Chapter 42
Alterations of Digestive Function in Children

Disorders of the Gastrointestinal Tract
Congenital Impairment of Motility
Cleft Lip and Cleft Palate
Cleft lip (harelip) and cleft palate (CLP) are developmental anomalies of the first branchial arch (Figure 42-1). The incidence of CLP is estimated at 1 in 1000 live births in the United States.1 Incidence is lower in black populations and higher in Asian populations.2 Cleft lip, with or without cleft palate, is more common in males and isolated cleft palate is more common in females. Both anomalies can be unilateral or bilateral, partial or complete and may also be associated with other malformations. Nonsyndromic (isolated) CLP is a malformation with an incomplete separation between nasal and oral cavities without any associated anomaly. Syndromic CLP is associated with other malformations (e.g., Crouzon syndrome [craniofacial dysostosis], Treacher Collins syndrome [mandibulofacial dysostosis], hemifacial microsomia).3
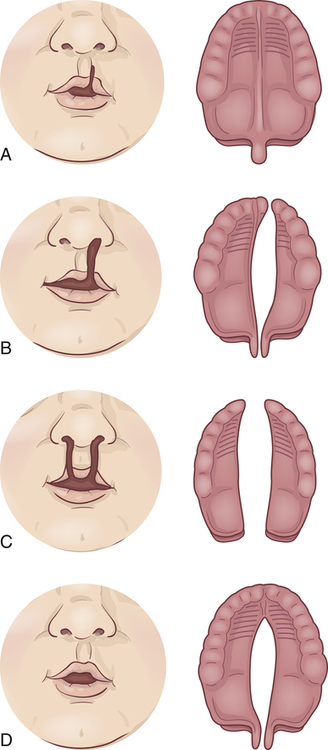
A, Notch in vermilion border. B, Unilateral cleft lip and palate. C, Bilateral cleft lip and cleft palate. D, Cleft palate.
In most cases, cleft lip and cleft palate are caused by multiple gene-environmental interactions, including maternal deficiency of B vitamins (B6, folic acid, and B12), maternal smoking and alcohol use, maternal hyperhomocysteinemia, and maternal diabetes mellitus as well as genetic variations of interferon regulatory factor-6, fibroblast growth factor, tumor growth factor-alpha, and other growth factors.4 (This phenomenon, called multifactorial inheritance, is discussed in Chapter 4.) Together these factors reduce the amount of neural crest mesenchyme that migrates into the area that will develop into the face of the embryo.5
Pathophysiology
Cleft lip (harelip) is caused by the incomplete fusion of the nasomedial and intermaxillary processes beginning during the fourth week of embryonic development,6 a period of rapid fetal growth. The cleft causes structures of the face and mouth to develop without the normal restraints of encircling lip muscles. The facial cleft may affect not only the lip but also the external nose, the nasal cartilages, the nasal septum, and the alveolar processes (bony ridge of maxilla that contains the tooth sockets).
Cleft palate is often associated with cleft lip but may occur without it. Cleft palate results from the failure of the primary palatal shelves, or processes, to fuse during the third month of gestation. The fissure may affect only the uvula and soft palate, or it may extend forward to the nostril and involve the hard palate and the maxillary alveolar ridge. It may be unilateral or bilateral, with the cleft occupying the midline posteriorly and as far forward as the alveolar process, where it deviates to the involved side. Clefts involving the palate only are usually but not necessarily in the midline. In some cases the vomer and nasal septum are partly or completely undeveloped. When these facial bones are involved, the nasal cavity may freely communicate with the oral cavity. Teeth in the cleft palate area may be missing or deformed.7
Clinical Manifestations
Feeding the infant with cleft lip usually presents no difficulty if the cleft lip is simple and the palate intact. Nursing at breast or bottle depends on suction developed by pressing the nipple against the hard palate with the tongue. Closure of the lips is not necessary, but the tongue must work harder if the lips cannot be pursed. A baby with cleft palate usually requires large, soft nipples with cross-cut openings and better tolerates feeding when in an upright position to prevent milk escaping through the nose. Although most infants with cleft palate can be successfully breast-fed, it may be impossible for some because of an unproductive suck and difficulty swallowing.8 An orthodontic prosthesis for the roof of the mouth may facilitate sucking for some infants.9
Evaluation and Treatment
Prenatal diagnosis is made by ultrasound; facial x-ray films confirm the extent of bone deformity. Soft tissue alterations are evaluated by history and physical examination.10 The nature and extent of the cleft, the infant’s condition, and the method of surgical correction proposed determine the course of treatment. Surgical correction is planned as soon as possible and may be in stages.11 The aim of surgery is to obtain an airtight closure of the palatal cleft and to preserve the mobility and length of the soft palate without compromising mid-facial growth.12 Children with cleft palate tend to have repeated infections of the paranasal sinuses and middle ear both before and after surgery, which require treatment. The child should be evaluated for hearing loss.13 Excessive dental decay is not unusual. Speech training and special attention by a prosthodontist and orthodontist are almost always required.14 Periconceptual B vitamins, folate, and folic acid intake and reduced tobacco and alcohol use may prevent orofacial clefts.5 Surgical correction facilitates feeding and normal growth and development.15
Esophageal Malformations
Congenital malformations of the esophagus are rare and occur in about 1 in 3000 live births, depending on geographic region.16 Esophageal atresia is a condition in which the esophagus ends in a blind pouch and is usually accompanied by a fistula between the esophagus and the trachea. This connection is called a tracheoesophageal fistula (TEF). Either defect can occur alone or in association with other defects (Figure 42-2). Many genes have been implicated.17
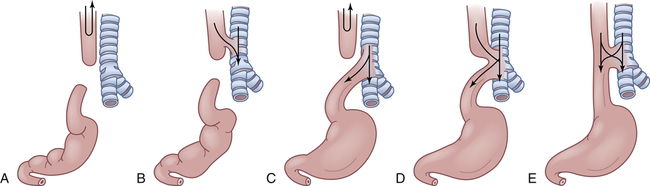
A, Simple esophageal atresia. Proximal esophagus and distal esophagus end in blind pouches, and there is no tracheal communication. Nothing enters the stomach; regurgitated food and fluid may enter the lungs. B, Proximal and distal esophageal segments end in blind pouches, and a fistula connects the proximal esophagus to the trachea. Nothing enters the stomach; food and fluid enter the lungs. C, Proximal esophagus ends in a blind pouch, and a fistula connects the trachea to the distal esophagus. Air enters the stomach; regurgitated gastric secretions enter the lungs through the fistula. D, Fistula connects proximal and distal esophageal segments to the trachea. Air, food, and fluid enter the stomach and the lungs. E, Simple tracheoesophageal fistula between otherwise normal esophagus and trachea. Air, food, and fluid enter the stomach and the lungs. Between 85% and 90% of esophageal anomalies are type C; 6% to 8% are type A; 3% to 5% are type E; and less than 1% are type B or D. Note: Type F, esophageal stenosis, is not shown.
