Chapter 32
Alterations of Cardiovascular Function

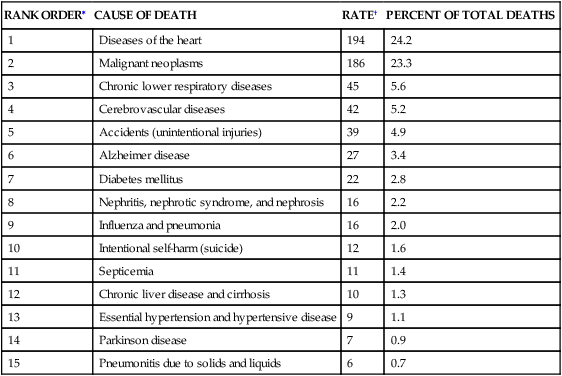
Cardiovascular disease is the leading cause of death worldwide (Table 32-1).1 The pathophysiology of heart disease is much more complicated than just structural and hemodynamic changes. Today the focus is on the genetic, neurohumoral, and inflammatory mechanisms that underlie tissue and cellular processes, such as endothelial injury, remodeling, stunning, reperfusion injury, and autoimmune disease.
TABLE 32-1
DEATH RATES AND PERCENT OF TOTAL DEATHS FOR THE 15 LEADING CAUSES OF DEATH IN THE UNITED STATES ALL RACES, BOTH SEXES, ALL AGES (FINAL DATA REPORT 2010)
| RANK ORDER∗ | CAUSE OF DEATH | RATE† | PERCENT OF TOTAL DEATHS |
| 1 | Diseases of the heart | 194 | 24.2 |
| 2 | Malignant neoplasms | 186 | 23.3 |
| 3 | Chronic lower respiratory diseases | 45 | 5.6 |
| 4 | Cerebrovascular diseases | 42 | 5.2 |
| 5 | Accidents (unintentional injuries) | 39 | 4.9 |
| 6 | Alzheimer disease | 27 | 3.4 |
| 7 | Diabetes mellitus | 22 | 2.8 |
| 8 | Nephritis, nephrotic syndrome, and nephrosis | 16 | 2.2 |
| 9 | Influenza and pneumonia | 16 | 2.0 |
| 10 | Intentional self-harm (suicide) | 12 | 1.6 |
| 11 | Septicemia | 11 | 1.4 |
| 12 | Chronic liver disease and cirrhosis | 10 | 1.3 |
| 13 | Essential hypertension and hypertensive disease | 9 | 1.1 |
| 14 | Parkinson disease | 7 | 0.9 |
| 15 | Pneumonitis due to solids and liquids | 6 | 0.7 |
∗Rank based on number of deaths.
†Rates per 100,000 population.
Data from Murphy SL, Xu J, Kochanek KD: Deaths: final data for 2010. National vital statistics reports; vol 61, no 4. Hyattsville, MD: National Center for Health Statistics, 2013. Available at www.cdc.gov/nchs/data/dvs/lcwk1_210.pdf. Accessed July 2, 2013.
Diseases of the Veins
Varicose Veins and Chronic Venous Insufficiency
Varicose veins are a common condition affecting more than 25 million Americans, women nearly twice as often as men.2 A varicose vein is a superficial vein in which blood has pooled. Varicose veins typically involve the saphenous veins of the legs and are distended, tortuous, and palpable (Figure 32-1). Varicose veins are caused by (1) trauma to the saphenous veins that damages one or more valves, or (2) gradual venous distention caused by the action of gravity on blood in the legs.
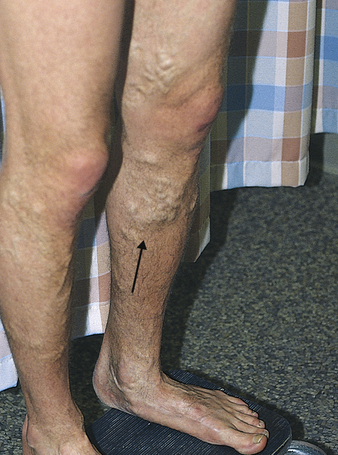
Veins are thin-walled, highly distensible vessels. Normally the muscular pump in the legs moves venous blood up toward the heart, and valves prevent backflow and pooling of blood (see Figures 31-35 and 31-31). In individuals who habitually stand for long periods, wear constricting garments, or cross the legs at the knees, distention progresses until the pressure in the vein damages venous valves, rendering the valves incompetent. Damaged valves cannot maintain normal venous pressure, which causes hydrostatic pressure in the vein to increase. As the vein distends further, it becomes tortuous, and edema develops in the extremity.
