Alphaviruses
Diane E. Griffin
The genus Alphavirus includes 29 species that can be classified antigenically into at least eight complexes (Table 23.1). Alphaviruses are geographically restricted in their distributions and have been found on all continents and on many islands. In nature, alphaviruses cycle between invertebrate insect vectors and vertebrate hosts. For most alphaviruses, the insect vectors are mosquitoes; however, other hematophagous arthropods, such as lice or mites, are vectors for a few. The vertebrate hosts are generally mammals or birds, although fish are hosts for the aquatic alphaviruses. In general, the pathogenic alphaviruses are divided into the viruses that cause human disease characterized by rash and arthritis, primarily found in the Old World, and viruses that cause encephalitis, primarily found in the New World. For many alphaviruses, no human or veterinary disease has been recognized. Larger mammals, such as humans and horses, that tend to develop severe or fatal disease are often dead-end hosts unimportant to the endemic virus transmission cycles but can be important for sustaining epidemics.
History
Records of diseases almost certainly attributable to alphaviruses date to the 18th and 19th centuries, when epidemics of fatal encephalitis in horses in the northeastern United States and outbreaks of arthritis in Southeast Asia were recognized and recorded.111,300,788 The first clear report of epidemic encephalitis comes from the summer of 1831, when 75 horses died in Massachusetts.300 Over the next 100 years, several local outbreaks of encephalitis in horses were noted along the Atlantic seaboard of the United States and in the Pampas region of South America.683 However, the first alphavirus to be cultured was western equine encephalitis virus (WEEV). This virus was isolated in 1930 from the central nervous system (CNS) tissues of 2 horses involved in an epidemic of equine encephalitis in the San Joaquin Valley of California.522 Eastern equine encephalitis virus (EEEV) was isolated from the brains of affected horses in New Jersey and Virginia in 1933.786 Both diseases occurred in summertime epidemics, suggesting an arthropod vector, and
in 1933, Kelser showed WEEV transmission by mosquitoes.388 In 1936, an epizootic of equine encephalitis occurred in the Guajira region of Venezuela; the virus isolated was not neutralized by antisera against EEEV or WEEV and was designated Venezuelan equine encephalitis virus (VEEV).420
in 1933, Kelser showed WEEV transmission by mosquitoes.388 In 1936, an epizootic of equine encephalitis occurred in the Guajira region of Venezuela; the virus isolated was not neutralized by antisera against EEEV or WEEV and was designated Venezuelan equine encephalitis virus (VEEV).420
Table 23.1 Alphaviruses, Abbreviations, Biological Features, and Association with Disease | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Summertime epidemics of polyarthritis were recognized in Australia and New Guinea in 1928,183,572 and subsequent outbreaks were reported in Northern Europe, Africa, and Southeast Asia.727 It is likely, of course, that the alphavirus-induced arthritic diseases are much older than these dates but were not clearly described or differentiated from more prevalent infections in these regions, such as dengue. This is considered to be particularly true of outbreaks of chikungunya virus (CHIKV) that have occurred in India and Southeast Asia over the past 200 years.111,788 Viruses associated with epidemic polyarthritis were eventually isolated, both from mosquitoes collected in the areas of human disease and later from humans. The first of these viruses was isolated in 1952 from a pool of Culex spp.
mosquitoes collected near Sindbis, Egypt.782 However, it was many years before Sindbis virus (SINV) was linked to human disease.493
mosquitoes collected near Sindbis, Egypt.782 However, it was many years before Sindbis virus (SINV) was linked to human disease.493
The first clear association of an alphavirus with arthritic disease came in 1953, when CHIKV was isolated in Tanzania from the blood of people with severe arthritis.668 During the next several years, several viruses that cause arthritis, often accompanied by a rash, were isolated in Africa, Australia, and South America.122,537,884 These viruses were added to the growing list of arthropod-borne (arbo) viruses, defined by the World Health Organization in 1967 as “viruses which are maintained in nature principally, or to an important extent, through biological transmission between susceptible vertebrate hosts by haematophagous arthropods; they multiply and produce viremia in the vertebrates, multiply in the tissues of arthropods, and are passed on to new vertebrates by the bites of arthropods after a period of extrinsic incubation”.621
In 1954, arboviruses were divided by Casals and Brown into three serologic groups—A, B, and C—based on cross-reactivity in hemagglutination inhibition (HI) tests.556,621 EEEV, WEEV, and VEEV constituted the group A arboviruses. A second cross-reacting set, including dengue, St. Louis encephalitis, and yellow fever viruses, constituted the group B arboviruses, and the nonreactive viruses were designated group C. As viruses became classified on the fundamental properties of the virion and the genome, the group A viruses became the Alphavirus genus within the Togaviridae family of enveloped RNA viruses.
