Chapter 30
Alterations of Hematologic Function in Children

Fetal and Neonatal Hematopoiesis
In neonates and young infants, hematopoietic marrow progressively fills the bony cavities of the entire axial skeleton (skull, vertebrae, ribs, sternum), the long bones of the limbs, and many intramembranous bones. (These structures are described in Chapter 45.) Fatty (yellow) marrow gradually replaces hematopoietic marrow in some bones. During childhood, hematopoietic tissue retreats centrally to the vertebrae, ribs, sternum, pelvis, scapulae, skull, and proximal ends of the femur and humerus.
In diseases characterized by hemolysis, erythrocyte production can increase as much as eight times the normal because erythropoietin causes hematopoietic marrow to increase in volume. Initially, hematopoietic marrow expands from the ends of the long bones toward the middle of the shafts, replacing fatty marrow. Next, blood cell production begins to occur outside the marrow cavities, especially in the liver and spleen. Extramedullary hematopoiesis is more likely to occur in children than in adults because the bony cavities of children already are filled with red marrow (Figure 30-1). This is why hemolytic disease causes especially pronounced enlargement of the spleen and liver in children.
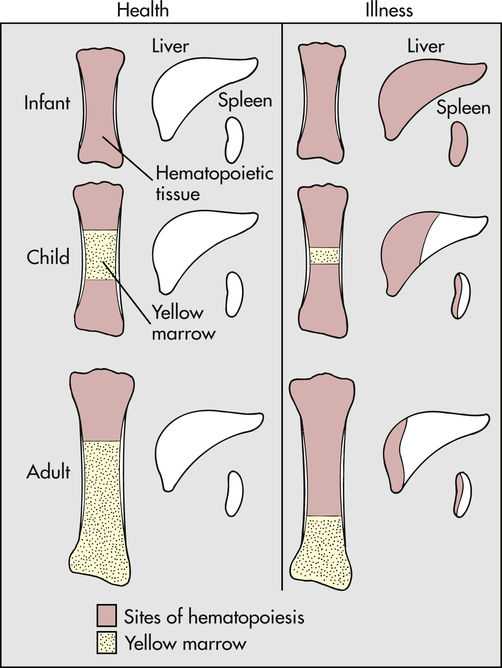
With normal maturation, red marrow is partly replaced by yellow marrow in the shafts of the long bones. In adults, red marrow is largely restricted to the proximal ends of the femur and humerus. In response to hemolysis, red marrow replaces yellow marrow in the long bones. In infants, whose long bones already are filled with red marrow, additional hematopoiesis takes place in the liver and spleen. In children and adults, red marrow can replace yellow marrow in response to hemolysis, necessitating less hematopoiesis in the liver and spleen.
A biochemically distinct type of hemoglobin is synthesized during fetal life. The three embryonic hemoglobins (Gower 1, Gower 2, and Portland) and the fetal hemoglobin (HbF) are composed of two α- and two γ-chains of polypeptides, whereas the adult hemoglobins (HbA and HbA2) are composed of two α-chains and two β-chains. (The structure of an adult hemoglobin molecule is illustrated in Figure 27-15, and types of hemoglobin are defined in Table 27-5.) Some unknown regulatory mechanism promotes γ-chain synthesis and inhibits β- and δ-chain synthesis in utero. This results in production of embryonic or fetal hemoglobin. After birth, γ-chain synthesis is inhibited, whereas β- and δ-chain synthesis is facilitated, resulting in production of adult hemoglobins.
During the first trimester nearly all of the hemoglobin in the fetus is embryonic, but some Hb A can be detected. Therefore, it is possible to identify as early as 16 to 20 weeks of gestation some disorders of adult hemoglobin, such as sickle cell anemia and thalassemia major. In the 6-month fetus, Hb F constitutes 90% of the total. This percentage then begins to decline. At birth, neonatal hemoglobin consists of 70% Hb F, 29% Hb A, and 1% Hb A2. Between 6 and 12 months of age, normal adult hemoglobin percentages are established (see Chapter 27).