Evidence has been emerging that varicose veins are not just the result of mechanical pressures. Altered connective tissue proteins, increased proteolytic enzyme activity, and decreased transforming growth factor-beta (TGF-β) in vein walls probably precede the development of valvular damage and the development of varicosities.2 Inflammatory changes that affect autonomic innervation of veins has also been implicated.3
Circulation to the extremities can become so sluggish that the metabolic demands of the cells for oxygen, nutrients, and waste removal are barely met. Any trauma or pressure can therefore lower the oxygen supply and cause cell death and necrosis (venous stasis ulcers). Infection can occur because poor circulation impairs the delivery of the cells and biochemicals for the immune and inflammatory responses. This same sluggish circulation makes infection following reparative surgery a significant risk. Treatment of varicose veins and CVI begins conservatively, and wound healing often occurs following noninvasive treatments, such as leg elevation, compression stockings, and physical exercise. New management techniques include endovenous ablation (radiofrequency and laser) and ultrasound-guided foam sclerotherapy. These techniques are as effective and safer than surgical ligation and vein stripping.4
Deep Venous Thrombosis
A thrombus is a blood clot that remains attached to a vessel wall (Figure 32-2). A detached thrombus is a thromboembolus. Venous thrombi are more common than arterial thrombi because flow and pressure are lower in the veins than in the arteries. Deep venous thrombosis (DVT) refers to clot formation in the large veins, primarily of the lower extremities and may result in venous thromboembolism (VTE) to the pulmonary circulation. The American Heart Association (AHA) estimates that more than 900,000 incident or recurrent VTE events occur annually in the United States, of which approximately one third are fatal.5 Three factors (triad of Virchow) promote venous thrombosis: (1) venous stasis (e.g., immobility, obesity, prolonged leg dependency [e.g., air travel], age, heart failure [HF]), (2) venous endothelial damage (e.g., trauma, medications), and (3) hypercoagulable states (e.g., inherited disorders, malignancy, pregnancy, oral contraceptives, hormone replacement, hyperhomocysteinemia, antiphospholipid syndrome).6 Virtually everyone who is hospitalized is at significant risk for DVT, especially those with orthopedic trauma or surgery, spinal cord injury, and obstetric/gynecologic conditions. Numerous genetic abnormalities are associated with an increased risk for venous thrombosis primarily related to states of hypercoagulability. These inherited abnormalities include factor V Leiden mutation; prothrombin mutations; and deficiencies of protein C, protein S, and antithrombin. Inherited hypercoagulability states should be suspected in individuals who develop thrombi in the absence of the usual risk factors.6,7
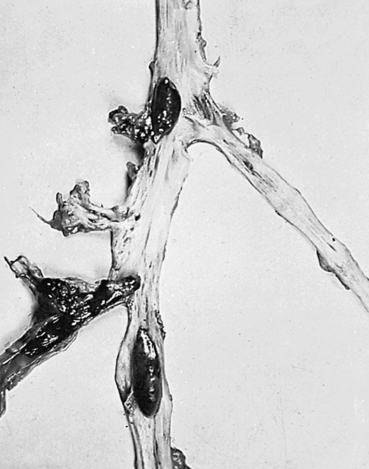
Most thrombi eventually dissolve without treatment, but untreated DVT is associated with a high risk of thromboembolization of a part of the clot from the leg to the lung (pulmonary embolism) (see Chapter 35).6 In up to one third of individuals with DVT, persistent venous outflow obstruction may lead to post-thrombotic syndrome (PTS), a frequent complication of DVT characterized by chronic, persistent pain; edema; and ulceration of the affected limb.8
Because DVT is usually asymptomatic and difficult to detect clinically, prevention for at-risk individuals is crucial. If possible, individuals should be mobilized as soon as possible after illness, injury, or surgery. Prophylactic treatment can include low-molecular-weight heparin, antithrombin agents, warfarin, or pneumatic devices.9,10 Unfortunately, even these measures may not prevent embolism in high-risk individuals such as those who have undergone hip replacement surgery.11 In individuals at high risk for pulmonary embolism, but for whom anticoagulation is contraindicated, placement of an inferior vena caval filter may be necessary to prevent pulmonary embolism.
Diagnosis is most often made by combining measurement of serum d-dimer concentration with lower extremity ultrasonography. d-dimer is an indirect measure of the presence of thrombosis that is very sensitive but is not specific. If the d-dimer is negative, DVT is ruled out. If it is positive, the diagnosis must be confirmed with ultrasonography.6,12 In selected individuals, computed tomography (CT) or magnetic resonance imaging (MRI) may be needed to make the diagnosis. If noninvasive testing is nondiagnostic, a venogram may be indicated.
DVT is treated with low-molecular-weight heparin, unfractionated intravenous heparin, antithrombin agents, or adjusted-dose subcutaneous heparin.6 Thrombolytic therapy may be used to dissolve the clot more quickly and reduce the risk of postphlebotic syndrome, especially when a large clot is located in a proximal vein.13 However, bleeding risk is increased, and many people have contraindications to the use of thrombolytics. Pharmacomechanical treatment involves catheter-mediated removal of clots that can be used in selected individuals and so far has a good safety record.14 DVT has a recurrence rate of 26% 5 years after discontinuation of anticoagulant therapy, especially if it was unprovoked and no identifiable underlying condition can be reversed.15 In these individuals aspirin therapy can reduce recurrence rates after discontinuation of anticoaglulants.16
Superior Vena Cava Syndrome
Superior vena cava syndrome (SVCS) is a progressive occlusion of the superior vena cava (SVC) that leads to venous distention in the upper extremities and head. The leading cause of SVCS is bronchogenic cancer (approximately 70% of cases), followed by lymphomas and metastasis of other cancers.17 Benign causes of SVCS include thrombosis, histoplasmosis, tuberculosis, mediastinal fibrosis, cystic fibrosis, and benign tumors, such as retrosternal goiter.18 Invasive therapies, including pacemaker wires, central venous catheters, and pulmonary artery catheters, can lead to acute and chronic SVCS.