Infectious Agents
Alphaviruses are enveloped plus-strand RNA viruses with icosahedral symmetry (Fig. 23.1). The virions are 60 to 70 nm in diameter and sensitive to ether and detergent. Cryo-electron microscopy structures are available for many.18,418,496,549,618,722,901,902,903 The RNA is contained within a capsid formed by a single protein arranged as an icosahedron with T = 4 symmetry. The nucleocapsid is enclosed in a lipid envelope derived from the host cell plasma membrane that contains the viral-encoded glycoproteins E1 and E2. These proteins form heterodimers that are grouped as trimers to form 80 knobs on the virion surface. Glycoproteins are arranged such that 240 copies of each interact with 240 copies of capsid protein.
The 49S genome is composed of a single-strand, nonsegmented, capped, and polyadenylated message sense RNA that is infectious. Complete sequence information is available for representatives of all currently known Alphavirus species.129,161,206,219,297,402,431,434,437,518,764 The genomes are 11 to 12 kb in size and are organized with the nonstructural proteins (nsPs) at the 5′ end and the structural proteins at the 3′ end. The nsPs are translated from genomic RNA and the structural proteins from a subgenomic RNA.765 Four nsPs function to replicate the viral RNA and produce the subgenomic RNA (see Chapter 22).
Five potential structural proteins (C, E3, E2, 6K, and E1) are encoded in the subgenomic RNA as a polyprotein, and an additional transframe protein (TF) is produced by –1 ribosomal frameshifting within the 6K coding region.141,215 The N-terminal portion of C is basic and is presumed to bind the viral genomic RNA, whereas the more conserved C-terminal portion interacts with other copies of the C protein to form the nucleocapsid and also interacts with the cytoplasmic tail of E2.765,901
E3 is a small, cysteine-rich glycoprotein that serves as a signal sequence for pE2 (the precursor containing E3 and E2), mediates proper folding of E2, and is necessary for pE2 to
heterodimerize with E1 for transport to the cell surface.468,599 As part of the pE2-E1 heterodimer, E3 prevents premature activation of E1.362,840,844 E3 is cleaved from E2 by furin in the trans-Golgi, remains associated with the spike under acidic conditions, and is usually shed when virions bud from the cell surface.737,904
heterodimerize with E1 for transport to the cell surface.468,599 As part of the pE2-E1 heterodimer, E3 prevents premature activation of E1.362,840,844 E3 is cleaved from E2 by furin in the trans-Golgi, remains associated with the spike under acidic conditions, and is usually shed when virions bud from the cell surface.737,904
 Figure 23.1. Three-dimensional (3D) reconstruction of the Venezuelan equine encephalitis virus (VEEV) virion. A: Radially colored 3D reconstruction of VEEV showing the E1 basal triangle (green) and E2 central protrusion (blue) for each spike. Scale bar: 10 nm. B: One asymmetric unit of the virus containing four unique copies of E1 (magenta), E2 (cyan), E3 (orange), and capsid (CP, blue). The cryo-electron microscopy densities for the viral membrane (yellow) and genomic RNA (green) are also displayed at slightly lower isosurface threshold. (Courtesy of Wah Chiu; reproduced with permission from Zhang R, Hryc CF, Cong Y, et al. 4.4A cryo-EM structure of an enveloped alphavirus Venezuelan equine encephalitis virus. EMBO J 2011;30:3854–3863.) |
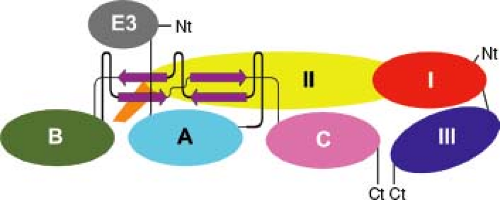 Figure 23.2. Domain structure of the glycoprotein spike. Schematic diagram of the E1pE2 heterodimer drawn “untwisted” to show the domains positioned with respect to one another and their connectivity. Domains of E1 (I, red; II, yellow; III, blue; fusion loop, orange) and pE2 (A, cyan; B, dark green; C, pink; E3, gray) are shown. Ct, C-terminus; Nt, N-terminus. (Courtesy of Felix Rey; reproduced with permission from Voss JE, Vaney MC, Duquerroy S, et al. Glycoprotein organization of chikungunya virus particles revealed by x-ray crystallography. Nature 2011;468:709–712.) |
The E2 glycoprotein is a transmembrane protein that has two or three N-linked carbohydrates and contains the most important epitopes for neutralizing antibody. E2 is organized into three immunoglobulin domains (A, B, and C) (Fig. 23.2). Domain A (residues 1–132) is at the center and top of the heterotrimer and has receptor and neutralizing antibody-binding sites. Domain B is at the spike tip, and C is toward the viral membrane.455,840 The intracytoplasmic portion interacts with the capsid and has a second stretch of hydrophobic amino acids and myristoylation sites that tether it to the inner surface of the membrane.
