyly
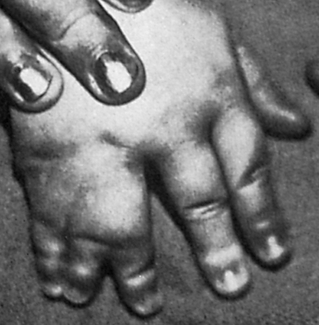
The most common congenital defect of the upper extremity is syndactyly, or webbing of the fingers (Figure 45-2). Simple webbing involves the soft tissue envelope alone and is best released surgically when the child is 6 months to 1 year of age. Complex syndactyly involves fusion of the bones and nails as well as the soft tissues; it may be associated with absence or anomaly of bony or neurovascular units. The primary goal in surgical correction of these defects is to achieve maximal function and appearance. Ideally, corrective surgery is deferred until the child is 1 to 2 years old and completed before the child enters school. Vestigial tabs, such as an extra digit, however, are best removed during the immediate neonatal period. Anomalies on the radial aspect of the arm, such as a foreshortened or absent radius, are often associated with abnormalities of blood, heart, or kidneys. Lateral or ulnar-sided defects are less often associated with systemic anomalies and are far more rare.
Developmental Dysplasia of the Hip
Developmental dysplasia of the hip (DDH), formerly known as congenital dislocation of the hip, is an abnormality in the development of the proximal femur, acetabulum, or both (Figure 45-3). Although most often present at birth, it may occur at any time in the newborn or infant period.
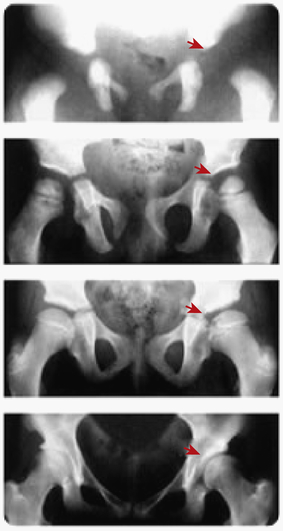
Developmental dysplasia of the hip (DDH) with residual acetabular dysplasia. Radiographs at birth, 3, 10, and 19 years (top to bottom) show persisting dysplasia.
Pathophysiology
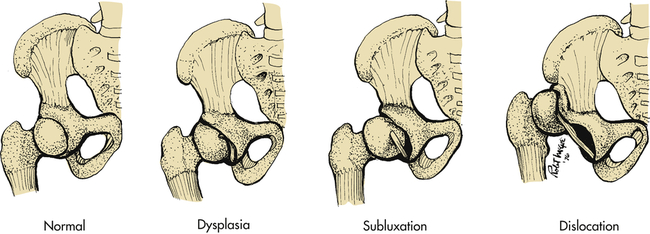
The hip can be described as subluxated (partial contact only), dislocated (no contact between femoral head and acetabulum), and acetabular dysplasia (the femoral head is located properly but the acetabulum is shallow) (Figure 45-4). The subluxated hip maintains contact with the acetabulum but is not well seated within the hip joint. The acetabulum is often dysplastic (or shallow) although the femur is often normal. The dislocatable hip is sometimes located but can be dislocated easily. The dislocated hip has no contact between the femoral head and the acetabulum. Some degree of acetabular dysplasia is present in almost all cases. Typically the acetabulum is shallow or sloping rather than cup shaped.