Pathophysiology
The pathogenesis of esophageal abnormalities is unknown. They are thought to arise from defective differentiation as the trachea separates from the esophagus during the fourth to sixth weeks of embryonic development. Defective growth of endodermal cells leads to atresia. Incomplete fusion of the lateral walls of the foregut leads to incomplete closure of the laryngotracheal tube and fistula formation.18
Clinical Manifestations
Polyhydramnios (excessive amniotic fluid) is reported to occur in 14% to 90% of mothers of affected infants.19 The blind end of the proximal esophagus has a capacity of only a few milliliters. Normally swallowed amniotic fluid is absorbed into the placental circulation, and it accumulates in the uterus if the fetus cannot swallow. As the infant with esophageal atresia swallows oral secretions, the pouch fills and overflows into the pharynx, resulting in drooling and occasionally in aspiration (see Figure 42-2, A and C).
If a fistula connects the trachea with the distal esophagus, the abdomen fills with air and becomes distended. The distention may be great enough to interfere with breathing (see Figure 42-2, C to E). If the fistula connects the proximal esophagus to the trachea, the first feeding after birth will be problematic (see Figure 42-2, B, D, and E). As the infant drinks, the blind end of the esophagus and the mouth fill with fluid. When the infant tries to take a breath, the fluid is aspirated into the lungs, which triggers protective cough and choke reflexes. Intermittent cyanosis may result. Plain water or glucose is recommended for the initial feeding to minimize the dangers associated with aspiration. If an abnormality of the esophagus is indicated, oral feedings are withheld until a diagnosis is confirmed.
Pulmonary complications are compounded by reflux of air and gastric secretions into the tracheobronchial tree through the fistula (see Figure 42-2, D and E), causing severe chemical irritation. The upper lobe of the right lung is most commonly involved because of its proximity to the tracheoesophageal (TE) fistula. Infants with esophageal atresia but no fistula have a scaphoid (boat-shaped), gasless abdomen. In fistula without atresia (see Figure 42-2, E), the usual symptoms are recurrent aspiration, pneumonia, and atelectasis that remains “silent” for days or even months. Late complications of esophageal atresia or tracheal esophageal fistula include stricture, reflux, dysphagia, chronic cough, and dyspnea on exertion.20
Other congenital anomalies are present in at least 50% of infants with esophageal defects. They are known as the VACTERAL association with the letters representing vertebral anomalies, anal atresia, cardiovascular malformations, TE fistula and/or esophageal atresia, renal anomalies, and limb anomalies.21
Evaluation and Treatment
Prenatal diagnosis may include ultrasound imaging but the findings of polyhydramnios and small stomach bubble are often nonspecific and associated with other problems.22 Esophageal atresia is usually diagnosed at birth, when attempts to pass a small-bore orogastric or nasogastric tube into the stomach fail. Imaging will show displacement of the tube.23
Initial postnatal treatment is to prevent aspiration. Surgery restores esophageal continuity, and the fistula is eliminated. Surgery is usually undertaken after birth, sometimes in stages. The child may continue to have problems with aspiration, gastroesophageal reflux, and esophagitis after surgical repair. The overall survival rate for infants with esophageal defects exceeds 90%.24
Pyloric Stenosis
Pyloric stenosis is an obstruction of the pyloric sphincter caused by hypertrophy of the sphincter muscle. It is one of the most common disorders of early infancy and affects infants between the ages of either 1 and 2 weeks or 3 and 4 months.25 The incidence of pyloric stenosis among males is approximately 5 in 1000, whereas among females it is only 1 in 1000.26 Whites are affected more often than blacks or Asians, and full-term infants are affected more often than premature infants. The cause is unknown but increased gastrin secretion by the mother in the last trimester of pregnancy raises the likelihood of pyloric stenosis in the infant. The overproduction of gastric secretions in the infant may be caused by stress-related factors in the mother. Exogenous administration of prostaglandin E is associated with an increased incidence of pyloric stenosis. There is an increased incidence of pyloric stenosis in those children who have a family member with pyloric stenosis, suggesting a genetic predisposition.27
Pathophysiology
The circular muscle of the pylorus is grossly enlarged because of an increase in cell size (hypertrophy) and an increase in cell number (hyperplasia).27 The mucosal lining of the pyloric opening is folded and the lumen is narrowed by the encroaching muscle. Because of the extra peristaltic effort necessary to force the gastric contents through the narrow pylorus, the muscle layers of the stomach may become hypertrophied as well.