The 6K protein serves as a signal peptide for E1, is cleaved from E1 and E2 by signal peptidase, and is important for budding, and small amounts are incorporated into the virion.238,461 The E1 protein has one or two N-linked carbohydrates, a short (one or two residue) intracytoplasmic tail, and a positionally conserved internal hydrophobic stretch of amino acids in the N-terminal portion that serves as the fusion peptide for virion entry into the cell. E1 is organized into three β-sheet-rich domains (I, II, and III) similar to the flavivirus E protein with the internal fusion loop at the tip of domain II253,443 (see Fig. 23.2). The function of TF has not yet been defined but can be incorporated into virions.215
Propagation and Assay in Tissue Culture
Initial isolations of alphaviruses were accomplished by intracerebral inoculation into suckling mice—a host very susceptible to infection with most alphaviruses.335,668,689,781,782,867,884 Many alphaviruses can also be isolated and propagated efficiently in primary chick embryo fibroblasts (CEFs) and in various continuous mammalian cell lines such as human epithelial (HeLa, MRC5) cells, baby hamster kidney (BHK) cells, monkey kidney (Vero) cells, and mouse fibroblast and neuroblastoma cells108,581 (Fig. 23.3).
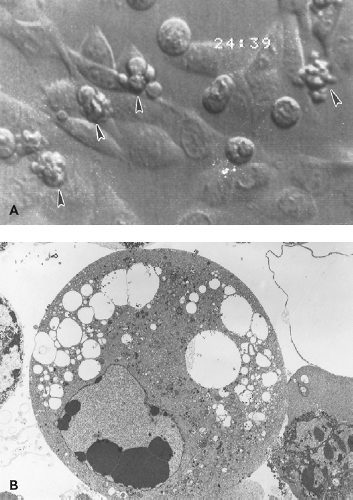 Figure 23.3. Effects of Sindbis virus infection on vertebrate cells. A: Cytopathic effects (CPE) of rounding, shrinkage, and cytoplasmic blebbing in baby hamster kidney (BHK) cells infected at a low multiplicity. B: Electron micrograph of an infected BHK cell showing chromosomal condensation and cytopathic vacuoles. |
Most alphaviruses will form plaques on susceptible mammalian or avian cells under an agar overlay. Mosquito cell lines also support replication, although often without cytopathic effect (CPE).635,761,763,803 The first plaque assay of an animal virus was performed using WEEV on CEF cells,190 and plaque assay remains a convenient and sensitive way to quantify infectious virus. Plaque size has been used to differentiate strains and is determined by the type of overlay used, by relative virus binding to negatively charged sulfated polysaccharides present in the overlay, and by replication efficiency.87,96,775
Biological Characteristics
Hemagglutination
Alphaviruses can hemagglutinate avian (e.g., goose, chicken) erythrocytes,581,684 and hemagglutination has been used as a method for quantifying virus and HI for measuring antiviral antibody.144 Hemagglutination requires prior exposure of the virus to acidic pH, depends primarily on the E1 glycoprotein, and reflects binding of the fusion domain of the E1 glycoprotein to lipids in the erythrocyte membrane.130,144,829,873 E2 also participates in hemagglutination because some monoclonal
antibodies (MAbs) specific for E2 also have HI activity.67,640 The HI test has been useful for determining antigenic relationships among alphaviruses.107
antibodies (MAbs) specific for E2 also have HI activity.67,640 The HI test has been useful for determining antigenic relationships among alphaviruses.107
Cellular Receptors
Binding of virus to the cell surface and entry into the cell is a multistep process that depends on virus glycoproteins E1 and E2, cell surface molecules, low pH in the endosome, and fusion of membrane lipids. Variations in any of these components will affect the efficiency of infection and the likelihood that any particular cell will become infected in vivo. Virus-specific attachment to cells is primarily a function of the E2 glycoprotein. The important role for E2 in initiating virus–cell interaction is evidenced by the ability of anti-E2 MAbs to inhibit binding to cells,96,474 of anti-idiotypic antibodies to E2-specific MAbs to recognize putative virus receptors on cells,823,852 and of amino acid changes in E2 to alter virus binding to cells of different types.189,438
Identification of specific alphavirus receptors has been difficult and may be complicated by experimental use of virus strains that are adapted to replicate in tissue culture.411 Because each alphavirus infects a wide range of hosts, often including birds, mammals, and mosquitoes, they must either use an evolutionarily well-conserved cell surface molecule or multiple molecules as receptors. None of the alphavirus receptors identified to date appears to be used exclusively, suggesting the possibility of several receptors. Alternatively, alphaviruses may use receptor-coreceptor combinations to achieve wide host range and the specific tropisms observed in vivo.