Diagnosis is made by chest x-ray, Doppler studies, CT, MRI, and ultrasound. With slow onset and the development of collateral venous drainage, SVCS is generally not a vascular emergency but rather an oncologic emergency.17 Treatment for malignant disorders can include radiation therapy, surgery, chemotherapy, and the administration of diuretics, steroids, and anticoagulants, as necessary. Treatment for nonmalignant causes may include bypass surgery using various grafts, thrombolysis (both locally and systemically), balloon angioplasty, and placement of intravascular stents.18
Diseases of the Arteries
Hypertension
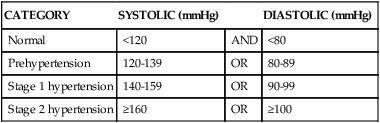
Hypertension is consistent elevation of systemic arterial blood pressure. Hypertension is the most common primary diagnosis in the United States—approximately 1 in 3 adults greater than 20 years of age has hypertension; this increases to nearly two thirds in those older than age 60. The prevalence of hypertension is nearly equal between men and women. Black adults have among the highest rates of hypertension in the world (44%). Approximately 80% of hypertensive adults are aware of their condition, 71% are using antihypertensive medication, but only 48% of those have their hypertension controlled.5 Hypertension is defined in the Seventh Report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure (JNC7)19 as a sustained systolic blood pressure of 140 mmHg or greater or a diastolic pressure of 90 mmHg or greater (Table 32-2). Normal blood pressure is associated with the lowest cardiovascular risk, whereas those who fall into the prehypertension category (which includes between 25% and 37% of the U.S. population) are at risk for developing hypertension and many associated cardiovascular complications unless lifestyle modification and treatment are instituted.20
TABLE 32-2
CLASSIFICATION OF BLOOD PRESSURE FOR ADULTS AGE 18 YEARS OR OLDER
| CATEGORY | SYSTOLIC (mmHg) | DIASTOLIC (mmHg) | |
| Normal | <120 | AND | <80 |
| Prehypertension | 120-139 | OR | 80-89 |
| Stage 1 hypertension | 140-159 | OR | 90-99 |
| Stage 2 hypertension | ≥160 | OR | ≥100 |
Data from the JNC 7 Report, JAMA 289(19):2560–2572, 2003.
Some individuals with hypertensive disease have isolated systolic hypertension. Isolated systolic hypertension (ISH) is elevated systolic blood pressure accompanied by normal diastolic blood pressure (less than 90 mmHg). ISH is becoming more prevalent in all age groups and is strongly associated with cardiovascular and cerebrovascular events.21
Approximately 95% of cases of hypertension have no known cause and therefore are diagnosed as primary hypertension.5 Secondary hypertension accounts for 5% of cases and is caused by altered hemodynamics associated with an underlying primary disease. Hypertension is a complex disorder that affects the entire cardiovascular system, and all types and stages of hypertension are associated with increased risk for target organ disease events, such as myocardial infarction (MI), kidney disease, and stroke.5
Factors Associated with Primary Hypertension
A combination of genetic and environmental factors is thought to be responsible for the development of primary hypertension. Genetic predisposition to hypertension is thought to be polygenic. The inherited defects are associated with renal sodium excretion, insulin and insulin sensitivity, activity of the sympathetic nervous system (SNS) and renin-angiotensin-aldosterone system (RAAS), and cell membrane sodium or calcium transport.22 In blacks, variants of the apolipoprotein L1 (APOL1) gene are associated with hypertension and renal disease.23
Risk factors associated with primary hypertension include: (1) family history of hypertension; (2) advancing age; (3) gender (men younger than 55 and women older than 70 years); (4) black race; (5) high dietary sodium intake; (6) glucose intolerance (diabetes mellitus); (7) cigarette smoking; (8) obesity; (9) heavy alcohol consumption; and (10) low dietary intake of potassium, calcium, and magnesium.19 Many of these factors are also risk factors for other cardiovascular disorders. In fact, hypertension, dyslipidemia, and glucose intolerance often are found together in a condition called metabolic syndrome (see Chapter 22).
Pathophysiology
Hypertension is caused by increases in cardiac output or total peripheral resistance, or both. (The many factors affecting cardiac output and peripheral resistance are described in Chapter 31.) Cardiac output is increased by any condition that increases heart rate or stroke volume, whereas peripheral resistance is increased by any factor that increases blood viscosity or reduces vessel diameter (vasoconstriction).
Primary Hypertension
Primary hypertension is the result of a complicated interaction between genetics and the environment that increase vascular tone (increased peripheral resistance) and blood volume, thus causing sustained increases in blood pressure. Multiple pathophysiologic mechanisms mediate these effects including the sympathetic nervous system (SNS), the RAAS, and natriuretic peptides. Inflammation, endothelial dysfunction, obesity-related hormones, and insulin resistance also contribute to both increased peripheral resistance and increased blood volume. Increased vascular volume is related to a decrease in renal excretion of salt, often referred to as a shift in the pressure-natriuresis relationship. This means that for a given blood pressure, individuals with hypertension tend to secrete less salt in their urine. The pathophysiology of primary hypertension is summarized in Figure 32-3.
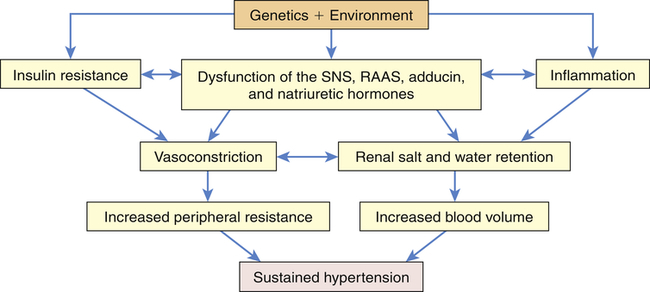
Numerous genetic vulnerabilities have been linked to hypertension and these, in combination with environmental risks, cause neurohumoral dysfunction (sympathetic nervous system [SNS], renin-angiotensin-aldosterone system [RAAS], adducin, and natriuretic hormones) and promote inflammation and insulin resistance. Insulin resistance and neurohumoral dysfunction contribute to sustained systemic vasoconstriction and increased peripheral resistance. Inflammation contributes to renal dysfunction, which, in combination with the neurohumoral alterations, results in renal salt and water retention and increased blood volume. Increased peripheral resistance and increased blood volume are two primary causes of sustained hypertension.
