Robin Beeman and Roberta J. Emerson
Alterations in Hemostasis and Blood Coagulation
KEY QUESTIONS
• How do platelets and factors of the clotting cascade contribute to hemostasis?
• How are laboratory tests used to differentiate the various coagulation disorders?
• What vascular alterations result in abnormalities of hemostasis?
• What are the common causes of platelet deficiencies, excesses, and dysfunction?
• What are the common causes of inherited and acquired disorders of coagulation?

http://evolve.elsevier.com/Copstead/
The term hemostasis means arrest of bleeding or prevention of blood loss after a blood vessel is injured. Hemostasis is accomplished via a complex interaction involving the vessel wall, circulating platelets, and plasma coagulation proteins. If hemostasis is inadequate, bleeding results; if hemostasis is excessive, inappropriate clotting or thrombosis results.
This chapter reviews the process of hemostasis and describes how that process is evaluated by means of clinical assessment and laboratory tests. The focus of this chapter is disorders of hemostasis and coagulation that result in bleeding. Disorders that result in thrombosis are discussed in Chapter 15.
The Process of Hemostasis
Stages of Hemostasis
Primary hemostasis, the initial response to vascular injury, involves the interaction between platelets and the endothelium of the injured blood vessel. The immediate response of the vessel to trauma is vasoconstriction to reduce blood loss. Although nervous reflex may play a part, this vasoconstriction results primarily from local myogenic spasm that may last from minutes to hours. The more trauma to the vessel, the greater the degree of vascular spasm.1
The second component of primary hemostasis is formation of a platelet plug. Platelets not only adhere to endothelial collagen exposed by injury but also aggregate (clump together) at the site of vessel injury. The formation of this platelet plug is usually completed within 3 to 7 minutes.
Secondary hemostasis involves the formation of a fibrin clot, or coagulation, at the site of injury to maintain the hemostasis already initiated. Clotting factors are activated via the intrinsic pathway or extrinsic pathway, and participate in a series of events that catalyze or facilitate the conversion of fibrinogen to fibrin.1 This process takes an average of 3 to 10 minutes.
Clot retraction, the final stage of clot formation, occurs when the components of the fibrin clot—the platelet plug, fibrin strands, and trapped red blood cells—are compressed or contracted to form a firm clot. This stage takes approximately 1 hour.
Platelets