If DDH is left untreated in the growing child, secondary changes occur. If the hip remains subluxated or dislocated, the acetabulum becomes increasingly shallow and the soft tissues shorten around the proximal femur. Subluxation leads to early osteoarthritis (OA), and it is now estimated that at least 60% of all OA of the hip is related to DDH. If the hip is dislocated, the bone acetabulum fills with soft tissue and a false acetabulum forms where the femoral head contacts the iliac crest. An apparent limb length inequity and hip muscle weakness occurs, leading to a waddling gait. Back pain and hip pain develop in adulthood. Adult reconstruction of a dislocated hip, even with an artificial hip, is very difficult.4
Clinical Manifestations
1. Asymmetry of gluteal or thigh folds
2. Limb length discrepancy (Galeazzi sign)
3. Limitation of hip abduction
4. Positive Barlow maneuver (hip reduced, but dislocatable) (Figure 45-5, A)
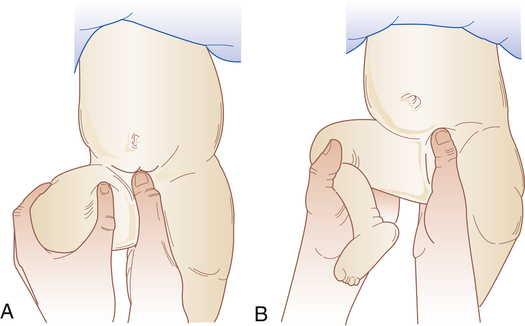
A, Barlow maneuver. With one hand pressing the symphysis in front and the sacral spine in back, lateral pressure is applied to the thigh with the thumb of the other hand while pressure is applied with the palm to the knee on the side being examined. The hip that has been flexed to 90 degrees is then adducted. A positive sign is a sensation of abnormal movement, indicating dislocation of the femoral head from the acetabulum. The hands are reversed for examining the other hip. This sign and Ortolani sign may be found only in the first weeks of life. B, Ortolani maneuver. Sign of jerking into correct position. After the Barlow maneuver (A), the hip should be abducted to about 80 degrees while the femur is lifted anteriorly with the fingers along the thigh. A positive sign is a sensation of a jerk or snap with reduction into the joint socket. (Adapted from Specht EE: Am Fam Physician 9:88–96, 1974.)
5. Positive Ortolani sign (hip dislocated, but reducible) (Figure 45-5, B)
6. Positive Trendelenburg gait (waddling)
Evaluation and Treatment
In the newborn period clinical examination is the most important diagnostic tool. Real-time ultrasound, in which the hip is examined while the ultrasound is performed, also is extremely valuable in the newborn period, especially in high-risk infants. The use of ultrasound allows visualization of the cartilaginous structures of the hip (the femoral head and the outer lip of the acetabulum), which are not seen on plain roentgenogram. Radiographs are used after age 6 months when the ossific nucleus of the femoral head appears.5
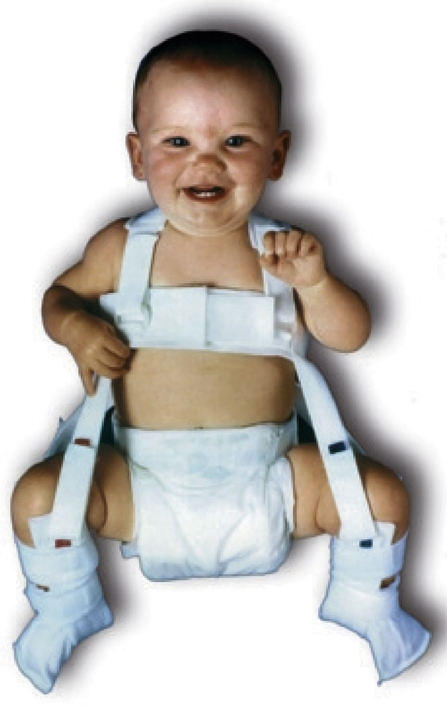
Treatment depends on the age of the child, severity of dysplasia, and duration of dysplasia. The earlier that treatment is begun, the better the result. In children less than 4 months of age, a Pavlik harness can brace the hip in abduction and flexion, and the acetabulum will remodel as the femoral head rests centered in the socket (Figure 45-6). With this treatment, up to 98% of children will have an excellent result. A “closed” reduction (without opening the joint) followed by spica or body casting for up to 3 months can be done in children up to 12 months of age. After 12 months, surgical intervention—including opening the joint and cutting and realigning the femur and/or acetabulum—may be required. As the child ages, the percentage of good outcomes decreases. Up to 70% of children treated surgically for DDH after age 3 develop early osteoarthritis.6 Early intervention before age 1 is critical for a good outcome; therefore, vigilance for this problem within the first year is essential.
Deformities of the Foot
Congenital Deformity
Congenital foot deformity is found in approximately 4% of all newborns, and metatarsus adductus accounts for 75% of these deformities (Table 45-1). Metatarsus adductus is a forefoot adduction deformity associated with a normal, plantigrade hindfoot and is believed to be secondary to intrauterine positioning. It is associated with developmental dysplasia of the hip in 20% of cases; consequently, the hips of these infants must be carefully evaluated. Metatarsus adductus is usually classified by two criteria: flexibility (passively correctable or rigid) and degree of deformity. The degree of deformity (mild, moderate, severe) is ascertained by the heel bisection line. A mild deformity is one in which the heel bisection line passes medial to the third toe; moderate, through the third or fourth toes; and severe, lateral to the fourth toe. Serial casts during the first 6 months of life are suggested for moderate to severe deformities and those deformities that appear less flexible. Casts are changed weekly for 6 to 12 weeks. By 6 years of age, 87% of children usually correct spontaneously, and 95% by 15 years of age. Even in those children with some residual deformity, functional symptoms are rare.