The SNS contributes to the pathogenesis of hypertension in many people. In the healthy individual the SNS contributes to the maintenance of adequate blood pressure and tissue perfusion by promoting cardiac contractility and heart rate (maintenance of adequate cardiac output) and by inducing arteriolar vasoconstriction (maintenance of adequate peripheral resistance). In individuals with hypertension, overactivity of the SNS can result from increased production of catecholamines (epinephrine and norepinephrine) or from increased receptor reactivity involving these neurotransmitters.24 Increased SNS activity causes increased heart rate and systemic vasoconstriction, thus raising the blood pressure. Efferent sympathetic outflow stimulates renin release, increases tubular sodium reabsorption, and reduces renal blood flow. Additional mechanisms of SNS-induced hypertension include structural changes in blood vessels (vascular remodeling), insulin resistance, increased renin and angiotensin levels, and procoagulant effects. The SNS is implicated in the cardiovascular and renal complications of hypertension, and new techniques such as renal denervation are being explored to treat hypertension.25,26 The role of the SNS in the pathogenesis of cardiovascular disease is summarized in Figure 32-4.
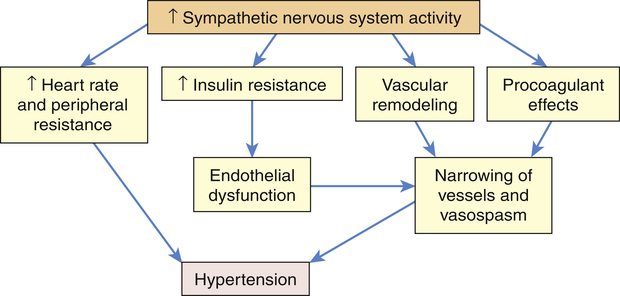
Increased activity of the sympathetic nervous system (SNS) not only increases heart rate and peripheral resistance but also causes vascular remodeling with narrowing and vasospasm of arteries. The SNS contributes to insulin resistance, which is associated with endothelial dysfunction and decreased production of vasodilators, such as nitric oxide. The SNS also has procoagulant properties, making vascular spasm and thrombosis more likely. All of these factors contribute to sustained increases in blood pressure.
In the healthy individual the RAAS provides an important homeostatic mechanism for maintaining adequate blood pressure and therefore tissue perfusion (see Chapter 31). In hypertensive individuals, overactivity of the RAAS contributes to salt and water retention and increased vascular resistance. High levels of angiotensin II contribute to endothelial dysfunction, insulin resistance, dyslipidemia, and platelet aggregation and play an important role in the complications associated with the metabolic syndrome.27,28 Further, angiotensin II mediates arteriolar remodeling, which is structural change in the vessel wall that results in permanent increases in peripheral resistance29 (see Figures 31-28 and 32-5). Angiotensin II is associated with end-organ effects of hypertension, including atherosclerosis, renal disease, and cardiac hypertrophy.30 Finally, aldosterone not only contributes to sodium retention by the kidney but also has other deleterious effects on the cardiovascular system.31 Medications, such as angiotensin-converting enzyme (ACE) inhibitors and angiotensin-receptor blockers (ARBs), oppose the activity of the RAAS and are effective in reducing blood pressure and protecting against target organ damage.32 A second RAAS has also been described. This system uses ACE 2 to create Ang 1-7, which has cardiovascular protective effects. Its discovery may lead to new and more effective medications33,34 (see What’s New? The Renin-Angiotensin-Aldosterone System (RAAS) and Cardiovascular Disease).
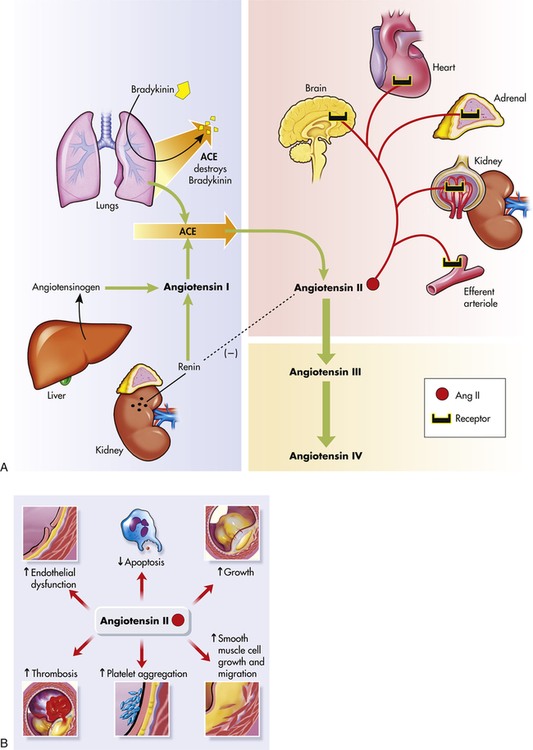
A, The shaded blue area is the classical pathway of biosynthesis that generates the renin and angiotensin I. Angiotensinogen is synthesized in the liver and is released into the blood where it is cleaved to form angiotensin I by renin secreted by cells in the kidneys. Angiotensin-converting enzyme (ACE) in the lung catalyzes the formation of angiotensin II from angiotensin I, and destroys the potent vasodilator, bradykinin. Further cleavage generates the angiotensins III and IV. The reddish shading shows the organs affected by angiotensin II including the brain, heart, adrenals, kidney, and the kidney’s efferent arterioles. The dashed line (on the left) shows the inhibition of renin by angiotensin II. B, Summary of angiotensin II effects on blood vessel structure and function leading to arteriosclerosis. (Redrawn from Goodfriend TL et al: N Engl J Med 334:2649–2654, 1996.)