Platelets have an integral role in hemostasis; thus, it is important to review their nature and function (Figure 14-1). A normal platelet count is between 150,000 and 400,000 platelets/mm3 of blood. Platelets, also known as thrombocytes, are the smallest of the formed elements in the blood. They are produced in the bone marrow from megakaryocytes, which are derived from the pluripotent stem cell. Most of the platelets are found in the circulation and about 25% are sequestered in the liver and spleen.3 Factors such as the stress response, epinephrine, and exercise may stimulate platelet production. The average life span of a platelet is 7 to 12 days. On completion of its life span, a platelet is eliminated from the circulation by the tissue macrophage system.1–3
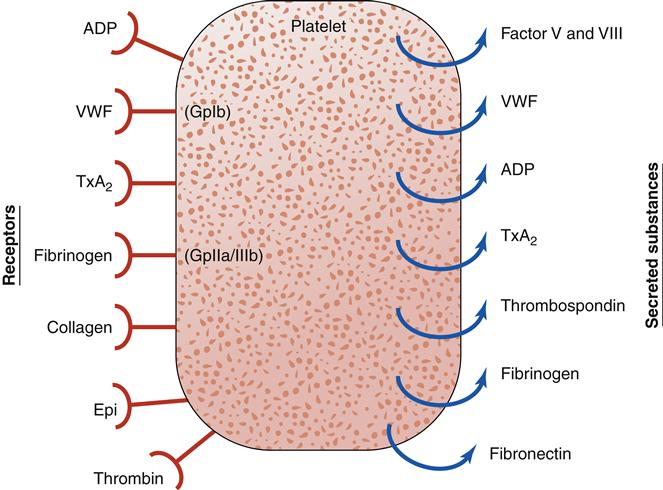
Platelets play a complex role in the process of hemostasis. Initially, platelets adhere to subendothelial collagen exposed by trauma (Figure 14-2). After adhesion, the platelets become activated and initiate degranulation, the release of α granules and dense bodies. α granules release platelet thrombospondin, fibrinogen, fibronectin, von Willebrand factor (VWF), and coagulation factors V and VIII. The dense granules release adenosine diphosphate (ADP), adenosine triphosphate (ATP), and serotonin. The presence of ADP and collagen encourages arachidonic acid formation, which leads to formation of thromboxane A2 (TxA2, a potent platelet aggregation agonist). Aspirin and other cyclooxygenase enzyme inhibitors can be used to block this cascade. Thromboxane A2 stimulates the glycoprotein IIb/IIIa (GpIIb/IIIa) receptors on platelets to be expressed and further promotes platelet adhesion. The glycoprotein IIb/IIIa blockers (e.g., eptifibatide) are useful antiplatelet agents.2–5
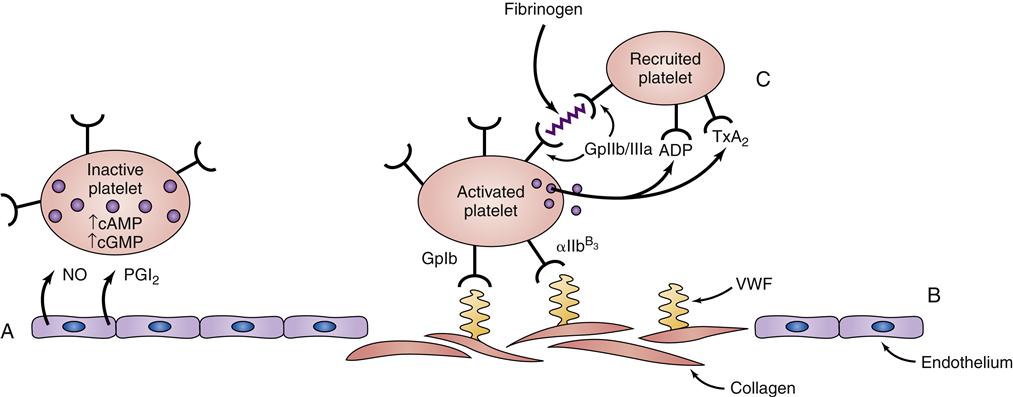
In addition to the major role platelets play in primary hemostasis, they are also involved in secondary hemostasis and clot retraction. Platelets catalyze interactions between activated coagulation factors, accelerating the conversion of prothrombin to thrombin. Platelets also have a role in clot retraction.
Blood Coagulation Factors
With the exception of tissue factor (factor III; tissue thromboplastin) and calcium, blood coagulation factors are plasma proteins that circulate in the bloodstream in an inactive state. These factors are listed in Table 14-1 according to the internationally standardized nomenclature. The factors are numbered in the order of their discovery, not the order in which they participate in the clotting cascade. Factors with both active and inactive forms are differentiated with the letter “a” after the Roman numeral to designate the active form.