The first alphavirus receptor to be identified was the major histocompatibility complex (MHC) class I molecule receptor for Semliki Forest virus (SFV) on mouse and human cells.315 This molecule is not absolutely required because cells lacking MHC molecules can still be infected with SFV.583 The high-affinity laminin receptor is a receptor for SINV on BHK cells and a potential receptor for SINV and VEEV on C6/36 mosquito cells.474,850 Again, this molecule appears to account for only a portion of the total virus interaction with the cells studied and does not contribute to SINV binding to avian cells.850 Ross River virus (RRV) uses the a1β1 integrin on HeLa cells for binding.426 SINV and VEEV can use natural resistance–associated macrophage protein (NRAMP) for binding to insect and mammalian cells.664 The type of cell in which the virus is grown can also influence initial virus–receptor interactions. For instance, SINV grown in mosquito cells is enriched in high-mannose carbohydrates that bind the C-type lectins DC-SIGN and L-SIGN on the surface of dendritic cells (DCs).410
Heparan sulfate (HS), a ubiquitously expressed glycosaminoglycan is an important initial binding molecule for certain strains of many alphaviruses. The use of HS is selected for by virus passage in vertebrate cells. However, wild-type strains of EEEV bind HS, so this property is not exclusively determined by passage in tissue culture.242 For these viruses, addition of heparin or lactoferrin, treatment of cells with heparinase, or the use of cells deficient in HS decreases binding to cells and plaque formation.59,96,242,312,409,411,426,740,843 Interaction with HS—a highly sulfated, negatively charged molecule—probably explains the effects of ionic strength and charge on the attachment of virus to cells.499,501,611 The heparin-binding domain of the E2 glycoprotein is on domain A and overlaps a neutralizing epitope (see Fig. 23.2). Changes toward more positively charged amino acids in the region around E2–70 increase the efficiency of attachment to cells in tissue culture.59,97,242,411
Entry requires endocytosis followed by a conformational change in the trimer of E1-E2 heterodimers induced by exposure to low pH.313,729,739,879 The E1 fusion peptide is protected in the virion heterotrimer by the B domain of E2, and this association is stabilized by E3.455,840 When exposed to acidic pH, E1 dissociates from E2 and forms stable E1 homotrimers in the presence of cholesterol-containing membranes. During this conformational change, the fusion peptide is exposed and inserted into the outer leaflet of the lipid bilayer.83,252,253 Fusion with the cell membrane to initiate infection depends on the presence of a membrane potential and sphingolipid.314,570,899
Effects on Vertebrate Cells
Alphaviruses replicate rapidly in most vertebrate cell lines with the release of progeny virus typically within 4 to 6 hours after infection. At the time of virus entry, there is an increase in permeability perhaps owing to pore formation by the E1, 6K, and/or TF proteins.492,871,872 Insertion of newly synthesized glycoproteins into the plasma membrane renders infected cells capable of adenosine triphosphatase (ATP)-dependent polykaryocyte formation on exposure to acid pH.389 Infection causes extensive CPE characterized by cell rounding, shrinkage, and cytoplasmic blebbing (see Fig. 23.3A), with the death of infected cells within 24 to 48 hours.449,635 Alphavirus-induced CPE has been linked to shut off of host cell transcription and translation,246 endoplasmic reticulum (ER) stress,574 and induction of apoptosis.20,26,257,270,351,449,656,699 The ability of alphaviruses to induce cell death, combined with the ability to express heterologous genes and activate natural killer (NK) cells, has led to their development as potential oncolytic agents.344,454,552,633,634,808,809,830
Shut off of host cell functions in Old World viruses (e.g., SINV, SFV) has been linked to the effects of nuclear nsP2 and in New World viruses (e.g., VEEV, EEEV) to the effects of C.14,32,247 Activation of protein kinase R (PKR) results in phosphorylation of eIF2a, which shuts down host protein synthesis without affecting translation of viral 26S subgenomic RNA.834 Alphavirus escape from eIF2a phosphorylation is owing to the presence of a hairpin loop structure in the subgenomic messenger RNA (mRNA) downstream of the initiation codon that allows the 40S ribosome to initiate translation in the absence of eIF2.185,804,834 This initiation process utilizes host cell factors ligatin and MCT-1/DFNR738 and does not depend on mTOR signaling.530 Alphavirus-induced inhibition of host protein synthesis can also be PKR-independent perhaps through late suppression of mTOR.265,530
The apoptotic process is associated with blebbing of the plasma membrane, condensation of nuclear chromatin, and formation of apoptotic bodies (see Fig. 23.3B). Viral proteins are concentrated in the surface blebs from which budding continues to occur.665 This process does not hamper, and may enhance, virus replication because inhibition of apoptosis usually decreases virus yield.445,458,699
The mechanism(s) by which alphaviruses induce apoptosis is not completely understood and likely differs with virus and type of target cell. Apoptosis of cultured cells can be initiated at the endosomal membrane during SINV fusion.364 Membrane-bound sphingomyelinases are activated releasing ceramide, an efficient inducer of cellular apoptosis.363,366 SFV-induced apoptosis requires RNA synthesis and accumulation777,828 and is
independent of p53.258,828 Other events in alphavirus-induced cell death often include early activation of poly(adenosine diphosphate [ADP] ribose) polymerase,564,596 activation of proapoptotic Bcl-2 family member proteins Bad or Bak,539 loss of mitochondrial membrane integrity, and release of cytochrome c.50,828 Cellular caspases are activated with cleavage of caspase-3 substrates and fragmentation of chromosomal DNA.825