The natriuretic hormones modulate renal sodium (Na+) excretion and require adequate potassium, calcium, and magnesium to function properly. The natriuretic hormones include atrial natriuretic peptide (ANP), brain natriuretic peptide (BNP), C-type natriuretic peptide (CNP), and urodilatin. These hormones induce diuresis; enhancement of renal blood flow and glomerular filtration rate, systemic vasodilatation, and suppression of aldosterone; and inhibition of the SNS.35 Dysfunction of these hormones, along with alterations in the RAA system and the SNS, cause an increase in vascular tone and a shift in the pressure-natriuresis relationship. When there is inadequate natriuretic function, serum levels of the natriuretic peptides are increased. In hypertension, increased ANP and BNP levels are linked to an increased risk for ventricular hypertrophy, atherosclerosis, and heart failure.36 Salt retention leads to water retention and increased blood volume, which contributes to an increase in blood pressure. Subtle renal injury results, with renal vasoconstriction and tissue ischemia. Tissue ischemia causes inflammation of the kidney and contributes to dysfunction of the glomeruli and tubules and promotes additional sodium retention.37 In addition to increasing dietary intake of potassium, calcium, and magnesium in order to enhance natriuretic peptide function, a drug that mimics the effect of one of these hormones (nesiritide) is used to treat heart failure.38 Newer and safer agonists are being studied.35
As mentioned, renal inflammation contributes to sodium retention. Inflammation also plays a role in the vascular dysfunction of hypertension. Endothelial injury and tissue ischemia result in the release of vasoactive inflammatory cytokines. Although many of these cytokines (e.g., histamine, prostaglandins) have vasodilatory actions in acute inflammatory injury, chronic inflammation contributes to vascular remodeling and smooth muscle contraction.39 Endothelial injury and dysfunction in primary hypertension is further characterized by decreased production of vasodilators, such as nitric oxide, and increased production of vasoconstrictors, such as endothelin.40 Endothelin blockade has been shown to reduce blood pressure and prevent proteinuria.41
Obesity is recognized as an important risk factor for hypertension in both adults and children and contributes to many of the neurohumoral, metabolic, renal, and cardiovascular processes that cause hypertension. Obesity causes changes in what are called the adipokines (leptin and adiponectin) and is associated with increased activity of the SNS and the RAAS.42,43 Obesity is linked to inflammation, small artery remodeling, endothelial dysfunction, and insulin resistance and an increased
risk for cardiovascular complications from hypertension (see What’s New? Obesity and Hypertension).
Finally, insulin resistance is common in hypertension, even in individuals without clinical diabetes. Insulin resistance is associated with decreased endothelial release of nitric oxide and other vasodilators.44 It also affects renal function and causes renal salt and water retention. Insulin resistance is associated with overactivity of the SNS and the RAAS. It is interesting to note that in many individuals with diabetes treated with drugs that increase insulin sensitivity, blood pressure often declines, even in the absence of antihypertensive drugs. The interactions between obesity, hypertension, insulin resistance, and lipid disorders in the metabolic syndrome result in a high risk of cardiovascular disease.28
It is likely that primary hypertension is an interaction between many of these factors leading to sustained increases in blood volume and peripheral resistance. The role of these mechanisms in increasing blood volume in the pathophysiology of primary hypertension is summarized in Figure 32-6.
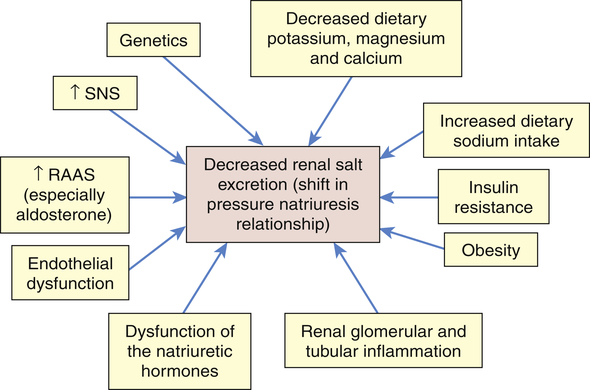
Numerous factors have been implicated in the pathogenesis of sodium retention in individuals with hypertension. These factors cause less renal excretion of salt than would normally occur with increased blood pressure. This is called a shift in the pressure-natriuresis relationship and is believed to be a central process in the pathogenesis of primary hypertension. RAAS, Renin-angiotensin-aldosterone system; SNS, sympathetic nervous system.