Evaluation and Treatment
The standard treatment for hypertrophic pyloric stenosis is a pyloromyotomy, in which the muscles of the pylorus are split and separated. The procedure can be completed with an open technique or with laparoscopy. The mortality associated with surgical correction is less than 0.5%. Fluid and electrolyte losses are managed before surgical intervention and children usually can tolerate feeding several hours after surgery.28
Intestinal Malrotation
During the tenth week of embryonic development, the emerging ileum and cecum normally rotate, so that the cecum moves into the lower right quadrant of the abdomen and is fixed there by the mesentery. Intestinal malrotation is a condition in which rotation does not occur and the colon remains in the upper right quadrant, where an abnormal membrane or band may press on and obstruct the duodenum. The obstructing band over the duodenum, called a periduodenal band (Ladd’s band), is one of the most significant findings in malrotation (Figure 42-3). Genetic factors associated with malrotation are being identified and most occur with other malformations or abnormalities.29 Associated abnormalities include 50% of children with duodenal atresia and 33% of those with jejunal atresia, as well as biliary or pancreatic malformations and heart defects.30
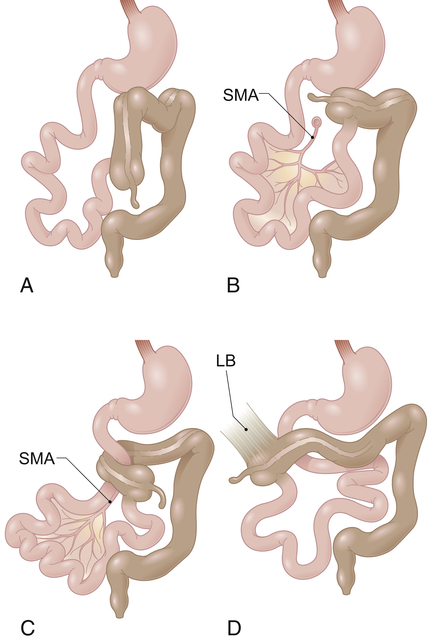
A, Nonrotation. B, Incomplete rotation. C, Midgut volvulus with duodenal obstruction and obstruction of superior mesenteric artery blood flow. D, Incomplete rotation with Ladd’s bands extending from the lateral and posterior abdominal wall to the subhepatic cecum. LB, Ladd’s band; SMA, superior mesenteric artery. (From Gilbert-Barness E et al: Potter’s pathology of the fetus, infant and child, ed 2, Philadelphia, 2007, Mosby.)
Pathophysiology
The small intestine lacks a normal posterior fixation in malrotation because it has only a rudimentary attachment near the origin of the superior mesenteric artery. Therefore, the entire mass can twist when the mobile loops of intestine from the duodenojejunal junction to the middle of the transverse colon twist on themselves. The twisting is termed volvulus. Intestinal twisting around the rudimentary mesentery angulates and obstructs the intestinal lumen and partly or completely occludes the superior mesenteric artery, causing infarction and necrosis of the entire midgut.
Clinical Manifestations
Although most cases of malrotation-associated volvulus and infarction develop during the neonatal period or infancy (younger than 1 year), some develop during childhood or even adulthood. In one study, 48% of individuals first diagnosed with malrotation were adults.31 In infants the obstruction causes intermittent or persistent bile-stained vomiting after feedings. Abdominal distention is limited initially to the epigastrium because only the stomach and duodenum are dilated. The degree of distention depends on the pressure of swallowed air and the degree of obstruction caused by the volvulus. Dehydration and electrolyte imbalance may occur rapidly because large amounts of pancreatic juice, bile, and gastric secretions are lost through vomiting. Fever usually ensues. Pain, scanty stools, diarrhea, bloody stools, and severe dehydration are associated with progressive volvulus, vascular compression, and infarction of the intestine in infants.32 Intermittent or partial volvulus may be seen in older children and adults. This may be asymptomatic (25% to 50% of the time) and discovered during unrelated abdominal surgery, or it may cause minor abdominal complaints, such as nausea after meals, recurrent episodes of vomiting, or abdominal pain.33
Evaluation and Treatment
Diagnosis of malrotation with volvulus and infarction is based on a review of the clinical manifestations, upper gastrointestinal contrast imaging, and explorative laparoscopy.34
Treatment includes laparoscopic or open surgery to reduce the volvulus.35 Necrotic bowel is resected and a primary anastomosis performed. An enterostomy may be created. Most children have a good outcome; however, there is risk for adhesion-related bowel obstruction in about 15% of cases.36
Meconium Ileus
Meconium ileus (MI) is intestinal obstruction caused by meconium formed in utero that is abnormally sticky and adheres firmly to the mucosa of the small intestine, resisting passage beyond the terminal ileum. The cause is usually a lack of digestive enzymes during fetal life. This meconium is also found to contain albumin, which is not normally found in meconium. MI is associated with cystic fibrosis.37 In the cases not associated with cystic fibrosis, genes other than CFTR mutations have been identified.38 The cause usually is unknown. Partial aplasia of the pancreas is an associated factor, however, and one fifth of infants with meconium ileus are premature or have a history of maternal hydramnios (excessive amniotic fluid). After intestinal atresia and malrotation with volvulus, meconium ileus is the most common cause of small intestinal obstruction in newborns.