TABLE 14-1
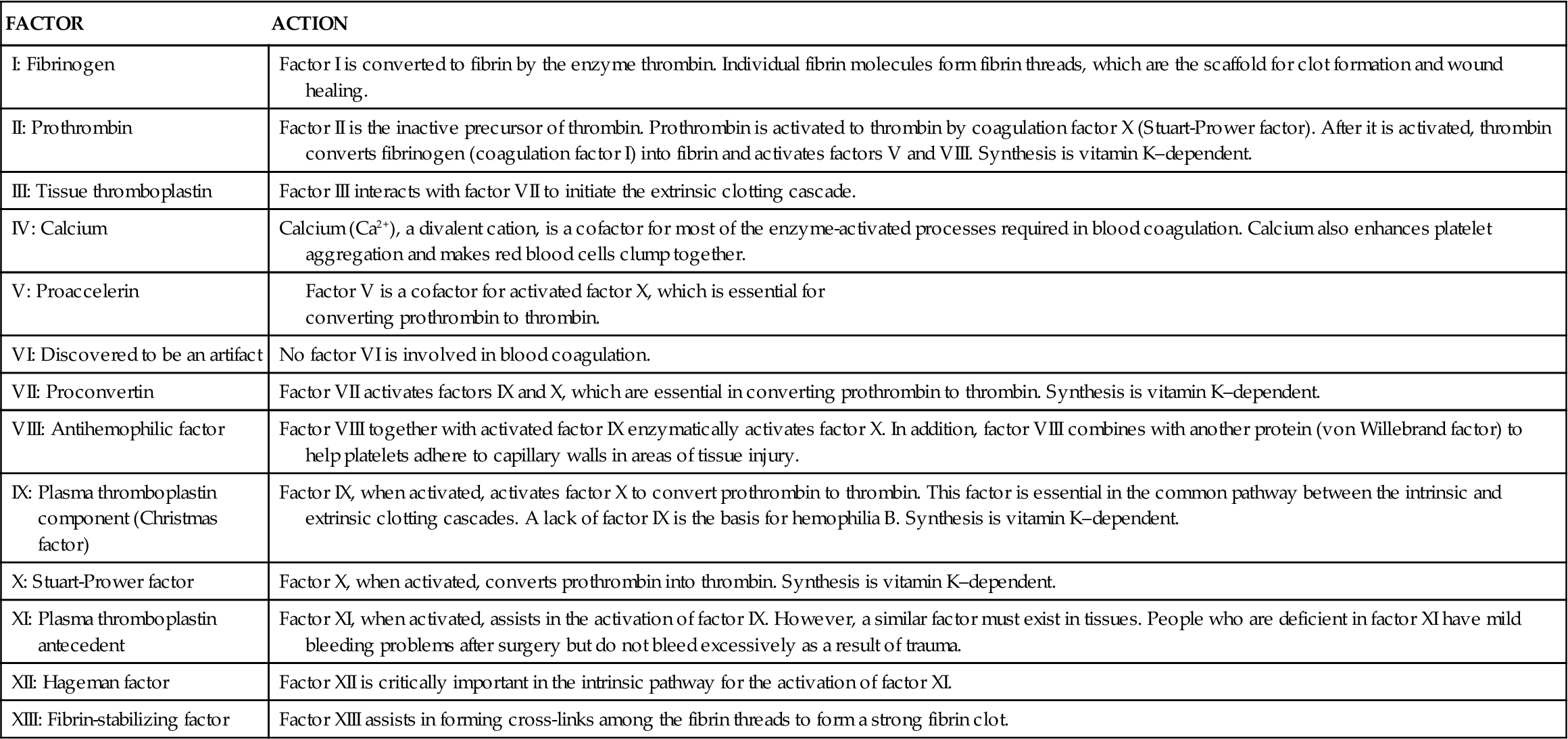
From Ignatavicius DD, Workman ML: Medical surgical nursing: patient-centered collaborative care, ed 7, Philadelphia, 2013, Saunders, p 860.
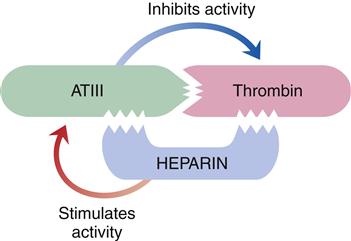
The liver is responsible for the synthesis of coagulation factors, with the exception of part of factor VIII. Factors II, VII, IX, and X; protein C; and protein S are dependent on vitamin K for synthesis and normal activity. Some of the coagulation proteins also can be synthesized by other cells, such as megakaryocytes and endothelial cells.4,5 Antithrombin III (ATIII) and protein C are protein complexes that promote anticoagulation. Antithrombin is a potent anticoagulant that binds to and inactivates free thrombin, preventing its binding and cleaving of fibrinogen. Protein C, a plasma protein that inactivates factors V and VIII, prevents clot formation. Protein S assists protein C in binding to phospholipase and stimulates release of tissue plasminogen activator, initiating fibrinolysis.4,5 Low-molecular-weight heparins and heparin work by enhancing the activity of antithrombin III (Figure 14-3).