TABLE 45-1
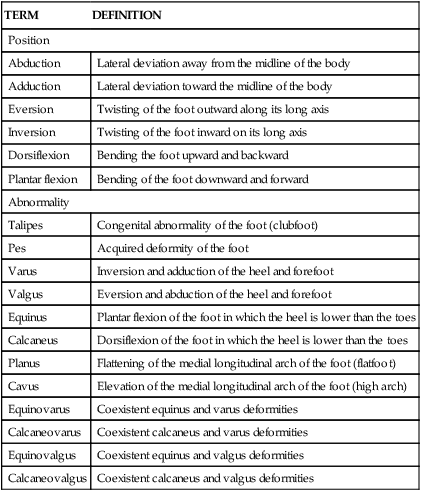
TERMS USED TO DESCRIBE FOOT ABNORMALITIES
| TERM | DEFINITION |
| Position | |
| Abduction | Lateral deviation away from the midline of the body |
| Adduction | Lateral deviation toward the midline of the body |
| Eversion | Twisting of the foot outward along its long axis |
| Inversion | Twisting of the foot inward on its long axis |
| Dorsiflexion | Bending the foot upward and backward |
| Plantar flexion | Bending of the foot downward and forward |
| Abnormality | |
| Talipes | Congenital abnormality of the foot (clubfoot) |
| Pes | Acquired deformity of the foot |
| Varus | Inversion and adduction of the heel and forefoot |
| Valgus | Eversion and abduction of the heel and forefoot |
| Equinus | Plantar flexion of the foot in which the heel is lower than the toes |
| Calcaneus | Dorsiflexion of the foot in which the heel is lower than the toes |
| Planus | Flattening of the medial longitudinal arch of the foot (flatfoot) |
| Cavus | Elevation of the medial longitudinal arch of the foot (high arch) |
| Equinovarus | Coexistent equinus and varus deformities |
| Calcaneovarus | Coexistent calcaneus and varus deformities |
| Equinovalgus | Coexistent equinus and valgus deformities |
| Calcaneovalgus | Coexistent calcaneus and valgus deformities |
Clubfoot: Equinovarus Deformity
Clubfoot describes a range of foot deformities in which the foot turns inward and downward. Technically called equinovarus, the heel is positioned varus (inwardly deviated) and equinus (plantar flexed) (Figures 45-7 and 45-8). The clubfoot deformity can be positional (correctable passively), idiopathic, or teratologic equinovarus (as a result of another syndrome, such as spina bifida). These three types are discussed in the following sections. Overall, the true positional equinovarus lends itself to rapid correction by application of serial casts. The idiopathic variety is treated by attempting cast correction, followed by surgical intervention of resistant deformities. Teratologic equinovarus nearly always requires surgical correction and/or muscle balancing procedures.
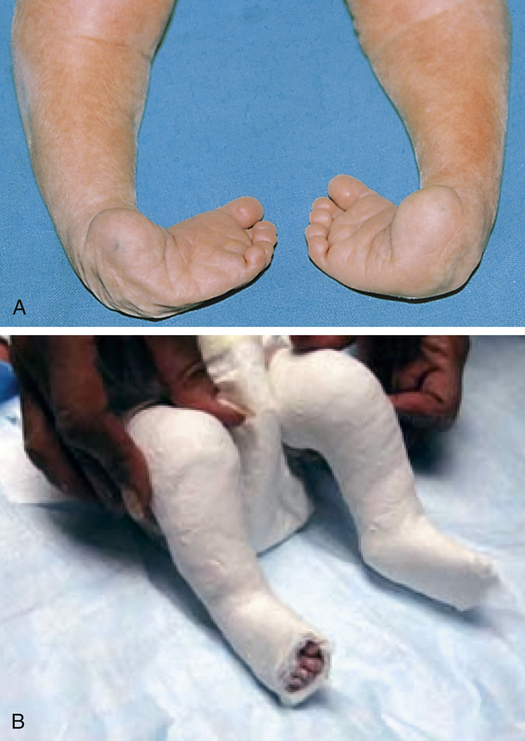
A, Infant with bilateral congenital talipes equinovarus. B, Ponseti casting. (A courtesy of Dr. a.E. Chudley, Section of Genetics and Metabolism, Department of Pediatrics and Child Health, Children’s Hospital and University of Manitoba, Winnipeg, Manitoba, Canada. In Moore KL, et al: The developing human, ed 9, Philadelphia, 2013, Saunders.)