Evaluation and Treatment
All women of reproductive age should be offered preconception and prenatal cystic fibrosis (CF) carrier screening. Prenatal diagnosis of MI can be made by ultrasound. Radiologic imaging is used to confirm the presence of MI.39 The sweat test is performed to detect or rule out cystic fibrosis. It is considered the “gold standard” and is accurate in 90% of infants. The treatment of choice for cases not complicated by volvulus or perforation is a hyperosmolar enema (e.g., metglutamine diatrizoate [Gastrografin]) performed under fluoroscopy. The fluid is drawn into the meconium mass, hydrating and softening it. Transient osmotic diarrhea follows. A warm saline enema containing 4% N-acetylcysteine may be given to help complete the evacuation. Surgical management includes tube enterostomy (a percutaneous drain), open enterostomy, or resection to remove the meconium mass. Survival of infants with meconium ileus is improving, with survival rates approaching 100%.40 The mortality increases if obstruction is complicated by peritonitis.41
Distal Intestinal Obstruction Syndrome
Distal intestinal obstruction syndrome (DIOS), formerly called meconium ileus equivalent, affects approximately 15% of children and adults with cystic fibrosis.42 DIOS is the partial or complete obstruction of the colon or the terminal ileum by abnormally viscous intestinal contents, particularly after episodes of dehydration or lack of pancreatic enzymes. It can occur in children or adults. The child displays signs and symptoms of intestinal obstruction. In most cases the obstruction is relieved by hypertonic enemas.43
Meckel Diverticulum
Meckel diverticulum is an outpouching of all layers of the small intestinal wall (usually in the ileum) and is the most common congenital malformation of the gastrointestinal tract. The rules of 2 are cited in association with Meckel diverticulum: it occurs in about 2% of the population, 2% develop complications usually before 2 years of age, there are 2 types of common ectopic tissue (gastric and pancreatic), and it is located within 2 feet of the ileocecal valve. Meckel diverticulum develops when there is failure to obliterate the omphalomesenteric duct, which normally leaves a fibrous band that connects the small intestine to the umbilicus during the first months of fetal development. Ectopic gastric mucosal cells are contained in the diverticuli and may cause peptic ulcer. Although most Meckel diverticuli are asymptomatic, the most common symptom is painless rectal bleeding. Intestinal obstruction, intussusception, and volvulus occur more commonly in adults. Diagnosis is made by symptom presentation and radionucleotide scintingraphy. The scan shows the gastric mucosal cells in the diverticuli. Treatment in those with symptoms is surgical resection.44
Congenital Aganglionic Megacolon
Congenital aganglionic megacolon (Hirschsprung disease) is a functional obstruction of the colon caused by the absence of the enteric ganglia along a variable length of the colon with inadequate motility. The incidence is approximately 1 in 5000 live births with an increased incidence in males, siblings of children with Hirschsprung disease, and children with Down syndrome or other congenital malformations.45 The exact cause is unknown but multiple interacting factors and a complex inheritance pattern with mutations in multiple genes are involved.46
Pathophysiology
Congenital aganglionic megacolon is caused by a malformation of the parasympathetic nervous system. It is characterized by abnormalities of the basement membrane and extracellular matrix and absence of the intramural ganglion cells in the enteric nerve plexuses (Meissner and Auerbach plexuses) along variable lengths of the colon, and there may be skipped segments. Lacking neural stimulation, the muscle layers of the colon wall fail to propel feces through the colon, leading to functional obstruction. In 80% of cases the aganglionic segment is limited to the rectosigmoid region (short-segment); in 3% the entire colon lacks ganglion cells and the ileum may be involved. The abnormally innervated colon impairs fecal movements, causing the proximal colon to become distended, hence the term megacolon (Figure 42-4).