Fibrin Clot
In normal hemostasis, the fibrin clot is produced through activation of the intrinsic or extrinsic pathway and, in turn, the common final pathway. Effective hemostasis is the result of interactions between all of these pathways and is commonly referred to as the coagulation cascade.
Figure 14-4 illustrates the coagulation cascade. The intrinsic pathway of coagulation begins when blood comes into contact with altered vascular endothelium or another negatively charged surface, such as glass. This contact phase of coagulation involves four factors: (1) factor XII, (2) high-molecular-weight kininogen (HMWK), (3) prekallikrein, and (4) factor XI. Factor XII is activated to factor XIIa, which in turn activates XI to XIa and prekallikrein to its active form, kallikrein (KAL). Kallikrein liberates bradykinin from HMWK. The release of bradykinin produces an initial vasodilation followed by release of angiotensin II and vasoconstriction. The major role of factor XIa is activation of factor IX to factor IXa in the presence of calcium. Factor IXa then activates factor X to factor Xa in the presence of factor VIII, calcium, and phospholipid. This activation usually takes place on the membrane of stimulated platelets. The common final pathway is initiated by factor Xa.
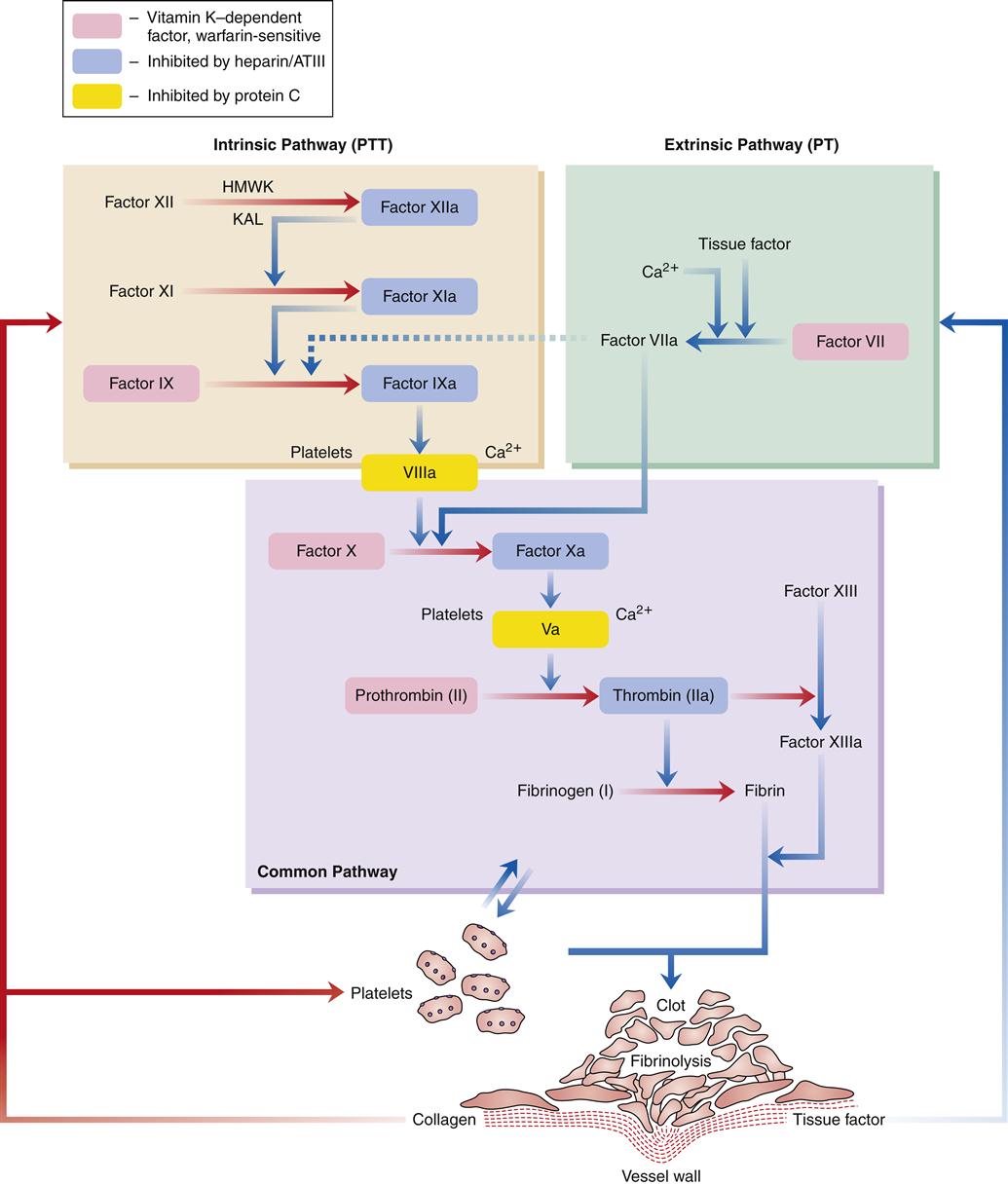
The extrinsic pathway of coagulation begins when the vascular wall is traumatized, as in a crush injury. Tissue factor (factor III) from injured tissue activates factor VII. Factor VIIa activates factor X to Xa, which in turn initiates the common final pathway. Factor VIIa also activates factor IX in the intrinsic system.