Secondary Hypertension
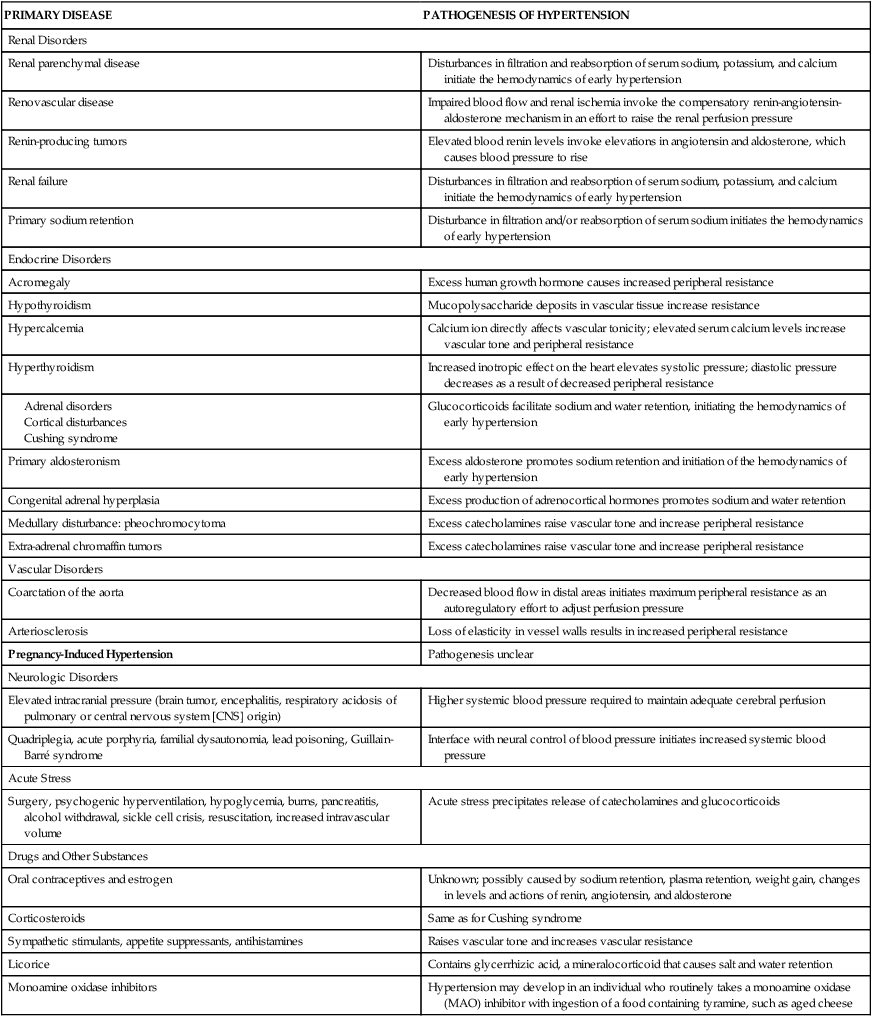
Secondary hypertension is caused by an underlying disease process that raises peripheral vascular resistance or cardiac output. If the cause is identified and removed before permanent structural changes occur, blood pressure returns to normal. In addition, medications are an important and often unrecognized cause of secondary hypertension.45 Table 32-3 summarizes the pathogenesis of major forms of secondary hypertension.
TABLE 32-3
PATHOGENESIS OF MAJOR FORMS OF SECONDARY HYPERTENSION BY CAUSE
| PRIMARY DISEASE | PATHOGENESIS OF HYPERTENSION |
| Renal Disorders | |
| Renal parenchymal disease | Disturbances in filtration and reabsorption of serum sodium, potassium, and calcium initiate the hemodynamics of early hypertension |
| Renovascular disease | Impaired blood flow and renal ischemia invoke the compensatory renin-angiotensin-aldosterone mechanism in an effort to raise the renal perfusion pressure |
| Renin-producing tumors | Elevated blood renin levels invoke elevations in angiotensin and aldosterone, which causes blood pressure to rise |
| Renal failure | Disturbances in filtration and reabsorption of serum sodium, potassium, and calcium initiate the hemodynamics of early hypertension |
| Primary sodium retention | Disturbance in filtration and/or reabsorption of serum sodium initiates the hemodynamics of early hypertension |
| Endocrine Disorders | |
| Acromegaly | Excess human growth hormone causes increased peripheral resistance |
| Hypothyroidism | Mucopolysaccharide deposits in vascular tissue increase resistance |
| Hypercalcemia | Calcium ion directly affects vascular tonicity; elevated serum calcium levels increase vascular tone and peripheral resistance |
| Hyperthyroidism | Increased inotropic effect on the heart elevates systolic pressure; diastolic pressure decreases as a result of decreased peripheral resistance |
| Glucocorticoids facilitate sodium and water retention, initiating the hemodynamics of early hypertension | |
| Primary aldosteronism | Excess aldosterone promotes sodium retention and initiation of the hemodynamics of early hypertension |
| Congenital adrenal hyperplasia | Excess production of adrenocortical hormones promotes sodium and water retention |
| Medullary disturbance: pheochromocytoma | Excess catecholamines raise vascular tone and increase peripheral resistance |
| Extra-adrenal chromaffin tumors | Excess catecholamines raise vascular tone and increase peripheral resistance |
| Vascular Disorders | |
| Coarctation of the aorta | Decreased blood flow in distal areas initiates maximum peripheral resistance as an autoregulatory effort to adjust perfusion pressure |
| Arteriosclerosis | Loss of elasticity in vessel walls results in increased peripheral resistance |
| Pregnancy-Induced Hypertension | Pathogenesis unclear |
| Neurologic Disorders | |
| Elevated intracranial pressure (brain tumor, encephalitis, respiratory acidosis of pulmonary or central nervous system [CNS] origin) | Higher systemic blood pressure required to maintain adequate cerebral perfusion |
| Quadriplegia, acute porphyria, familial dysautonomia, lead poisoning, Guillain-Barré syndrome | Interface with neural control of blood pressure initiates increased systemic blood pressure |
| Acute Stress | |
| Surgery, psychogenic hyperventilation, hypoglycemia, burns, pancreatitis, alcohol withdrawal, sickle cell crisis, resuscitation, increased intravascular volume | Acute stress precipitates release of catecholamines and glucocorticoids |
| Drugs and Other Substances | |
| Oral contraceptives and estrogen | Unknown; possibly caused by sodium retention, plasma retention, weight gain, changes in levels and actions of renin, angiotensin, and aldosterone |
| Corticosteroids | Same as for Cushing syndrome |
| Sympathetic stimulants, appetite suppressants, antihistamines | Raises vascular tone and increases vascular resistance |
| Licorice | Contains glycerrhizic acid, a mineralocorticoid that causes salt and water retention |
| Monoamine oxidase inhibitors | Hypertension may develop in an individual who routinely takes a monoamine oxidase (MAO) inhibitor with ingestion of a food containing tyramine, such as aged cheese |
From Cheng J et al: PLoS Pathog May 5(5), 2009. (Epub ahead of print.); Kaplan NM: Clinical hypertension, ed 8, Baltimore, 2002, Lippincott Williams & Wilkins.