Postnatal Changes in the Blood
Blood cell counts tend to rise above adult levels at birth and then decline gradually throughout childhood. Table 30-1 lists normal ranges during infancy and childhood. The immediate rise in values is the result of accelerated hematopoiesis during fetal life, increased numbers of cells that result from the trauma of birth, and cutting of the umbilical cord. These events surrounding the birth also are accompanied by the presence of large numbers of immature erythrocytes and leukocytes (particularly granulocytes) in peripheral blood (see Chapter 27). As the infant develops over the first 2 to 3 months of life, the numbers of these immature blood cells decrease.
TABLE 30-1
HEMATOLOGIC VALUES DURING INFANCY AND CHILDHOOD
| Age | Hemoglobin (g/dl) | Hematocrit (%) | Reticulocytes (%)Mean | MCV (fl) Lowest | Leukocytes (WBC/mm3) | Neutrophils (%) | Lymphocytes (%)Mean∗ | Eosinophils (%)Mean | Monocytes (%)Mean | ||||
| Mean | Range | Mean | Range | Mean | Range | Mean | Range | ||||||
| Cord blood | 16.8 | 13.7-20.1 | 55 | 45-65 | 5 | 110 | 18,000 | (9000-30,000) | 61 | (40-80) | 31 | 2 | 6 |
| 2 wk | 16.5 | 13-20 | 50 | 42-66 | 1 | 12,000 | (5000-21,000) | 40 | 63 | 3 | 9 | ||
| 3 mo | 12 | 9.5-14.5 | 36 | 31-41 | 1 | 12,000 | (6000-18,000) | 30 | 48 | 2 | 5 | ||
| 6 mo to 6 yr | 12 | 10.5-14 | 37 | 33-42 | 1 | 70-74 | 10,000 | (6000-15,000) | 45 | 48 | 2 | 5 | |
| 7-12 yr | 13 | 11-16 | 38 | 34-40 | 1 | 76-80 | 8,000 | (4500-13,500) | 55 | 38 | 2 | 5 | |
| Adult | |||||||||||||
| Female | 14 | 12-16 | 42 | 37-47 | 1.6 | 80 | 7,500 | (5000-10,000) | 55 | (35-70) | 35 | 3 | 7 |
| Male | 16 | 14-18 | 47 | 42-52 | 80 | ||||||||
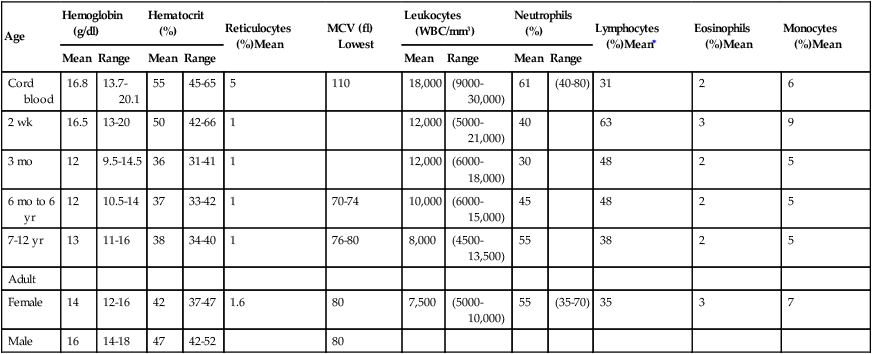
fl, Femtoliters; MCV, mean corpuscular volume; WBC, white blood cells.
From Behrman R et al, editors: Nelson textbook of pediatrics, ed 17th, Philadelphia, 2004, Saunders.
Erythrocytes
The hypoxic intrauterine environment stimulates erythropoietin production in the fetus. This accelerates fetal erythropoiesis, producing polycythemia (excessive proliferation of erythrocyte precursors) of the newborn. After birth the oxygen from the lungs saturates arterial blood, and the amount of oxygen delivered to the tissues increases. In response to the change from a placental to a pulmonary oxygen supply during the first few days of life, levels of erythropoietin and the rate of blood cell formation decrease. The very active rate of fetal erythropoiesis is reflected by the large numbers of immature erythrocytes (reticulocytes) in the peripheral blood of full-term neonates. After birth the number of reticulocytes decreases about 50% every 12 hours, so it is rare to find an elevated reticulocyte count after the first week of life. A decrease in extramedullary hematopoiesis also occurs at this time. In the peripheral blood the erythrocyte count drops for 6 to 8 weeks after birth. During this period of rapid growth the rate of erythrocyte destruction is greater than that in later childhood and adulthood. In full-term infants, normal erythrocyte life span is 60 to 80 days; in premature infants it may be as short as 20 to 30 days; and in children and adolescents, it is the same as that in adults—120 days. (Mechanisms of hemolysis are described in Chapter 27.)