independent of p53.258,828 Other events in alphavirus-induced cell death often include early activation of poly(adenosine diphosphate [ADP] ribose) polymerase,564,596 activation of proapoptotic Bcl-2 family member proteins Bad or Bak,539 loss of mitochondrial membrane integrity, and release of cytochrome c.50,828 Cellular caspases are activated with cleavage of caspase-3 substrates and fragmentation of chromosomal DNA.825
Apoptotic death is accelerated by glycoprotein-induced ER stress,50,234,574 sphingomyelinase deficiency,567 low levels of extracellular Ca++,825 expression of Bax,574 and activation of Bid.828 Alphavirus-induced apoptosis can be slowed or prevented by expression of ceramidase,363 altered Ras signaling,365,413 expression of p21WAF1/CIP1,338 expression of Bcl-2 family member and interacting proteins,270,449,451,458,539,552,574,699,826,828 expression of the ER stress protective protein Parkin,574 mutation of nsP2,246,574 phosphorylation of PKCδ,910 inhibition of constitutive expression of NFkB,462 and caspase inhibition.565,695,828
Alphavirus-induced vertebrate cell death can also occur by caspase-independent, nonapoptotic mechanisms. Alphaviruses efficiently shut down protein, ribosomal RNA (rRNA) and mRNA synthesis in infected cells,246,264,510,556 deplete nicotinamide adenine dinucleotide (NAD) and energy stores,199,825 and induce dysfunction of Na+K+ adenosine triphosphatase (ATPase) causing loss of membrane potential and changes in intracellular cation concentrations.51,827 Genome replication without structural protein synthesis can induce cell death.828 For Old World viruses, expression of nsP2 alone is cytotoxic, and this property is independent of its protease activity and can be separated from the effects of nsP2 on host transcription.187,233,246,247,264,507,605,777 Although immature neurons die by apoptosis, mature neurons are more resistant to apoptotic cell death.257,307 This resistance is attributable to an intrinsic ability to suppress virus replication.120,835 Mature motor neurons, infected by virulent strains of virus, die by a necrotic process and are not protected from death by Bcl-2 family member proteins.307,393 Autophagic clearance of viral proteins may promote neuronal survival.588
Persistent infection can occasionally be established in mammalian cell cultures in vitro. Mouse fibroblasts producing interferon (IFN), or BHK cells with a high concentration of defective interfering particles, can establish SINV persistent infection.345,869 Infection with SINV or SINV replicons that have mutations in the C-terminal methyl transferase–like domain of nsP2 results in reduced viral RNA synthesis, decreased CPE, and persistent infection in some vertebrate cell lines.7,187,233,235,507,605 Persistent infection can also be established if the cell infected is resistant to virus-induced apoptosis.90,449,824,835
Effects on Invertebrate Cells
Studies of alphavirus infection of cell lines derived from Aedes albopictus (e.g., C6/36, U4.4) and Aedes aegypti (e.g., Aag2) larvae demonstrate differences in alphavirus replication between vertebrate and invertebrate cells. The time course of virus replication is similar; however, frequently, virus matures within vesicular structures and virions are released by exocytosis rather than at the plasma membrane.523,763 Virions are relatively deficient in cholesterol and have detectable differences in structure.296,310,754 There is only a modest effect on host gene expression,225,696 and persistent noncytopathic infection, or death by a nonapoptotic process,383 is more likely than in vertebrate cells.
The uncloned lines derived from Ae. albopictus larvae contain many types of cells with properties representative of different mosquito tissues. Lytic infection occurs in clones that support high levels of virus replication,523,803 whereas persistent infection is associated with a short period of relatively high virus replication followed by a decrease in virus production and often in the numbers of cells in the culture that are producing virus.164,547,548 The decrease in virus production is not associated with activation of the signal transducer and activator of transcription (STAT), immune deficiency (IMD), or toll insect innate response pathways but is associated with decreased processing of the nonstructural polyprotein and expression of the protease inhibitor TEPII.225,547 Lytic infection can also be induced by viruses engineered to express death-inducing insect proteins such as reaper or michelob_x.849 Transcriptional analysis of persistently infected cells shows an increase in mRNAs associated with vesicle formation and the Notch signaling pathway.547
RNA interference (RNAi) is an important insect defense mechanism, and SFV infection of U4.4 or Aag2 cells leads to production of virus-derived small interfering RNAs (viRNAs) derived from replicative double-stranded (dsRNA) that are unevenly distributed across the genome and variable in efficiency for mediating antiviral RNAi.736 This RNAi signal can spread from cell to cell and inhibit replication.37 The RNAi pathway is defective in C6/36 cells74; however, the IMD, and not the toll innate response pathway, can suppress SINV replication in these cells.41
Superinfection Exclusion
Vertebrate and invertebrate cells infected with one alphavirus cannot be productively infected with the same, or a closely related, alphavirus at a later time. Exclusion is established after translation of the nsP genes of the first virus to enter. The superinfecting genome can be translated but not replicated,762 probably owing to the presence of the transacting nsP2 protease that prematurely cleaves the replicase polyprotein required for minus-strand synthesis.384
Antigenic Composition