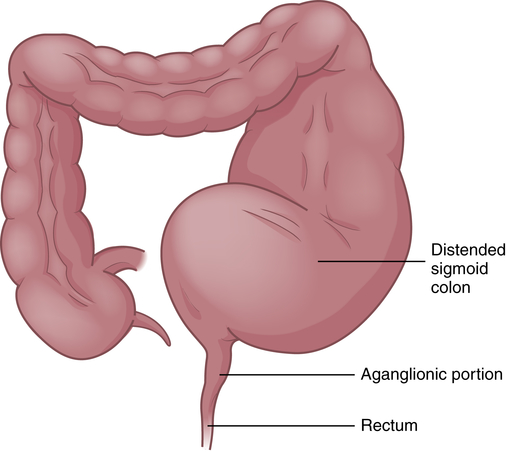
The ganglia normally develop from an advancing neural crest between the muscle layers (tunica muscularis) in the submucosal area (muscularis mucosae) of the intestinal wall. In cases of congenital megacolon, neurologic development is blocked and large, nonmyelinated fibers develop in place of these ganglion cells. The segment of colon that lacks ganglion cells has a relatively normal lumen caliber and wall thickness. In the segment of the colon proximal to it, the lumen is dilated and the muscle hypertrophied. Therefore, the abnormal portion of the colon appears to be normal and the normal portion appears to be diseased.47
Clinical Manifestations
In the neonate there is delayed passage of meconium and abdominal distention, with or without bilious vomiting. Bowel dilation stretches and partly occludes the encircling blood and lymphatic vessels, causing edema, ischemia, infarction of the mucosa, and significant outflow of fluid into the bowel lumen. Copious, liquid stools result. In severe cases infarction and destruction of the mucosa enable enteric microorganisms to penetrate the bowel wall. Frequently, gram-negative sepsis occurs, accompanied by fever and vomiting. Severe and rapid electrolyte changes may take place, causing collapse and rapid death.48
Evaluation and Treatment
Hirschsprung disease in usually diagnosed in the newborn period and should be suspected with failure to pass meconium within 24 to 48 hours after birth. Radiocontrast enema, anorectal manometry, and rectal suction biopsy are screening tools for the diagnosis of Hirschsprung disease. Serial manometry measurements may be required in neonates.49 This test has uncovered ultrashort-segment Hirschsprung disease in older children with a history of constipation.50 The definitive diagnosis is made by rectal suction biopsy showing an absence of ganglion cells in the submucosa of the colon.51
The involved segment is usually resected within the first few months of life using a “pull-through” procedure that anastomoses the proximal and distal segments of remaining bowel or rectum. Laparoscopic or open approaches may be used. In skipped segment disease, the area of normal bowel may be preserved.52 For children with short-segment Hirschsprung disease, enemas are given to relieve impaction, and laxatives with a dietary and bowel training program are used in preference to surgical intervention.50 The child is not treated for diarrhea.
In general, the prognosis of congenital megacolon is satisfactory for children who undergo surgical treatment. Bowel training may be prolonged; however, most children achieve bowel continence before puberty whereas others have long-term constipation or fecal incontinence.53
Anorectal Malformations
Congenital anorectal malformations (ARMs) are rare birth defects of unknown etiology that range from mild anal stenosis, which is corrected by simple dilation, to complex deformities, such as anal or rectal agenesis, atresia, and rectourethral fistula (Figure 42-5). Deformities that cause complete obstruction are known collectively as imperforate anus. The cause is unknown.54 ARM occurs in approximately 1 in 2500 to 1 in 5000 newborn babies.55
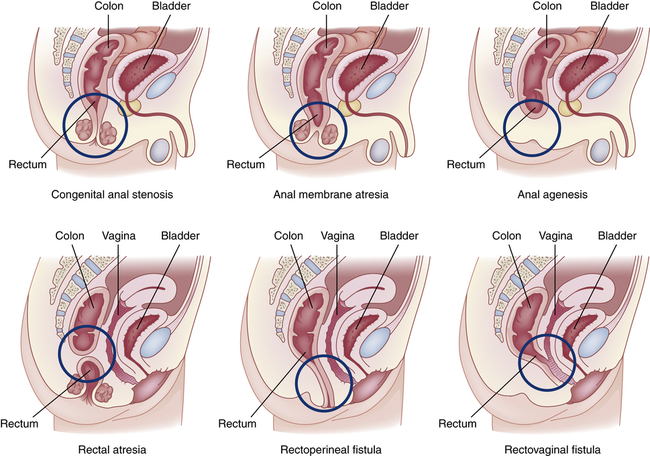
Approximately 40% of infants with ARMs have other developmental anomalies, including Down syndrome, congenital heart disease, renal and urologic abnormalities, cryptorchidism, esophageal atresia, and malformations of the spine.56 Imperforate anus may not be obvious. It can be detected by gentle insertion of a rectal tube; x-ray films show dilations throughout the intestinal tract. Anal stenosis can be treated by dilations, but all other anorectal malformations require surgical correction. The overall death rate is approximately 10%. More than 90% of children with a low (anal) anomaly and intact sacrum achieve bowel continence with stenosis as a common complication. Children with very high anomalies or anomalies associated with genitourinary fistulae have more difficulty achieving continence.57,58
Acquired Impairment of Motility
Intussusception
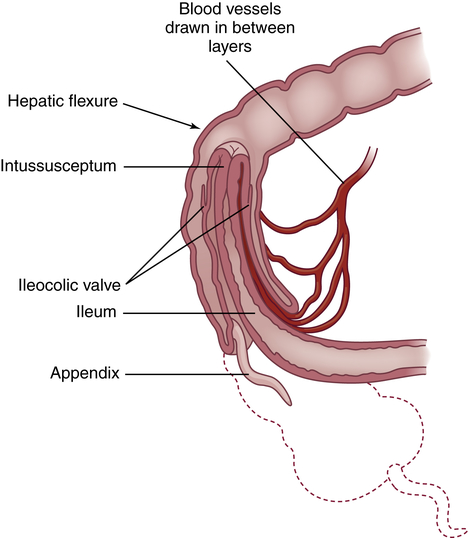
Intussusception is the telescoping or invagination of one portion of the intestine into another. It is the most common cause of acquired intestinal obstruction in infants between 5 and 10 months of age, although it can occur at any age. The ileum usually invaginates the cecum and part of the ascending colon by collapsing through the ileocecal valve (ileocolic intussusception). It can occur anywhere from the duodenum to the rectum. Intussusception is more common in males and can be idiopathic, can be associated with lead points (polyps or tumors, Meckel diverticulum, intestinal adhesions) or cystic fibrosis, or can develop immediately after abdominal surgery.59
Pathophysiology
Most commonly, the proximal portion of the intestine, the intussusceptum, collapses into the distal portion, the intussuscipiens, in the direction of peristaltic flow (Figure 42-6). As this occurs, the intussusceptum drags its mesentery into the enveloping lumen. Initially, the mesentery is constricted, obstructing venous return. Compression of the mesenteric vessels between the two layers of intestinal wall and at the U-shaped angle at either end of the intussusceptum leads within hours to venous stasis, engorgement, edema, exudation, and further vascular compression. Unless the intussusception is treated, bleeding, necrosis, bowel perforation, and gangrene ensue. The tension of the mesentery on the intussusceptum tends to arch the bowel in a curve with its center at the mesenteric root. Edema and compression obstruct the flow of chyme through the intestine.