Leukocytes and Platelets
The lymphocytes of children tend to have more cytoplasm and less compact nuclear chromatin than do the lymphocytes of adults. The significance of these differences is unknown. One possible explanation is that children tend to have more frequent viral infections, which are associated with atypical lymphocytes. Even minor infections, in which the child fails to exhibit clinical manifestations of illness, or administration of immunizations may result in lymphocyte changes.1
At birth the neutrophil count is very high and rises further during the early days of life.2 After 2 weeks neutrophil counts fall to within or below normal adult ranges. By approximately 4 years of age the neutrophil count is the same as that of an adult. White children have slightly higher counts than black children.3
Eosinophil count is high in the first year of life and is higher in children than in teenagers or adults.4 Monocyte counts are high in the first year of life and then decrease to adult levels. No relationship between age and basophil count has been found. Platelet counts in full-term neonates are comparable to platelet counts in adults and remain so throughout infancy and childhood.5
Disorders of Erythrocytes
The most dramatic form of acquired congenital hemolytic anemia is hemolytic disease of the newborn (HDN), also termed erythroblastosis fetalis. HDN is an alloimmune disorder in which maternal blood and fetal blood are antigenically incompatible, causing the mother’s immune system to produce antibodies against fetal erythrocytes. Fetal erythrocytes that have been bound to maternal antibodies are recognized as foreign or defective by the fetal mononuclear phagocyte system and are removed from the circulation by phagocytosis, usually in the fetal spleen. (For a complete discussion of HDN, see p. 1059.) Other acquired hemolytic anemias—some of which begin in utero—include those caused by infections or the presence of toxins.
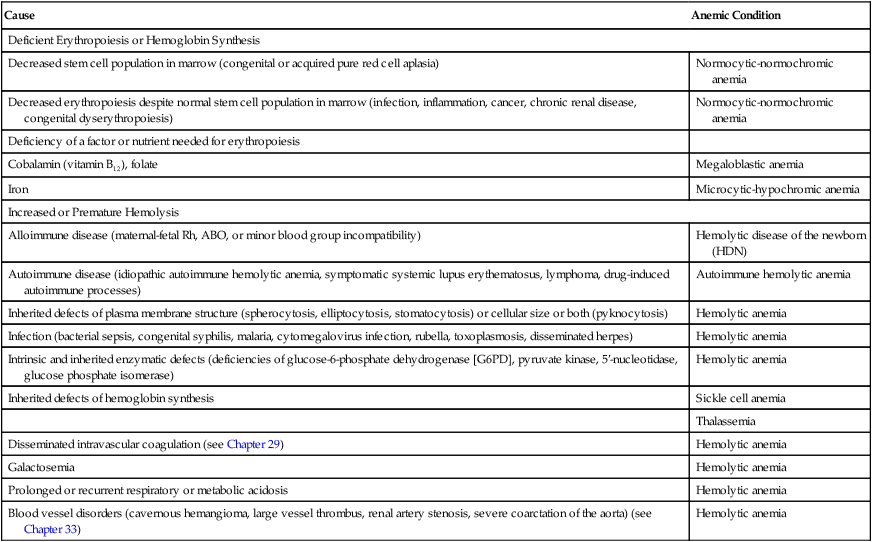
The inherited forms of hemolytic anemia result from intrinsic defects of the child’s erythrocytes, any of which can lead to erythrocyte destruction by the mononuclear phagocyte system. Structural defects include abnormal red blood cell size and abnormalities of plasma membrane structure (spherocytosis). Intracellular defects include enzyme deficiencies, the most common of which is G6PD deficiency, and defects of hemoglobin synthesis, which manifest as sickle cell disease or thalassemia, depending on which component of hemoglobin is defective. These and other causes of childhood anemia, some more common than others, are listed in Table 30-2.