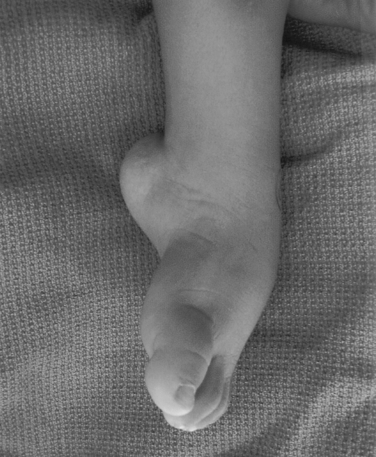
Idiopathic clubfoot displaying forefoot adduction (toward midline of body) and supination (upturning) and hindfoot equinus (pointed downward). Note skin creases along arch and back of heal.
Idiopathic Congenital Equinovarus
Idiopathic equinovarus occurs in approximately 1 of every 1000 live births, with males being affected twice as often as females. Historically, these deformities were treated by posteromedial release, a surgical procedure that lengthened all tight structures and opened the capsule of all tight joints in the foot. However, since 1998 the casting technique developed by Ignacio Ponseti (see Figure 45-7, B) has been used; the technique involves six to eight above-knee casts, left on for 5 to 7 days each, followed by a percutaneous tendoachilles lengthening procedure performed with local anesthesia. The child then uses special braces at night until 3 years of age. Noncompliance with braces leads to increased recurrence and need for additional casting or surgery. Nearly 30% of children may need an anterior tibialis transfer around age 3.7 Studies comparing operative posteromedial release with Ponseti techniques show better long-term results with less invasive Ponseti method8 (Box 45-1).
Teratologic Equinovarus
The most common causes of teratologic equinovarus are either neuromuscular (such as spina bifida) or syndromic, as in arthrogryposis or osteochondrodysplasia (such as diastrophic dwarfism). The teratologic clubfoot, unlike the idiopathic type, more often fails to be corrected with Ponseti casting and may require operative intervention. The surgery is often more extensive than that for an idiopathic clubfoot, and revision surgery is also more common (Box 45-1).
Abnormal Density or Modeling of the Skeleton
Osteogenesis Imperfecta
Osteogenesis imperfecta (OI) (brittle bone disease) is a spectrum of disease caused by genetic mutation in the gene that codes for type I collagen, the main component of bone and blood vessels. The disorder was first described in 1840 as a syndrome in newborns that consisted of osteoporosis with fractures and skeletal deformities. The Sillence classification is based on both models of inheritance and clinical findings (Table 45-2). In the most severe form of this disorder, the child is usually stillborn or dies soon after birth, although some survive into childhood. OI in its more severe forms is evident at birth because fractures and deformity have occurred in utero. The less severe forms may not become evident until the child begins to walk. Some children with this milder form then experience numerous fractures and can be mistaken for nonaccidental trauma until the diagnosis is made.