The common final pathway of coagulation is initiated by factor X, which is activated by both the intrinsic and extrinsic pathways. Factor Xa, in the presence of factor V, calcium, and phospholipid, converts prothrombin (factor II) to thrombin. This conversion is facilitated by the presence of activated platelets. Thrombin then cleaves fibrinogen to form an insoluble fibrin clot. Thrombin also activates factor XIII, which promotes fibrin stabilization. The clot is further stabilized by clot retraction. Thrombin also helps to perpetuate the clotting cascade by continuing to activate factors V and VIII.
Fibrinolysis
At the same time the fibrin clot is forming, the process of fibrinolysis or clot dissolution is initiated (Figure 14-5). Factor XII, HMWK, kallikrein, and thrombin are involved in the release of plasminogen activators. The plasminogen activators cleave plasminogen, a plasma protein that has been incorporated into the fibrin clot, to its active form, plasmin. Plasmin digests fibrinogen and fibrin and inactivates blood coagulation factors V and VIII. Fibrin split products, or fibrin degradation products, result from the dissolution of the fibrin clot.
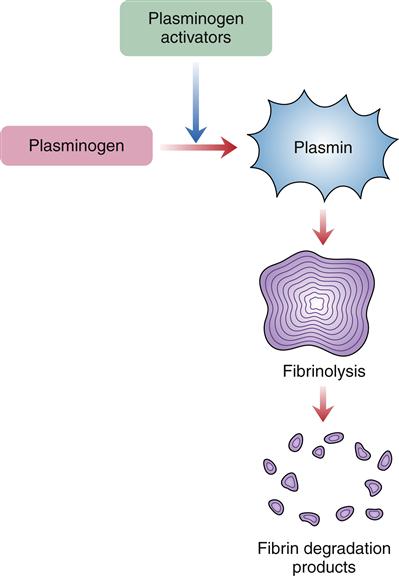
Plasmin, activated from plasminogen, enzymatically cleaves fibrin proteins in the clot. This results in fibrin split products, which can be measured.
The control of fibrinolysis is complex. The Kupffer cells of the liver and macrophages located in the spleen and bone marrow clear the circulation of activated clotting factors and fibrin degradation products. Antiplasmins that inhibit plasmin exist to prevent inappropriate fibrinolysis. All these factors and mechanisms are present to create a balance between clot production and clot dissolution.