TABLE 30-2
| Cause | Anemic Condition |
| Deficient Erythropoiesis or Hemoglobin Synthesis | |
| Decreased stem cell population in marrow (congenital or acquired pure red cell aplasia) | Normocytic-normochromic anemia |
| Decreased erythropoiesis despite normal stem cell population in marrow (infection, inflammation, cancer, chronic renal disease, congenital dyserythropoiesis) | Normocytic-normochromic anemia |
| Deficiency of a factor or nutrient needed for erythropoiesis | |
| Cobalamin (vitamin B12), folate | Megaloblastic anemia |
| Iron | Microcytic-hypochromic anemia |
| Increased or Premature Hemolysis | |
| Alloimmune disease (maternal-fetal Rh, ABO, or minor blood group incompatibility) | Hemolytic disease of the newborn (HDN) |
| Autoimmune disease (idiopathic autoimmune hemolytic anemia, symptomatic systemic lupus erythematosus, lymphoma, drug-induced autoimmune processes) | Autoimmune hemolytic anemia |
| Inherited defects of plasma membrane structure (spherocytosis, elliptocytosis, stomatocytosis) or cellular size or both (pyknocytosis) | Hemolytic anemia |
| Infection (bacterial sepsis, congenital syphilis, malaria, cytomegalovirus infection, rubella, toxoplasmosis, disseminated herpes) | Hemolytic anemia |
| Intrinsic and inherited enzymatic defects (deficiencies of glucose-6-phosphate dehydrogenase [G6PD], pyruvate kinase, 5′-nucleotidase, glucose phosphate isomerase) | Hemolytic anemia |
| Inherited defects of hemoglobin synthesis | Sickle cell anemia |
| Thalassemia | |
| Disseminated intravascular coagulation (see Chapter 29) | Hemolytic anemia |
| Galactosemia | Hemolytic anemia |
| Prolonged or recurrent respiratory or metabolic acidosis | Hemolytic anemia |
| Blood vessel disorders (cavernous hemangioma, large vessel thrombus, renal artery stenosis, severe coarctation of the aorta) (see Chapter 33) | Hemolytic anemia |
Acquired Disorders
Iron Deficiency Anemia
Iron deficiency anemia is the most common blood disorder of infancy and childhood, with the highest incidence occurring between 6 months and 2 years of age. Incidence is not related to gender or race, but socioeconomic factors are important because they affect nutrition, for example, the risk of iron deficiency anemia in children of single, homeless women.6 However, greater use of iron-fortified products has decreased the prevalence of anemia in low-income infants.7 Iron deficiency anemia is a common disorder in children because of their extremely high need for iron for normal growth to occur.
Between 4 years of age and the onset of puberty, dietary iron deficiency is uncommon. During adolescence, however, it is relatively common, especially in menstruating females. Rapid growth, together with the average adolescent’s dietary habits, causes iron depletion. (Mechanisms of iron depletion are described in Chapter 27.)
Evaluation and Treatment
The most definitive test for differentiating iron deficiency from other microcytic anemias is the absence of iron stores in the bone marrow. However, measurement of serum ferritin iron concentration, transferrin saturation, iron-binding capacity, and serum transferrin receptors often is an adequate diagnostic tool and often prevents proceeding to actual bone marrow evaluation. Evaluation and treatment of iron deficiency anemia in children are similar to evaluation and treatment in adults (see Chapter 27). Oral administration of simple ferrous salts usually is satisfactory, and additional vitamin C helps promote absorption.8 Administration of supplementary trace metals or other vitamins is not necessary. Iron in a liquid form should be administered through a straw because it can stain teeth. If malabsorption is the cause of the anemia (or if oral administration has not been successful), iron dextran is given intravenously. Iron therapy is continued for at least 2 months after erythrocyte indexes have returned to normal in order to replenish iron stores.9
Hemolytic Disease of the Newborn
HDN can occur only if antigens on fetal erythrocytes differ from antigens on maternal erythrocytes. The antigenic properties of erythrocytes are determined genetically: they may be type A, B, or O and may or may not include Rh antigen D. Erythrocytes that express Rh antigen D are Rh-positive; those that do not are Rh-negative. The frequency of Rh negativity is higher in whites (15%) than in blacks (5%), and is rare in Asians. Maternal-fetal incompatibility exists if mother and fetus differ in ABO blood type or if the fetus is Rh-positive and the mother is Rh-negative. (The antigenic properties of erythrocytes are described in Chapter 9.)