All alphaviruses are related and share common antigenic sites, as revealed by HI and complement fixation (CF) tests with polyclonal immune sera107 and by cytotoxic T-cell lysis of infected cells.465,550 Antigenic cross-reactivities may confer some cross protection and interfere with sequential alphavirus immunizations.106,181,277,465,508 These cross-reactivities formed the basis for the original classification into the group A arboviruses and continue to be a valuable means for initial identification and classification of alphaviruses.107 Closely related viruses within a serogroup form a complex. Seven broad antigenic complexes have been identified within the alphavirus serogroup: Barmah Forest, eastern equine encephalitis (EEE), Middelburg (MID), Ndumu (NDU), Semliki Forest, Venezuelan equine encephalitis (VEE), and western equine encephalitis (WEE).427,626,806 A few viruses remain unclassified and will probably form one or more additional complexes (see Table 23.1). The Barmah Forest, EEE, MID, and NDU complexes each contain only a single virus, whereas the Semliki Forest, VEE, and WEE complexes include several viruses. Viruses within each complex can be subtyped using reactivity with MAbs, kinetic HI, or neutralization assays.98
Antibodies to E1 are more likely to cross-react with other alphaviruses than are antibodies to E2.67,343,892 This is
consistent with the documented sequence conservation in the E1 protein. Competitive binding assays using MAbs have identified approximately seven epitopes on the E1 glycoproteins of SINV, SFV, WEEV, and VEEV.67,343,519,662,703 Most E1 epitopes are not exposed on the virion surface but are present on the surface of infected cells or on acid-exposed virions.15,282,320,519,703 These transitional epitopes map to domain III in a region buried at the spike interfaces840 (Fig. 23.4). The in vitro biological activities of antibodies to E1 include HI, neutralization of virus infectivity, and inhibition of fusion.15,67,282,343,703 Neutralizing epitopes map to domains I, II, and III.282,455
consistent with the documented sequence conservation in the E1 protein. Competitive binding assays using MAbs have identified approximately seven epitopes on the E1 glycoproteins of SINV, SFV, WEEV, and VEEV.67,343,519,662,703 Most E1 epitopes are not exposed on the virion surface but are present on the surface of infected cells or on acid-exposed virions.15,282,320,519,703 These transitional epitopes map to domain III in a region buried at the spike interfaces840 (Fig. 23.4). The in vitro biological activities of antibodies to E1 include HI, neutralization of virus infectivity, and inhibition of fusion.15,67,282,343,703 Neutralizing epitopes map to domains I, II, and III.282,455
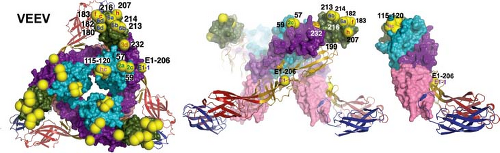 Figure 23.4. Neutralization escape mutations and positions affecting host range and tissue tropism mapped on the Venezuelan equine encephalitis virus spike. Neutralizing antibody escape mutations are displayed as yellow spheres on the spike with epitope written in the spheres. E1 is shown in ribbons and E2 in surface rendering using the colors defined in Figure 23.2. The left panel shows a top-down view of the spike, the middle panel shows the spike from the side, and the right panel shows the far right dimer from the middle panel. (Courtesy of Felix Rey; reproduced with permission from Voss JE, Vaney MC, Duquerroy S, et al. Glycoprotein organization of chikungunya virus particles revealed by x-ray crystallography. Nature 2011;468:709–712.) |
Antibodies to E2 are usually alphavirus specific. Competitive binding assays using MAbs identify four to five epitopes on the E2 glycoproteins of SINV, SFV, RRV, and VEEV.67,394,519,586,662 In vitro biological activities of antibodies to E2 include HI, neutralization of virus infectivity, and blocking of virus binding to the cell surface.662 Many anti-E2 MAbs have both neutralizing and HI activity, suggesting that these functions overlap.282 Neutralization escape mutants, naturally occurring variants, lgt11 expression libraries, site-directed mutagenesis, and recombinant viruses have been used to identify amino acids contributing to the various epitopes on E2 and have identified two major neutralizing sites704,758,851 that have been mapped onto the crystal structure of the E1-E2 heterodimer and trimerized spike and are in domain B8,282,367,455,840 (see Fig. 23.4). This is an exposed hydrophilic region that often includes an N-linked carbohydrate. There are linear, as well as conformational, determinants in this region because these MAbs frequently react in Western blots and recognize l fusion proteins, and antibodies to peptides from this region are protective against challenge.851
The second neutralizing epitope on E2 appears to be primarily conformational and is in domain A. This region is responsible for binding to HS and is obscured if pE2 is not cleaved.96,675,840 Visualization by cryo-electron microscopy of the binding of HS and MAbs to this epitope on SINV and RRV identifies the domain A knob on the glycoprotein spike.742,902
Evolution and Phylogeny
Alphaviruses, which replicate in arthropods, birds, reptiles, fish, and mammals, derive from a single unknown protoalphavirus and are part of the alphavirus superfamily of viruses. Viruses in this superfamily, including many RNA plant viruses, have a similar genetic organization and replicase proteins but diverse coat proteins.417,766 Among alphaviruses, amino acids important in secondary structure (e.g., cysteines and those close to one another in adjacent β sheets and a helices) have been conserved for the glycoproteins E1, E2, and E3, consistent with a similar three-dimensional structure of the virion for all.219 The most variable regions of the genome are in the C-terminus of nsP3 and the N-terminus of C.219 Highly conserved regions in the nsP1 and nsP4 genes have allowed development of primers to detect a broad range of alphaviruses by reverse transcription polymerase chain reaction (RT-PCR).201,290 Sequence information from the entire genome generally groups the viruses similarly to that derived by antigenic analysis (Fig. 23.5) and has detected at least one recombination event.297 Criteria for species demarcation of alphaviruses combine genetic, ecological, and antigenic information. Species generally have distinct transmission cycles and differ by more than 23% at the nucleotide level and 10% in amino acid sequence when E1 genes are compared.