Evaluation of Hemostasis and Coagulation
Data obtained from clinical assessment and laboratory tests facilitate the identification and evaluation of a hemostatic abnormality. Evaluation of a patient for a bleeding tendency is indicated in the following circumstances: when there is a personal or family history of bleeding; during active bleeding that is unresponsive to standard interventions; as part of screening before surgery; and for ongoing evaluation of anticoagulation therapy. A bleeding tendency may be inherited or acquired, and may result from defects in blood vessels, platelets, or coagulation factors. The purpose of the evaluation process is to determine if a problem exists and to ascertain the underlying cause so that appropriate management can be initiated.
Clinical Assessment
Both the family history and the personal history are important in the evaluation of a bleeding problem (Table 14-2). A family history of bleeding in males is often linked to one of the types of hemophilia, which accounts for the majority of serious inherited coagulation problems.4,5 The location, severity, duration, and setting in which bleeding occurs are also important clues to the type of defect that is present. Bleeding associated with vascular or platelet defects usually occurs immediately after trauma (e.g., dental extraction), involves skin or mucous membranes, and is brief. Delayed bleeding or bleeding into muscles or joints is more typical of a coagulation defect.5–8
TABLE 14-2
CLUES FROM PATIENT HISTORY REGARDING BLEEDING DISORDERS
| CLUE FROM PATIENT HISTORY | POSSIBLE CAUSE |
| Family history of bleeding in both males and females | von Willebrand disease |
| Family history of bleeding in males | Hemophilia A or B |
| Newly acquired bruising | Drugs (especially aspirin and NSAIDs, anticoagulant therapy), thrombocytopenia |
| Excessive bleeding/bruising during/after surgery | Mild-severe deficiency of coagulation factors, von Willebrand disease; thrombocytopenia, drug ingestion |
| Bleeding following initial hemostasis | Factor XIII deficiency |
Systemic diseases, such as renal failure, liver disease, systemic lupus erythematosus, and malignancies, may be associated with a bleeding problem. Medication history, including use of over-the-counter medications, is another important aspect in the evaluation of a hemostatic defect. A common cause of acquired bleeding problems is drug ingestion. Specific drugs that alter hemostasis include aspirin and aspirin-containing preparations, nonsteroidal antiinflammatory agents, some antibiotics, anticoagulants, alcohol, and chemotherapeutic and thrombolytic agents.
Many of the physical findings of bleeding are manifested in the skin and mucous membranes. The individual may appear pale or jaundiced. Pallor is associated with a marked decrease in hemoglobin level; jaundice is associated with liver or gallbladder disease, possible coagulation disorders, and excessive red blood cell destruction.
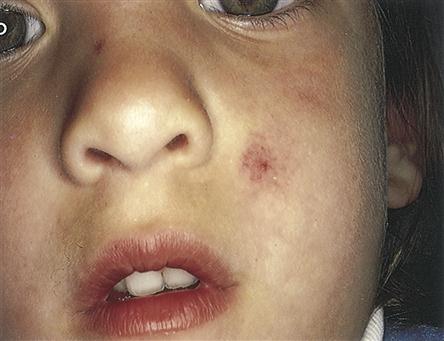
Petechiae are flat, pinpoint, nonblanching red or purple spots caused by capillary hemorrhages in the skin and mucous membranes (Figure 14-6). Petechiae are commonly seen with vascular and platelet disorders. They are usually present on dependent areas of the body, such as the legs, or on areas constricted by tight clothing. Not all petechiae indicate a bleeding problem. Petechiae found on other body areas not constricted by tight clothing, such as the abdomen or thorax, may be associated with infectious disease or other pathophysiologic sources. Petechiae may be seen in the newborn as a result of the trauma of delivery, not as a result of a bleeding problem.