Pathophysiology
If the mother and fetus have antigenically incompatible erythrocytes, HDN will result if the mother’s blood contains preformed antibodies against fetal erythrocytes or produces them on exposure to fetal erythrocytes, if sufficient amounts of antibody (usually immunoglobulin G [IgG]) cross the placenta and enter fetal blood, and if IgG binds with sufficient numbers of fetal erythrocytes to cause widespread antibody-mediated hemolysis or splenic removal. (Antibody-mediated red blood cell destruction is discussed in Chapter 9.)
IgG-coated fetal erythrocytes are destroyed through extravascular hemolysis, primarily by mononuclear phagocytes in the spleen. As hemolysis progresses, the fetus becomes anemic. Erythropoiesis accelerates, particularly in the liver and spleen, and immature nucleated cells (erythroblasts) are released into the bloodstream, hence the name erythroblastosis fetalis (Figure 30-2). The degree of anemia depends on the length of time the antibody has been in the fetal circulation, antibody concentration, and the ability of the fetus to compensate for increased hemolysis. Unconjugated (indirect) bilirubin, which is formed during breakdown of hemoglobin, is transported across the placental barrier into the maternal circulation and is excreted by the mother. Hyperbilirubinemia occurs in the neonate after birth because excretion of lipid-soluble unconjugated bilirubin through the placenta no longer is possible.
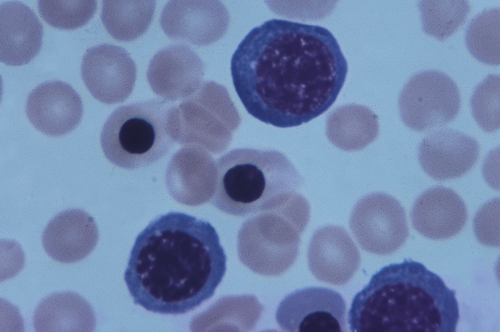
This micrograph shows immature red blood cells not normally found in blood. Large purple cells are erythroblasts; nucleated red blood cells are normoblasts. Normal red blood cells also are shown (× 500). (Copyright Ed Reschke.)
Rh incompatibility is more likely than ABO incompatibility to cause severe or even life-threatening anemia, death in utero, or damage to the central nervous system (CNS). Severe anemia alone can cause death as a result of cardiovascular complications (see Chapter 28). Extensive hemolysis also results in increased levels of unconjugated bilirubin in the circulation. If bilirubin levels exceed the liver’s ability to conjugate and excrete bilirubin, some of it is deposited in the brain, causing cellular damage and eventually death, if exchange transfusions are not administered.
Evaluation and Treatment
Routine evaluation for HDN includes the Coombs test. The indirect Coombs test measures antibody in the mother’s circulation and indicates whether the fetus is at risk for HDN. The direct Coombs test measures antibody already bound to the surfaces of fetal erythrocytes and is used primarily to confirm the diagnosis of antibody-mediated HDN. Determining prior history of fetal hemolytic disease, as well as diagnostic tests, may help predict the severity of the disorder. Diagnostic measures include maternal antibody titers, fetal blood sampling, amniotic fluid spectrophotometry, and ultrasound fetal assessment.10
The key to treatment of HDN resulting from Rh incompatibility lies in prevention (immunoprophylaxis). Rh immune globulin (RhoGAM), a preparation of antibody against Rh antigen D administered within 72 hours of exposure to Rh-positive erythrocytes, ensures that the mother will not produce antibody against the D antigen, and the next Rh-positive baby will be protected (Figure 30-3). The injected antibodies remain in the mother’s bloodstream long enough to prevent her immune system from producing its own anti-Rh antibodies but not long enough to affect subsequent offspring. The mother must be given Rh immune globulin injections after the birth of each Rh-positive baby and after a miscarriage. The mother must be especially careful not to receive a transfusion containing Rh-positive blood, because this would stimulate production of anti-Rh antibodies. In many hospitals Rh immune globulin is given prophylactically at 28 weeks to all pregnant Rh-negative women with Rh-positive partners. Various international recommendations suggest that antenatal anti-D prophylaxis should be administered to unsensitized Rh(D)-negative women as a complement to postpartum prophylaxis.11