The origin of the alphaviruses is unclear. Partial genome sequencing has suggested origins both in the Americas and in the Old World.107,267,434,626,859 Recently, comparison of whole genome sequences from all known alphaviruses has suggested an origin from the louse-borne aquatic alphaviruses219 (see Fig. 23.5). All of these scenarios require repeated movement across the globe to explain the current virus distributions.
Like other RNA viruses, alphaviruses undergo genetic change primarily by accumulation of point mutations in the genomic RNA; however, deletions and duplications also occur.1,217 Mutation occurs at a rate that is slower (1–7 × 10−4 substitutions/nucleotide/year) than is estimated for other RNA viruses,93,146 presumably because fitness must be maintained in both insect vectors and vertebrate hosts.147 Recombination
between alphaviruses can be demonstrated in vitro but is infrequent and usually puts the chimeric virus at a replicative disadvantage.93,146 However, successful recombination has occurred at least occasionally in nature because WEEV resulted from recombination between EEE- and Sindbis-like viruses (see Fig. 23.5) at the junction of E3 and E2, an event that is estimated to have occurred thousands of years ago.219,421,766
between alphaviruses can be demonstrated in vitro but is infrequent and usually puts the chimeric virus at a replicative disadvantage.93,146 However, successful recombination has occurred at least occasionally in nature because WEEV resulted from recombination between EEE- and Sindbis-like viruses (see Fig. 23.5) at the junction of E3 and E2, an event that is estimated to have occurred thousands of years ago.219,421,766
 Figure 23.5. Alphavirus phylogenetic tree produced using the E2, 6K, and E1 structural protein genes and Bayesian methods with midpoint rooting. The tree includes representatives from all alphavirus species, and the dashed line indicates the recombination between ancestors of Sindbis and eastern equine encephalitis viruses that led to the western equine encephalitis virus (WEEV) group. Roman numerals indicate major subtypes of some species, and scale indicates 50% nucleotide sequence divergence. All posterior probabilities were 1 except as indicated; nodes with a diamond had posterior probabilities less than 0.9, and nodes with a star had no posterior support. A similar tree utilizing full-length sequences without the WEEV recombinant group showed a similar topology except Middelburg virus was basal to chikungunya and o’nyong-nyong viruses, and Una, Mayaro, Semliki Forest, and Bebaru viruses were grouped separately together with Getah, Sagiyama, and Ross River viruses.219 (Courtesy of Scott Weaver; from Forrester NL, Palacios G, Tesh RB, et al. 2012. Genome-scale phylogeny of the alphavirus genus suggests a marine origin. J Virol 2012;86(5):2729–2738.) |
Alphaviruses replicate in and are transmitted horizontally by a wide range of invertebrate, primarily mosquito, species. However, each virus usually has a principal or preferred vector for the enzootic cycle. Most alphaviruses can infect various vertebrates but have birds, mammals, or fish as their primary amplifying and reservoir hosts (see Table 23.1). The specific invertebrate vector and vertebrate host used by an alphavirus will contribute significantly to determining the geographic distribution of that virus. Experiments modeling evolution in vitro show that fewer mutations accumulate if replication alternates between vertebrate and invertebrate cells, although diversity, fitness, and adaptability are greater with serial passage.149,150,153,276,856 Experiments employing in vivo passage show that serial passage in mosquitoes increases mosquito infection and that passage in vertebrates produces higher viremias; however, alternately passaged viruses do not change fitness.149 It is hypothesized that short transmission seasons and host mobility influence alphavirus genetic diversity and evolution in a geographic region.146,858 Viruses using avian enzootic hosts
(e.g., EEEV, WEEV, SINV) extend over wide geographic regions and evolve as a few highly conserved genotypes, whereas viruses using mammalian enzootic hosts with a more limited range of dispersal (e.g., RRV, VEEV) evolve within multiple geographically restricted genotypes.490,688
(e.g., EEEV, WEEV, SINV) extend over wide geographic regions and evolve as a few highly conserved genotypes, whereas viruses using mammalian enzootic hosts with a more limited range of dispersal (e.g., RRV, VEEV) evolve within multiple geographically restricted genotypes.490,688
Some strains of alphaviruses associated with epidemics or epizootics are antigenically and biologically distinguishable from enzootic strains. Phylogenetic evidence indicates that, at least for VEEV and WEEV, the virulent epizootic strains evolve by mutation from avirulent viruses being maintained in the enzootic cycle.23,61,629,657
Pathogenesis and Pathology in Vertebrates
Excellent and well-studied model systems exist for several alphaviruses, and much of our detailed knowledge about alphavirus pathogenesis comes from investigations in mice. Information from these models will be combined where appropriate with information from studies of humans with alphavirus-induced disease to deduce the pathogenesis of infection. Specifics will be covered in sections on the individual viruses.