Clinical Manifestations
The affected infant suddenly develops abdominal pain, becomes irritable (colicky), and flexes the knees. Vomiting occurs soon afterward. A single normal stool may be passed, evacuating the colon distal to the apex of the intussusception. Following the passage of normal stool, 60% of infants pass “currant jelly” stools, which appear dark and gelatinous because of their blood and mucus content. In one study, less than one third of children had this clinical triad of vomiting, colicky abdominal pain, and bloody stools.60 Most infants have a tender, sausage-shaped abdominal mass. Abdominal tenderness and distention develop as intestinal obstruction becomes more acute.
Evaluation and Treatment
Diagnosis is based on clinical manifestations and onset of symptoms. Ultrasound of the abdomen, computed tomography (CT), and magnetic resonance imaging (MRI) are commonly completed for diagnosis.61 More than 82% of children have positive ultrasound results in ileocolic and jejunointestinal intussusception. Reduction is an emergency procedure involving hydrostatic pressure generated by an air or a barium enema given using fluoroscopic guidance. This technique is successful most of the time with about a 10% to 13% recurrence rate. Surgical reduction is done on children who fail or, in rare cases, have perforation.62,63 Untreated intussusception in infants is nearly always fatal. Most infants recover if the intussusception is reduced within 24 hours. Spontaneous reduction of intussusception may occur in symptomatic or asymptomatic children.64
Gastroesophageal Reflux Disease
Pathophysiology
GERD in children may be related to delayed maturation of the lower esophageal sphincter or impaired hormonal or neurotransmitter response mechanisms (i.e., vasoactive intestinal peptide and nitric oxide). Factors that maintain lower esophageal sphincter integrity in children include location of the gastroesophageal junction in a high-pressure zone within the abdomen, mucosal gathering within the sphincter, and the angle at which the esophagus is inserted into the stomach. Reflux persists if any of these pressure-maintaining factors is altered. Irritation of the mucosa by acidic gastric contents results in inflammation of the esophageal epithelium (esophagitis) and stimulation of the vomiting reflex.65
GERD may be a factor in the stimulation of reactive airway disease and otitis media with effusion in some children but the exact relationships are yet to be defined.66,67 The relationship between apnea of prematurity caused by GERD and laryngospasm is controversial.68
Eosinophilic esophagitis is differentiated from GERD (see Chapter 41) and can occur in children. It is thought to be an atopic disease involving immediate as well as delayed hypersensitivity reactions to food ingestion. A mast cell, eosinophil, and T lymphocyte infiltrate are associated with inflammation of the entire esophagus that is nonresponsive to acid-suppression therapy. Eosinophilic inflammation may lead to progressive subepithelial fibrosis with esophageal strictures, narrowing, and dysphagia. The esophageal mucosa can appear normal in children at endoscopy. Dysphagia, food impaction, and vomiting are common symptoms and other atopic diseases, such as asthma and eczema, may be present. Treatment includes elimination diets and oral corticosteroids.69,70
Clinical Manifestations
Vomiting may be forceful and must be differentiated from pyloric stenosis. Aspiration pneumonia develops in one third of infants with gastroesophageal reflux (GER). In cases that persist into childhood, chronic cough, wheezing, hoarseness, and recurrent pneumonia are common.71,72 Repeated vomiting leads to inadequate retention of nutrients, adversely affecting growth and weight gain. Esophagitis from exposure of the esophageal mucosa to acidic gastric contents is manifested by pain, bleeding, and eventually stricture formation and abnormal motility. Approximately 10% to 25% of children with GERD also have iron deficiency anemia caused by frank or occult blood loss.73
Evaluation and Treatment
The clinical manifestations are often adequate to confirm a diagnosis of GERD. However, irritability, crying, feeding refusal, and regurgitation can be common problems in infants and not specific to GERD. Esophageal pH monitoring with a probe for 24 hours and endoscopy with biopsy are routinely used for diagnosis.74–76 Mild GER resolves without treatment. Thickened feedings may help some infants77; however, this has not been shown to be consistently helpful. Techniques for managing infant reflux include small, frequent feedings; prolonged feeding duration and slower flow rate; and frequent burping.78,79
Medications are used to treat erosive esophagitis and prokinetic agents must be used with caution.80 If no improvement is seen with medical management or the child has life-threatening events with reflux, an antireflux surgical procedure, including gastropexy and fundoplication, is performed. A fundoplication recreates a valve by wrapping the fundus of the stomach around the lower esophagus and can be completed using laparoscopic techniques.65, 81
Impairment of Digestion, Absorption, and Nutrition
Cystic Fibrosis
Cystic fibrosis of the pancreas, which is also called mucoviscidosis or fibrocystic disease of the pancreas, is a genetically transmitted disease (mutation of the long arm of chromosome 7) that involves many organs and systems and usually causes death in childhood or young adulthood. It is the most common cause of chronic suppurative lung disease in children and is the most common life-threatening inherited disease in the white population. This section focuses on the deficiency of pancreatic enzymes. (Chapter 36 discusses the pulmonary consequences of cystic fibrosis.)