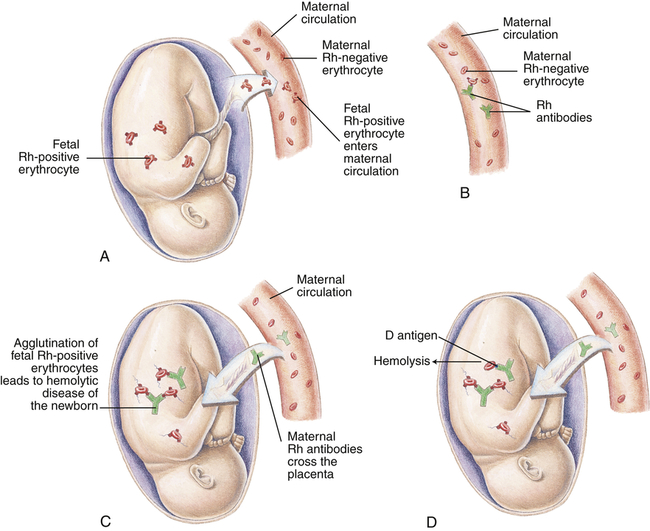
A, Before or during delivery, Rh-positive erythrocytes from the fetus enter the blood of an Rh-negative woman through a tear in the placenta. B, The mother is sensitized to the Rh antigen and produces Rh antibodies. Because this usually happens after delivery, there is no effect on the fetus in the first pregnancy. C, During a subsequent pregnancy with an Rh-positive fetus, Rh-positive erythrocytes cross the placenta, enter the maternal circulation, and (D) stimulate the mother to produce antibodies against the Rh antigen. The Rh antibodies from the mother cross the placenta, using agglutination and hemolysis of fetal erythrocytes, and HDN develops. (Modified from Seeley RR, Stephens TD, Tate P: Anatomy and physiology, ed 3, St Louis, 1995, Mosby.)
Anemia in Critically ILL Children
Anemia is a common occurrence in critically ill children (see Chapter 49). The causes are numerous and include decreased erythropoietin activity, poor iron use by the body, and blood loss from diverse conditions and consequences of treatment. A topic of discussion is anemia has been shown to worsen patient outcomes presumably because of decreased oxygen delivery. Controversial is whether transfusion of blood products, particularly packed RBCs, improve outcomes because of problems related to blood storage. New research is ongoing and needed to understand these problems, the development of new blood transfusion strategies, and blood substitutes.11a
Inherited Disorders
A number of inherited and intrinsic erythrocyte defects are known to cause increased hemolysis (see Table 30-2). These defects may be associated with enzymatic abnormalities that disrupt metabolic processes and prevent normal biochemical balance within the cell, with alterations of hemoglobin structure or synthesis, or with plasma membrane defects accompanied by changes in erythrocyte size or shape.
Glucose-6-Phosphate Dehydrogenase Deficiency
Glucose-6-phosphate dehydrogenase (G6PD) deficiency is an inherited, X-linked recessive disorder, most fully expressed in homozygous males, although partial expression and a carrier state are possible in heterozygous females. (X-linked inheritance is discussed in Chapter 4.) The deficiency is present in 10% of blacks and tends to occur in Sephardic Jews, Greeks, Iranians, Chinese, Filipinos, and Indonesians, with a frequency ranging from 5% to 40%.
Hereditary Spherocytosis
Pathophysiology
Transmitted as an autosomal dominant trait, HS represents approximately new mutations in about 25% of cases. The defect is believed to be caused by an undefined abnormality in the erythrocyte membrane. Affected cells are overly permeable to sodium and acquire a particular characteristic structure (Figure 30-4). An increased concentration of intracellular sodium is believed to lead to increased use of adenosine triphosphate (ATP) to drive the so-called cation pump. Early aging or destruction of erythrocytes is believed to result from metabolic overwork and loss of erythrocyte membrane.
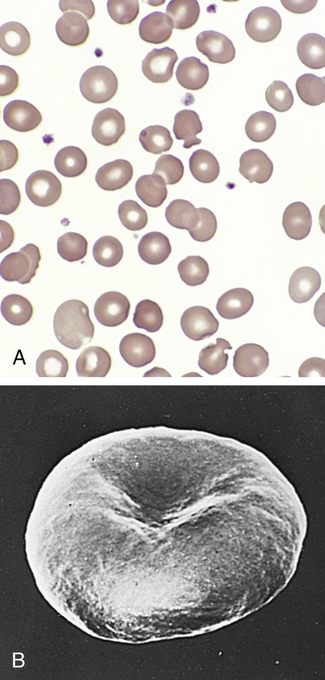
A, Blood smear from individual with hereditary spherocytosis (Wright stain). B, Scanning electron micrograph. (A from Hoffbrand AV, Pettit JE, Vyas P: Color atlas of clinical hematology, ed 4, Philadelphia, 2009, Mosby. B courtesy Dr. M. Bessis. In Miale JB, editor: Laboratory medicine: hematology, ed 6, St Louis, 1982, Mosby.)