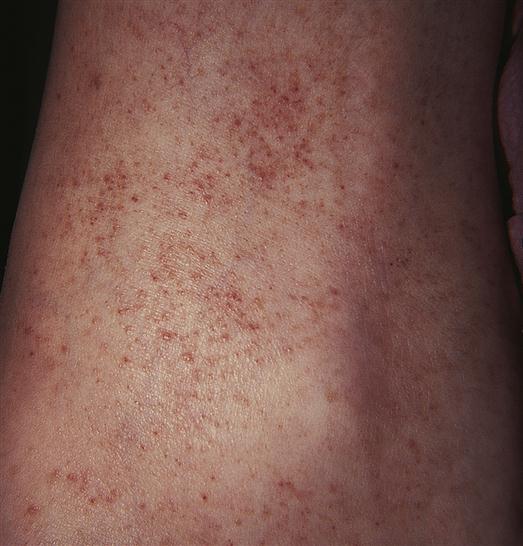
When petechiae occur in groups or patches, the term purpura is used (Figure 14-7). Purpuric lesions are often pruritic (itchy). Fever and malaise may be present, as may effusions into joints or viscera, manifested by joint or abdominal pain.
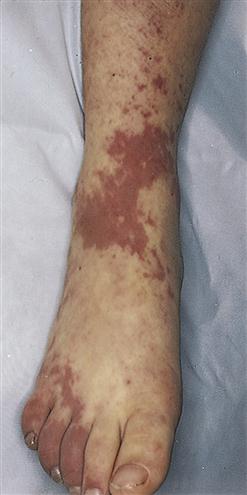
Ecchymosis occurs when blood escapes into the tissues, producing a bruise (Figure 14-8). If the area is raised, it is called a hematoma. Hemarthrosis, manifested by swelling and pain, is bleeding into a joint. Large ecchymoses, hematomas, and hemarthroses are seen in coagulation disorders.
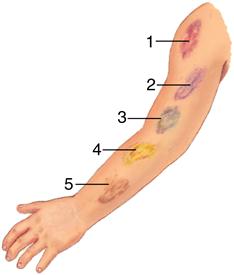
A large patch of capillary bleeding into tissues. Color in a light-skinned person is first red-blue or purple (1) immediately after or within 24 hours of trauma and generally progresses to blue to purple (2), blue-green (3), yellow (4), and brown to disappearing (5). (From Jarvis C: Physical examination and health assessment, ed 6, Philadelphia, 2012, Saunders.)
Telangiectasia is a lesion created by dilation of capillaries and small arteries, typically on the lips, tongue, tips of the fingers and toes, and sometimes in visceral vessels (Figure 14-9). These thin, dilated, tortuous vessels are red to violet in color, blanch with pressure, and tend to bleed with minimal trauma. Spider telangiectasia branch into the subcutaneous and dermal layers of the skin and are often associated with liver disease.
A fiery red, star-shaped marking with a solid circular center. Capillary radiations extend from the central arterial body. With pressure, note a central pulsating body and blanching of extended legs. Develops on face, neck, or chest; may be associated with pregnancy, chronic liver disease, or estrogen therapy, or may be normal. (From Hurwitz S: Clinical pediatric dermatology: a textbook of skin disorders in childhood and adolescence, ed 2, Philadelphia, 1993, Saunders, p 266.)