Pathophysiology
The pathophysiologic triad that is the hallmark of CF includes: (1) pancreatic enzyme deficiency, which causes maldigestion; (2) overproduction of mucus in the respiratory tract and inability to clear secretions, which cause progressive chronic obstructive pulmonary disease; and (3) abnormally elevated sodium and chloride concentrations in sweat. Exocrine secretions tend to be abnormally thick and precipitate in the glandular ducts, obstructing flow. Almost all clinical manifestations of CF are a result of overproduction of extremely viscous mucus and pancreatic enzyme deficiency. The full spectrum of involvement is summarized in Table 42-1.
TABLE 42-1
PATHOPHYSIOLOGY, CLINICAL MANIFESTATIONS, AND COMPLICATIONS OF CYSTIC FIBROSIS
| ORGAN INVOLVED | SECRETORY DYSFUNCTION | CLINICAL MANIFESTATIONS | COMPLICATIONS |
| Sweat glands | Elevated concentrations of sodium and chloride in sweat | Hyponatremia; hypochloremia | Heat prostration; shock |
| Intestine | |||
| Newborn | Viscid meconium | Meconium ileus with intestinal obstruction | Meconium peritonitis |
| Older child and adult | Inspissated (dried out) mucofecal masses (intestinal sludging) | Partial intestinal obstruction with severe cramping pains | Gastroesophageal reflux Volvulus (obstruction), intussusception (prolapse) Distal intestinal obstruction syndrome |
| Pancreas (enzyme deficiency) | Inspissation and precipitation of pancreatic secretions, causing obstruction of pancreatic ducts | Absence of pancreatic enzymes, causing malabsorption of food; fatty, bulky stools | Hypoproteinemia; iron deficiency anemia; malnutrition |
| Decreased vitamins A, D, E, and K absorption Growth failure | Vitamins A, D, E, and K deficiency and rectal prolapse Decreased bone density and risk of fractures in adolescents and adults | ||
| Insulin deficiency | Glucose intolerance | Diabetes mellitus | |
| Liver | Inspissation and precipitation of bile in biliary system | Focal biliary cirrhosis; shrunken, “hobnail” liver | Portal hypertension with esophageal varices, hematemesis and hypersplenism Steatorrhea from lack of bile salts |
| Salivary glands | Inspissation and precipitation of secretions in small ducts of submaxillary and sublingual salivary glands | Mild patchy fibrosis of salivary glands | None |
| Paranasal structures | Viscid mucus | Retention of mucus; clouding seen on sinus roentgenograms | Mucopyoceles (pus accumulations) with nasal deformity or orbital cavity extension |
| Nose | Nasal polyps | Obstruction of nasal airflow | None |
| Lungs | Viscid mucus in bronchioles and bronchi | Obstruction of bronchioles causing bronchiolectasis, bronchiectasis, and chronic lung infection | Hemoptysis; pneumothorax; cor pulmonale; atelectasis; chronic bacterial infection; respiratory failure |
| Reproductive tract | |||
| Male | Viscid genital tract secretions during embryologic development, causing failure of formation of normal vas deferens | Sterility | None |
| Female | Distention of endocervical epithelial cells with cytoplasmic mucin | Decreased fertility | Polypoid cervicitis (cervical inflammation) while taking oral contraceptives |
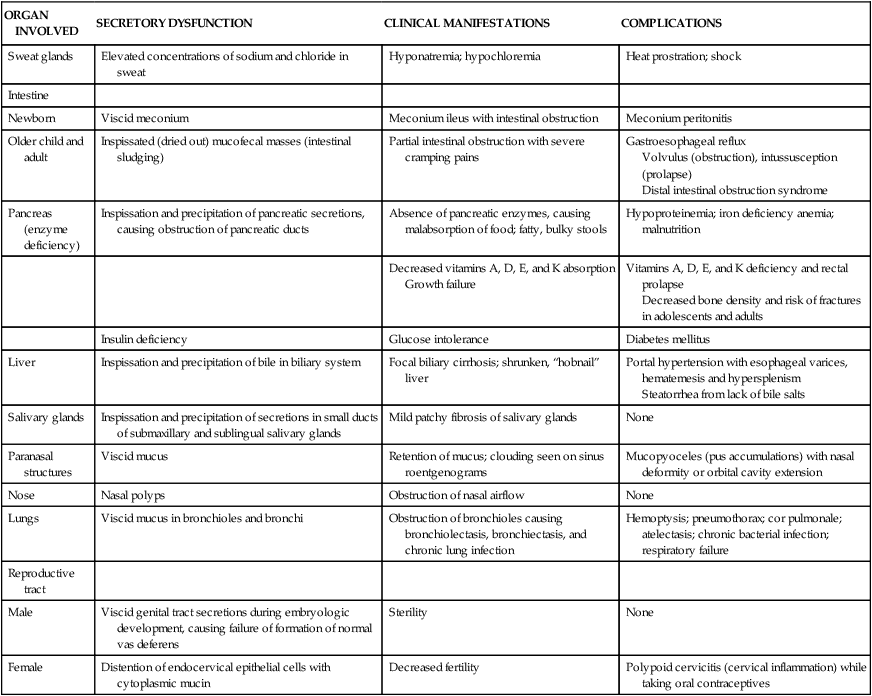
Data from Gelfond D, Borowitz D: Clin Gastroenterol Hepatol 11(4):333–342, 2013; Javier RM, Jacquot J: Joint Bone Spine (5):445–450, 2011; Rudolph CD et al: Rudolph’s pediatrics, ed 21, New York, 2003, McGraw-Hill.