TABLE 45-2
SILLENCE CLASSIFICATION OF OSTEOGENESIS IMPERFECTA SYNDROMES
| TYPE | TRANSMISSION | MAIN BIOCHEMICAL DEFECT | ORTHOPEDIC | MISCELLANEOUS |
| IA | AD | Decreased production of type I collagen | Mild to moderate bone fragility, osteoporosis, normal stature | Blue sclera, hearing loss, easy bruising, dentinogenesis imperfecta absent |
| IB | AD | Short stature | More severe in IA with dentinogenesis imperfecta | |
| II | AD, AR, and mosaic | Substitutions of glycyl residue in X1 or X2 chains in triple helix | Multiple intrauterine fractures, extreme bone fragility | Usually lethal in perinatal period, delayed ossification of skull, intrauterine growth restriction |
| IIA | Long bones broad, crumpled; ribs broad with continuous beading | |||
| IIB | Long bones broad, crumpled; ribs discontinuous or beading | |||
| IIC | Long bones thin, fractured; ribs thin, beaded | |||
| IID | Severely osteoporotic with generally well-formed skeleton; normal-shaped vertebrae and pelvis | |||
| III | AD and AR | Abnormal type I collagen | Progressive deforming phenotype, severe bone fragility with fractures | Hearing loss, short stature, blue sclerae becoming less blue with age, shortened life expectancy, dentinogenesis imperfecta, relative macrocephaly with triangular facies |
| IVA | AD | Shortened pro-α (I)-chains | Mild to moderate bone fragility, osteoporosis, bowing of long bones, scoliosis | Light sclerae, normal hearing, normal dentition, dentinogenesis imperfecta absent |
| IVB | AD | Dentinogenesis imperfecta present |
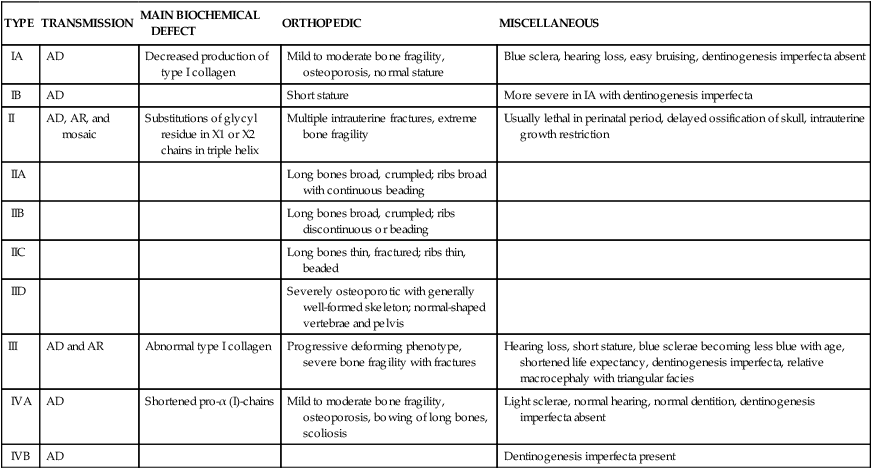
AD, Autosomal dominant; AR, autosomal recessive.
Reproduced with permission from Vaccaro AR, editor: Orthopaedic knowledge update 8, p 248, Rosemont, IL, 2005, American Academy of Orthopaedic Surgeons.
The prevalence rate of the most common form is about 1 in 30,000. Inheritance is usually autosomal dominant but can be autosomal recessive. At least four syndromes have been identified that have various clinical manifestations and prognoses (see Table 45-2).
Pathophysiology
The major errors in OI lie in the synthesis of collagen, a triple helix with two matching alpha chains and one beta chain. Collagen is present in bone, cartilage, eye tissue, skin, and the vascular system. The severity of the OI phenotype and the related anomalies of the eye, dentition, or vascular system are all dependent on the severity of the genetic anomaly and the part of the triple helix that is affected.9 (Genes are discussed in Chapter 4.)
Evaluation and Treatment
Type II OI is often terminal in the perinatal period, and therefore little is known about appropriate treatment for the few children who survive. For other types of OI, careful positioning and handling of the newborn help prevent fractures. Beyond the neonatal period, various orthopedic measures are applied, such as prompt splinting of fractures and correction of deformities arising from the progressive bowing or bending of the skeleton by intramedullary rodding of the bones (Figure 45-9). Newer, telescoping rods, which grow with the child, have been shown to reduce the reoperative rate by 30%.10 Scoliosis is present in up to 50% of Sillence III cases and often requires surgery. A multicenter study of a bisphosphonate therapy showed promising results in type III OI, with marked improvements of bone density (up to 30%). Despite these results, there is concern that the healing of fractures and surgical intervention can be more difficult. More study is needed to address the efficacy and safety of these types of drugs. Genetic counseling for affected families should aim at primary prevention.