Other significant findings indicative of a bleeding disorder include blood (bright red, rusty, or black) in drainage or excreta, such as feces (hematochezia or melena), urine (hematuria), vomitus (hematemesis), nasal drainage (epistaxis), gastric drainage, or sputum (hemoptysis). Excessive menstrual bleeding may occur (menorrhagia). Acute abdominal or flank pain may indicate internal bleeding. Hypovolemia from bleeding may produce a shock state and present as hypotension, tachycardia, pallor, altered mentation, and decreased urine output. The two sites at which bleeding is most life threatening are the oropharynx (resulting in airway compromise) and within the brain tissue. One of the leading causes of death in patients experiencing severe disorders of coagulation is intracerebral hemorrhage.5
Laboratory Tests
Many laboratory tests are available to aid in the diagnosis of hemostasis problems (Table 14-3). Basic screening includes a complete blood cell count (CBC), including a platelet count and peripheral blood smear, bleeding time, prothrombin time (PT) or international normalized ratio (INR), activated partial thromboplastin time (aPTT), and thrombin time. These screening tests evaluate both primary and secondary hemostasis. The CBC determines if anemia is present, the platelet count determines the number of platelets, and the peripheral smear indicates the number and gross morphologic characteristics of platelets. The bleeding time evaluates vascular status and platelet function. The PT and INR assess the extrinsic pathway of coagulation, and the aPTT assesses the intrinsic pathway. Reporting prothrombin activity as a percentage of PT in seconds can pose difficulty in the adjustment of anticoagulation therapy because the PT varies with each laboratory and the reagent used at that lab. Laboratories have tried to compensate for this variation by using the ratio of the patient’s value to the laboratory’s control value, which again varied with the reagent. The INR is a standardized PT value used worldwide that controls for this reagent variability. Thrombin time measures the time needed to convert fibrinogen to fibrin; this reflects the quantity and quality of fibrinogen as well as the influence of any inhibitors. The D-dimer assay reflects fibrinolysis.
TABLE 14-3
SELECT LABORATORY TESTS USED TO ASSESS BLEEDING
| TEST | NORMAL VALUE∗ | PURPOSE OR SIGNIFICANCE |
| Platelet count | 150,000-400,000/mm3 | Determines number of platelets; decreased in ITP, anemias, DIC, infection, chemotherapy; increased in leukemia, cancer, splenectomy |
| Bleeding time | 3-10 min | Assesses platelet and vascular response; increased in thrombocytopenia, vascular defects, severe liver disease, DIC, von Willebrand disease, aspirin ingestion |
| Prothrombin time | 10-14 sec; 100% | Evaluates extrinsic pathway of coagulation; increased in vitamin K deficiency, hemorrhagic disease of the newborn, liver disease, DIC, anticoagulant therapy. Evaluates all coagulation factors except VIII and XII. |
| International normalized ratio | Evaluates extrinsic pathway of coagulation (as prothrombin time); provides uniformity worldwide, independent of reagents | |
| Activated partial thromboplastin time | 33-45 sec | Evaluates intrinsic pathway of coagulation; increased in hemophilia, vitamin K deficiency, liver disease, DIC, circulating anticoagulants, heparin therapy |
| Thrombin time | 15 sec, or control + 5 sec | Measures conversion of fibrinogen to fibrin; increased in DIC, liver disease, low fibrinogen <100 mg/dl, multiple myeloma |
| Fibrinogen | 200-400 mg/dl | Measures fibrinogen level; decreased in liver disease, DIC |
| Fibrin split products or fibrin degradation products† | <3 μg/ml | Measures by-products from breakdown of fibrin clot; increased in DIC, hypoxia, leukemia, thromboembolic disorders |
| Clot retraction† | Approximate measure of platelet function; decreased in thrombocytopenia, von Willebrand disease | |
| Platelet aggregation† | Visible aggregates form in <5 min | Measures rate and percentage of aggregation; decreased in mononucleosis, ITP, von Willebrand disease, leukemia, aspirin ingestion, thrombasthenia, Bernard-Soulier syndrome |
| Tourniquet test (Rumpel-Leede test, capillary fragility test) † | No petechiae or occasional petechiae | Evaluates vascular fragility and platelet function; positive test in thrombocytopenia, vascular purpuras, thrombasthenia |
| Euglobulin lysis time† | No lysis of fibrin clot at 37° C for 3 hr; clot is observed for 24 hr | Assesses fibrinolysis; increased lysis in DIC, incompatible blood transfusion, cirrhosis, cancer, obstetric complications |
| Plasma D-dimer assay | <200 ng/ml | Assesses fibrinolysis; increased in DVT, pulmonary embolism (highly nonspecific), DIC (high negative predictive value) |
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