Complicated Hypertension
Chronic hypertension damages the walls of systemic blood vessels. Within the walls of arteries and arterioles, smooth muscle cells undergo hypertrophy and hyperplasia with associated fibrosis of the tunica intima and media in a process called vascular remodeling (Figure 32-7). Endothelial dysfunction, angiotensin II, catecholamines, insulin resistance, and inflammation contribute to this process. Once significant fibrosis has occurred, reduced blood flow and dysfunction of the organs perfused by these affected vessels is inevitable. Target organs for hypertension include the kidney, brain, heart, extremities, and eyes (these effects are summarized in Table 32-4).
TABLE 32-4
PATHOLOGIC EFFECTS OF SUSTAINED, COMPLICATED PRIMARY HYPERTENSION
| SITE OF INJURY | MECHANISM OF INJURY | POTENTIAL PATHOLOGIC EFFECT |
| Heart | ||
| Myocardium | Increased workload combined with diminished blood flow through coronary arteries | Left ventricular hypertrophy, myocardial ischemia, left heart failure |
| Coronary arteries | Accelerated atherosclerosis (coronary artery disease) | Myocardial ischemia, myocardial infarction, sudden death |
| Aorta | Weakened vessel wall | Aneurysms, acute aortic syndromes |
| Kidneys | Renin and aldosterone secretion stimulated by reduced blood flow | Retention of sodium and water, leading to increased blood volume and perpetuation of hypertension |
| Inflammation and ischemia | Tissue damage that compromises filtration | |
| High pressures in renal arterioles | Nephrosclerosis leading to renal failure | |
| Brain | Reduced blood flow and oxygen supply; weakened vessel walls, accelerated atherosclerosis | Transient ischemic attacks, cerebral thrombosis, aneurysm, hemorrhage, acute brain infarction |
| Eyes (retinas) | Reduced blood flow | Retinal vascular sclerosis |
| High arteriolar pressure | Exudation, hemorrhage | |
| Arterial vessels of lower extremities | Reduced blood flow and high pressures in arterioles, accelerated atherosclerosis | Intermittent claudication, arterial thrombosis, gangrene |
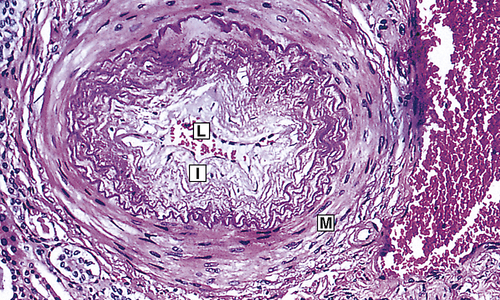
Fibrous intimal proliferation (I) with reduction in lumen vessel caliber (radius) (L) and normal media (M). (From Stevens A, Lowe JS, Scott I: Core pathology, ed 3, London, 2009, Mosby.)
Cardiovascular complications include left ventricular hypertrophy, angina pectoris, congestive heart failure (left heart failure), CAD, MI, and sudden death. Myocardial hypertrophy in response to hypertension is mediated by several neurohormonal substances, including the SNS and angiotensin II. Hypertrophy is characterized by changes in the myocyte proteins, apoptosis of myocytes, and deposition of collagen in heart muscle, which causes it to become thickened, scarred, and less able to relax during diastole leading to diastolic heart failure.46 In addition, the increased size of the heart muscle increases demand for oxygen delivery over time, contractility of the heart is impaired, and the individual is at increased risk for systolic heart failure. Vascular complications include the formation, dissection, and rupture of aneurysms (outpouchings in vessel walls); intermittent claudication; and gangrene resulting from vessel occlusion. Renal complications are parenchymal damage, nephrosclerosis, renal arteriosclerosis, and renal insufficiency or failure.47
Microalbuminuria (small amounts of protein in the urine) is an early sign of impending renal dysfunction and significantly increased risk for cardiovascular events.48
Changes in the vascular beds can be estimated by viewing the arterioles of the retina. Complications specific to the retina include retinal vascular sclerosis, exudation, and hemorrhage.49 Cerebrovascular complications are similar to those of other arterial beds and include transient ischemia, stroke, cerebral thrombosis, aneurysm, and hemorrhage. Chronic hypertension also has been linked to cognitive decline with aging.50–52
Evaluation and treatment
A single elevated blood pressure reading does not mean that a person has hypertension. Diagnosis requires the measurement of blood pressure on at least two separate occasions averaging two readings at least 2 minutes apart, with the individual seated, the arm supported at heart level, after 5 minutes rest, with no smoking or caffeine intake in the past 30 minutes.19 Some individuals benefit from 24-hour ambulatory blood pressure monitoring because of better correlation with end-organ damage and the ability to screen out “white coat hypertension” (elevated blood pressure that occurs only in a clinic setting) and “masked hypertension” (normal blood pressure in the clinic setting but elevated elsewhere).53,54 Ambulatory measurement also detects those who fail to have a nocturnal decrease in blood pressure and who may be at higher cardiovascular risk. It is especially recommended for individuals with drug resistance, hypotensive symptoms with medications, episodic hypertension, and autonomic dysfunction.54
Treatment of primary hypertension depends on its severity. Figure 32-8 illustrates an overview of the JNC7 recommendations.19 Treatment begins with reducing or eliminating risk factors. Lifestyle modification can prevent hypertension from developing in those individuals who fall into the prehypertension category, may control the blood pressure in stage I hypertension, and can enhance the effects of drug treatment for those with more significant blood pressure elevation. The usual dietary recommendations are to restrict sodium intake to 2.4 g/day, to increase potassium intake, to restrict saturated fat intake, and to adjust calorie intake as required to maintain optimum weight. The Dietary Approaches to Stop Hypertension (DASH) diet is recommended.55,56 Although it has been widely recommended since 1998, the DASH diet continues to be studied with beneficial effects extending to reduced coronary risks, increased insulin sensitivity, and improved lipid profiles, especially in those with diabetes.57 An exercise program that promotes endurance and relaxation usually is recommended. Physical training increases stroke volume, which has the effect of lowering heart rate and hence systolic blood pressure, and should consist of regular aerobic physical activity. Relaxation is expected to reduce levels of circulating catecholamines, which has the effect of reducing vascular tone and blood pressure. Individuals are counseled to stop smoking to eliminate vasoconstrictor effects of nicotine.