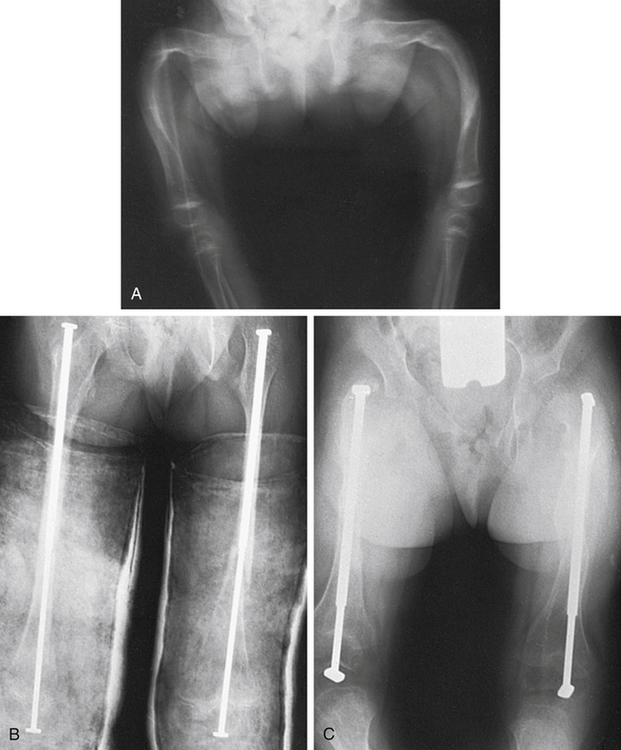
A, Severe deformity of both femurs. B, Same individual after multiple osteotomies with telescoping medullary rod fixation. C, Same individual 4 years later demonstrating growth of femurs, no recurrence of deformity, and elongation of rods. (Plaster casts are in place for immobilization of tibial osteotomies.) (From Crenshaw AH, editor: Campbell’s operative orthopaedics, ed 8, vol 3, St Louis, 1992, Mosby.)
Rickets
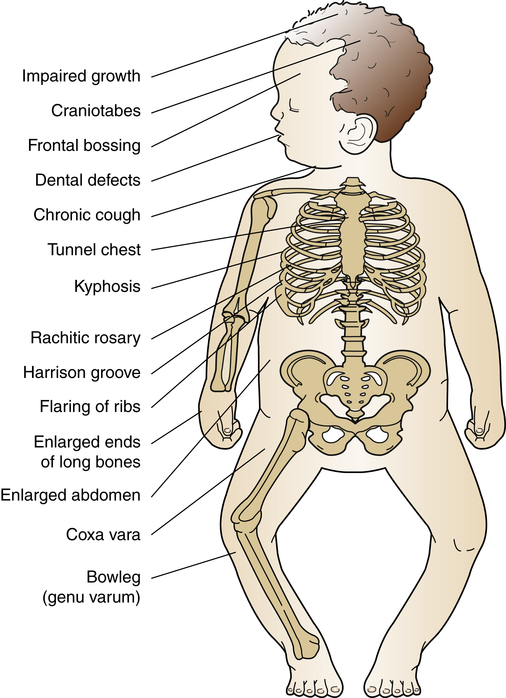
Rickets is a disorder in which growing bone fails to become mineralized (ossified), resulting in “soft” bones and skeletal deformity (Figure 45-10). Rickets results from either insufficient vitamin D, insensitivity to vitamin D, wasting of vitamin D by the kidney, or inability to absorb vitamin D and calcium in the gut. The most common form is X-linked hypophosphatemic rickets in industrialized nations. In addition to the severe form of metabolic rickets, dietary and lifestyle changes in the United States have led to widespread vitamin D deficiency in children.11 Although unprotected exposure to ultraviolet rays is not suggested, children still need 15 to 20 minutes per week of true sun exposure to activate vitamin D, the mineral necessary for absorption and metabolism of calcium and phosphate. In one recent study up to 90% of normal American children had a low vitamin D level, especially children of color. This can lead to early fracture or slow bone healing after fracture.12
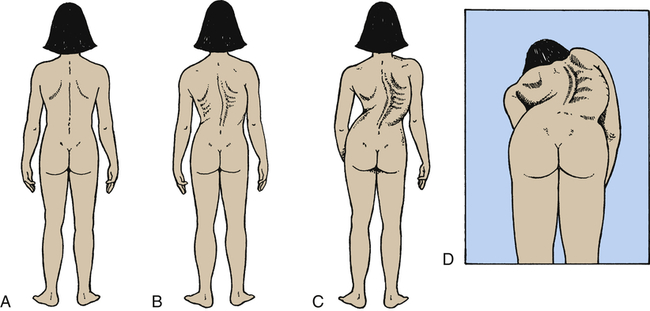
Scoliosis
Scoliosis is a rotational curvature of the spine most obvious in the anteroposterior plane (Figure 45-11). It can be classified as nonstructural or structural. Nonstructural scoliosis results from a cause other than the spine itself, such as posture, leg length discrepancy, or pain. Structural scoliosis is curvature of the spine associated with vertebral rotation. Nonstructural scoliosis can become structural if the underlying cause is not found and treated.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