Pancreatic function may range from normal to completely ablated. Approximately 85% of patients have pancreatic insufficiency. Obstruction of the pancreatic ducts with thick mucus blocks the flow of pancreatic enzymes and causes degenerative and fibrotic changes in the pancreas. Pancreatic damage eventually can affect the beta cells, resulting in diabetes mellitus (10% to 25%). The incidence of diabetes mellitus and cirrhosis (13% to 17%) in this population has increased as larger numbers of people with cystic fibrosis have moved into young and middle adulthood. Severe problems with maldigestion of proteins, carbohydrates, and fats occur because of insufficient secretion of pancreatic enzymes. Failure to thrive, growth failure, malabsorptive symptoms, metabolic abnormalities, trace element deficiencies, fat-soluble vitamin alterations, electrolyte imbalances, and decreased bone mineral density can occur early in the disease.82–84
Evaluation and Treatment
To determine the extent of pancreatic function 72-hour stool fat measurements are used. Stools also may be examined for absence of pancreatic enzymes, particularly fecal elastase, trypsin, and chymotrypsin. To optimize treatment, the carbon-13 (13C) mixed triglyceride breath test offers a simple, noninvasive way of assessing the need for pancreatic enzyme supplementation in children with cystic fibrosis.85 Pancreatic replacement enzymes are administered before or with meals and high-calorie, high-protein diets with frequent snacks and vitamin supplements are used to treat malnutrition.86 However, anorexia is not uncommon in this group secondary to pulmonary disease and frequently large sputum output. To combat the worsening problem of growth failure in children with cystic fibrosis, nasogastric or gastrostomy tube feedings are used to supplement oral intake and promote weight gain. Monitoring of growth and body mass index is critical to treatment evaluation.87,88
Gluten-Sensitive Enteropathy
Gluten-sensitive enteropathy, formerly called celiac sprue or celiac disease, is an autoimmune disease of the small intestinal villous epithelium when there is ingestion of the cereal protein gluten (gliadin) found in wheat, rye, barley, and oats in genetically susceptible individuals. The disease has a prevalence of about 1% worldwide and about 0.71% in the United States; many cases are undiagnosed.89 Although gluten-sensitive enteropathy is widely perceived as a malabsorption syndrome of childhood, the diagnosis is usually made in adulthood. It is not known why disease presentation varies so widely.90 Numerous genes have been identified and other autoimmune diseases can be associated with the autoantibodies of gluten-sensitive enteropathy, including type 1 diabetes mellitus, autoimmune thyroiditis, and Addison disease.91
Pathophysiology
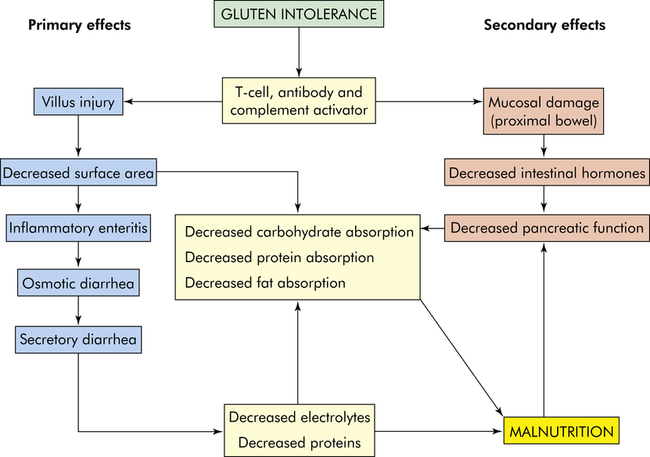
The disease is complex and involves the interaction of genetic, immune, and environmental factors. Both cellular immunity and humoral immunity are implicated.92 The major pathophysiologic characteristic is T-cell–mediated autoimmune injury to the intestinal epithelial cells.91 Transglutaminase 2 (TG2) and endomysial autoantibodies closely correlate with the acute phase of the disease in the presence of gluten.93 T-cell infiltration results in mucosal cell destruction with inflammation, atrophy, and flattening of villi in the upper small intestine (Figures 42-7 and 42-8). The atrophy is caused by accelerated shedding of epithelial cells from the villi. To compensate for this loss, epithelial cell production increases, causing hypertrophy of the crypts of Lieberkühn. Increased cell production is not sufficient to keep pace with cell loss, and the cells are not mature enough to sustain absorptive functions. The microvilli and brush border disappear, leaving patches of bald mucosa. The loss of mucosal surface area and brush-border enzymes leads to severe malabsorption. The pathologic process is most pronounced in the duodenum and jejunum. The ileum may be spared. The severity of disease correlates with the length of the small intestinal mucosa involved.94,95
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