Clinical Manifestations
The presenting signs of HS are anemia, jaundice, and splenomegaly. Anemia may be mild or absent in some cases depending on physiologic compensation. If this is the case the reticulocyte count will be elevated. Splenomegaly is usually mild. HS can present at any age, from the neonatal period until older adulthood. More severe types of HS present during the newborn period when the infant develops signs of hemolytic anemia and hyperbilirubinemia.12 These children therefore may have life-threatening anemia with clinical symptoms ranging from difficulty feeding, circumoral pallor, tachycardia, nasal flaring, diaphoresis, and lethargy. They also are at increased risk for gallstones because of the presence of extra bile pigment. Infection (specifically parvovirus),13 fever, and stress stimulate the spleen to destroy more red blood cells than usual, leading to a worsening anemia in an already baseline anemic child.
Evaluation and Treatment
It is important to ascertain family history of spherocytosis. Laboratory findings include spherocytes in the peripheral blood smear, elevated reticulocyte count (with or without anemia), indirect hyperbilirubinemia, and a positive osmotic fragility test. An osmotic fragility test is performed by placing red blood cells in a saline solution for 24 hours. Spherocytes do not tolerate saline solutions, thus causing them to burst more readily than normal red blood cells. Treatment of HS is based on disease severity. Although some children with HS will have severe anemia, blood transfusions are rarely required. Treatment before the age of 5 years consists of daily folic acid supplementation to increase production of healthy red blood cells. In the past, splenectomy was the first line of treatment. Currently, however, splenectomy is only recommended for those children more than 5 years of age with severe disease or those who develop symptomatic gallstones. Partial splenectomy, in which only a portion of the spleen is removed, is being performed on children with HS in an attempt to decrease the risk of postsplenectomy complications.14
Sickle Cell Disease
Sickle cell disease (SCD) is a group of disorders characterized by the presence of an abnormal form of hemoglobin—hemoglobin S (HbS)—within the erythrocytes. Hb S is formed by a genetic mutation in which one amino acid (valine) replaces another (glutamic acid) (Figure 30-5, A). Hb S, also known as sickle hemoglobin, reacts to deoxygenation and dehydration by solidifying and stretching the erythrocyte into an elongated sickle shape. This change causes a variety of pathologic consequences, including hemolytic anemia.
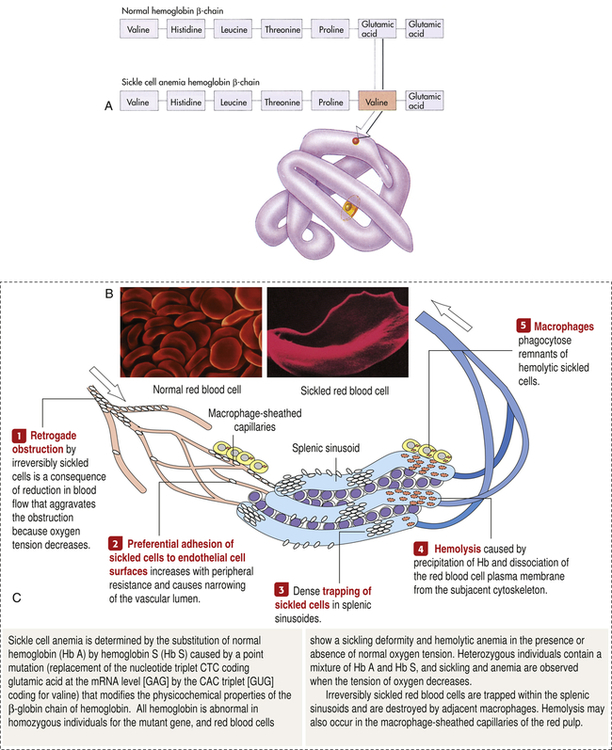
A, Sickle cell hemoglobin is produced by a recessive allele of the gene encoding the β-chain of the protein hemoglobin. It represents a single amino acid change—from glutamic acid to valine at the sixth position on the chain. In this model of a hemoglobin molecule, the position of the mutation can be seen near the end of the upper arm. B, Color-enhanced electron micrograph shows normal erythrocytes. C, Illustration of the characteristic shape of a red blood cell containing the abnormal hemoglobin. (A from Raven PH, Johnson GB: Biology, ed 3, St Louis, 1992, Mosby. B copyright Dennis Kunkel Microscopy, Inc. C from Miale JB: Laboratory medicine: hematology, ed 6, St Louis, 1982, Mosby.)