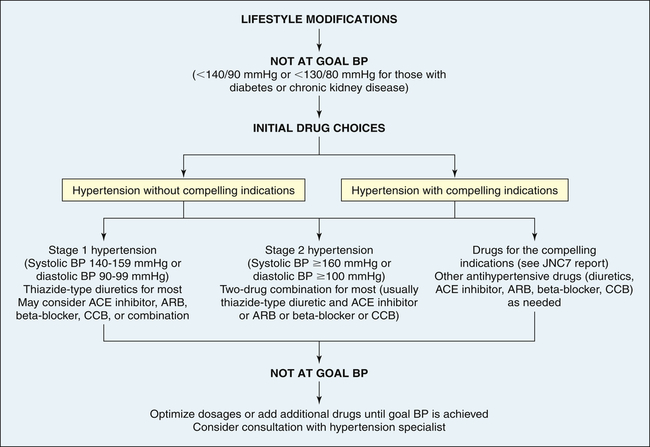
ACE, Angiotensin-converting enzyme; ARB, angiotensin-receptor blocker; BP, blood pressure; CCB, calcium channel blocker. (Data from Chobanian AV et al: JAMA 289:2560–2572, 2003.)
Pharmacologic treatment of hypertension reduces the risk of end-organ damage and prevents major diseases, such as myocardial ischemia and stroke. Thiazide diuretics and beta-blockers have been shown to be safe and effective medications for lowering blood pressure and preventing the cardiovascular complications of hypertension for many individuals. However, these medications are associated with lipid disorders and glucose intolerance.58 Many individuals will have “compelling indications” for choosing a particular antihypertensive as a first-line medication. For example, individuals with heart failure or those who have chronic kidney disease, are postmyocardial infarction, or have had recurrent stroke should begin antihypertensive treatment with an ACE inhibitor, ARB, or aldosterone antagonist.19 It is widely anticipated that the new report from the National Committee on the Prevention, Detection, Evaluation and Treatment of High Blood Pressure (JNC8) will have a greater emphasis on managing the RAAS in hypertension as a first-line intervention; however, diuretics and beta-blockers will continue to be effective choices for many individuals.59 If a person requires two drugs for blood pressure control, the recommendation is combinations of thiazide diuretics and other antihypertensives, such as beta-blockers and ACE inhibitors. As described on page 1133, a new development is the use of renal denervation, which is being investigated for the treatment of selected individuals with severe or refractory hypertension.25,60
Orthostatic (Postural) Hypotension
Orthostatic (postural) hypotension means a decrease in systolic and diastolic arterial blood pressure on standing. The American Autonomic Society (AAS) and the American Academy of Neurology (AAN) define orthostatic hypertension as a systolic blood pressure decrease of at least 20 mmHg or a diastolic blood pressure decrease of at least 10 mmHg within 3 minutes of standing up. It can be categorized as arteriolar, venular, or mixed.61 Compensatory changes during standing normally increase sympathetic activity mediated through stretch receptors (baroreceptors) in the carotid sinus and the aortic arch (see Chapter 31). This reflex response to shifts in volume caused by postural changes leads to a prompt increase in heart rate and constriction of the systemic arterioles, which maintains a stable blood pressure. These compensatory mechanisms are not effective in maintaining a stable blood pressure in individuals with orthostatic hypotension.
Chronic orthostatic hypotension can be categorized as (1) secondary to a specific disease, and (2) idiopathic or primary. The diseases that cause secondary orthostatic hypotension are endocrine disorders (e.g., adrenal insufficiency, diabetes mellitus), metabolic disorders (e.g., porphyria), or diseases of the central or peripheral nervous system (e.g., intracranial tumors, cerebral infarcts, Wernicke encephalopathy, peripheral neuropathies). Cardiovascular autonomic neuropathy is a common cause of orthostatic hypotension in diabetes and is a serious and often overlooked complication. Severe chronic autonomic failure may result from multiple system atrophy (MSA), in which there are multiple central nervous system degenerative changes, and Parkinson disease. Individuals with these disorders also may exhibit supine hypertension, altered drug sensitivity, hyperresponsiveness of blood pressure to hypo/hyperventilation, sleep apnea, and other neurologic disturbances.62,63
Idiopathic, or primary, orthostatic hypotension is the term for hypotension in which there is no known initial cause. It affects men more often than women and usually occurs between the ages of 40 and 70 years. Up to 18% of the older adult population may be affected by chronic orthostatic hypotension.61 It is a significant risk factor for falls and associated injuries and has been associated with an increased risk for cardiovascular events. In addition to cardiovascular symptoms, impotence and bowel and bladder dysfunction often are found in this type.
Orthostatic hypotension often is accompanied by dizziness, blurring or loss of vision, and syncope or fainting. To assess hypotensive episode frequency, severity, and correlation with symptoms, 24-hour blood pressure monitoring is recommended. No curative treatment is available for idiopathic orthostatic hypertension. In the secondary form, postural hypotension improves when the underlying disorder is corrected. Several treatments can help acute and chronic orthostatic hypotension, including liberalization of salt intake, raising the head of the bed, thigh-high stockings, volume expansion with mineralocorticoids, and vasoconstrictors such as midodrine.61
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