SCD is an inherited autosomal recessive disorder that is expressed as sickle cell anemia, sickle cell–thalassemia disease, or sickle cell–Hb C disease, depending on mode of inheritance (Table 30-3). (See Chapter 4 for a discussion of genetic inheritance of disease.) Sickle cell anemia, a homozygous form, is the most severe. Sickle cell–thalassemia disease and sickle cell–Hb C disease are heterozygous forms in which the child simultaneously inherits another type of abnormal hemoglobin from one parent. Sickle cell trait, in which the child inherits Hb S from one parent and normal hemoglobin (Hb A) from the other, is a heterozygous carrier state that rarely has clinical manifestations. All forms of SCD are lifelong conditions. Bone marrow or stem cell transplants can cure sickle cell anemia. However, they are currently not an option for most children because it is often difficult to find well-matched stem cell donors.
TABLE 30-3
INHERITANCE OF SICKLE CELL DISEASE∗
| Hemoglobin (Hb) Inherited from First Parent | Hemoglobin Inherited from Second Parent | Form of Sickle Cell Disease in Child |
| Hb S (an abnormal Hb) | Hb S | Sickle cell anemia: homozygous inheritance in which the child’s Hb is mostly Hb S, with the remainder fetal hemoglobin (Hb F) |
| Hb S | Defective or insufficient α- or β-chains of Hb A (alpha- or beta-thalassemia) | Sickle cell: thalassemia disease (heterozygous inheritance of Hb S and alpha- or beta-thalassemia) |
| Hb S | Hb C or D (both abnormal Hb) | Sickle cell: Hb C (or D) disease (heterozygous inheritance of Hb S and either Hb C or Hb D) |
| Hb S | Normal Hb (mostly Hb A) | Sickle cell trait, the carrier state (heterozygous inheritance of Hb S and normal Hb) |
∗See Chapter 27 for a description of normal fetal and adult hemoglobins.
SCD tends to occur in people with origins in equatorial countries, particularly central Africa, the Near East, the Mediterranean, and parts of India. In the United States, about 1 out of 500 black children and 1:36,000 Hispanic-American born have sickle cell anemia. Most infants with SCD born in the United States are now identified by routine neonatal screening. Sickle cell trait occurs among about 1:12 blacks and 1:100 Hispanic-Americans. It is estimated that 2.5 million Americans are heterozygous carriers for the sickle cell trait.15 The sickle cell trait may provide protection against lethal forms of malaria, a genetic advantage to carriers who reside in endemic regions for malaria (Mediterranean and African zones) but no advantage to carriers living in the United States.
Pathophysiology
Deoxygenation is probably the most important variable in determining the occurrence of sickling. The degree of deoxygenation required to produce sickling varies with the percentage of Hb S in the cells. Sickle trait cells will sickle at oxygen tensions of about 15 mmHg, whereas those from an individual with SCD will begin to sickle at about 40 mmHg. Hb S that is not bound with oxygen forms aggregates of semisolid gel that become stacked within the erythrocyte, stretching it into an elongated crescent (see Figure 30-5, C; Figure 30-6). Sickled erythrocytes are stiff and cannot change shape as easily as normal cells when they pass through the microcirculation. (The reversible deformability of erythrocytes is described in Chapter 27.) As a result, sickled erythrocytes tend to plug the blood vessels, causing vascular occlusion, pain, and organ infarction. Sickled cells undergo hemolysis in the spleen or become sequestered there, causing blood pooling and infarction of splenic vessels. The anemia that follows triggers erythropoiesis in the marrow and, in extreme cases, in the liver.16
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