Entry
The primary mode of alphavirus transmission to vertebrates is through the bite of an infected insect, most often a mosquito. Mosquitoes salivate during feeding and deposit virus-infected saliva extravascularly.819 Saliva virus titers are highest early after the mosquito is infected and decline, along with transmission rates, after 1 to 2 weeks; however, mosquitoes remain infected for life.525,833 The high-mannose glycans on virus from mosquitoes inhibits induction of IFN by myeloid DCs,718 and proteins in saliva further facilitate transmission by skewing the host cellular immune response toward Th2 cytokines.794
Sites of Primary Replication
The initial sites of virus replication vary with the virus and host. Mice have received the most extensive study. After subcutaneous inoculation, viruses may infect skeletal muscle or fibroblasts at the local site (e.g., EEEV, WEEV, SFV, RRV, SINV, and Getah virus) or be taken up by and infect Langerhans cells in the skin (e.g., VEEV)287,325,466,553 (Fig. 23.6). Langerhans cells and DCs transport virus to lymph nodes draining the site of inoculation that also may become infected.242,487 In vitro, human DCs are susceptible to infection with VEEV but not to infection with CHIKV or EEEV241,573,702,707,753; thus, the importance of DC infection after mosquito inoculation is likely to differ with the infecting virus.
Spread
Alphaviruses induce a substantial plasma viremia in their amplifying hosts and in hosts susceptible to disease (Fig. 23.7). The ability to mount and sustain a viremia depends on the continued efficient production of virus, delivery of virus into the vascular system, and slow clearance from the blood. Animal studies have shown that small plaque viruses are generally less virulent because they are cleared more rapidly from the circulation than are large plaque viruses.354,360,623 This phenomenon is related to the ability of small plaque viruses to bind HS and thus to be rapidly removed from the circulation by the highly sulfated glycosaminoglycans in the liver.97 Ability to invade target organs depends in part on the duration and height of the viremia but also on other characteristics of the virus important for tissue invasion.484
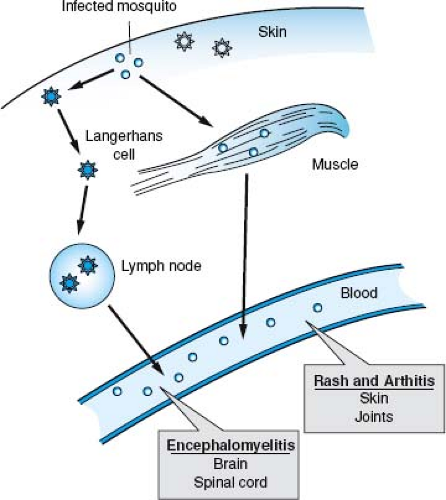 Figure 23.6. Basic steps in alphavirus infection of vertebrates. Virus is delivered extravascularly by an infected mosquito and infects local muscle cells or Langerhans cells in the skin. Langerhans cells can carry virus to local lymph nodes, where further replication may occur. Virus is delivered to the blood and spreads to target tissues such as skin, joints, and the central nervous system, in addition to distant muscle and lymphatic tissue. |
Cell and Tissue Tropism
Viruses that replicate initially in skeletal muscle and lymph nodes near the site of inoculation often spread through the bloodstream to more distant skeletal muscles and other lymphatic tissues. In addition, cardiac myocytes, osteoblasts, brain and spinal cord neurons, and brown fat cells are secondary sites of replication for many alphaviruses in mice9,466,535,554 (see Fig. 23.6). Getah virus causes polymyositis.325,554,713 EEEV, WEEV, SFV, and SINV cause encephalitis44,242,350,466,838; RRV and CHIKV cause myositis and arthritis244,543,907; and VEEV causes lymphoid depletion and encephalitis.260,359,845
In humans, the skin is a target for alphaviruses that cause a rash,230,493 the joints for alphaviruses that cause arthritis,229,328,543,772 muscle for alphaviruses that cause myalgia,543,590,907 and the nervous system for alphaviruses that cause encephalitis.245,760 RRV and SINV have been recovered from skin biopsies.423,424,493 RRV replicates in skin basal epidermal and eccrine duct epithelial cells.230 CHIKV is found in fibroblasts and macrophages.328,428 Human synovial cells support RRV infection in vitro,373 and RRV RNA is detected in synovial biopsy specimens.747 Joint fluid taken from humans with acute arthritis has
not yielded infectious virus; however, viral antigen and RNA can be detected in macrophages, and these cells support RRV and CHIKV replication in vitro.328,373,854
not yielded infectious virus; however, viral antigen and RNA can be detected in macrophages, and these cells support RRV and CHIKV replication in vitro.328,373,854
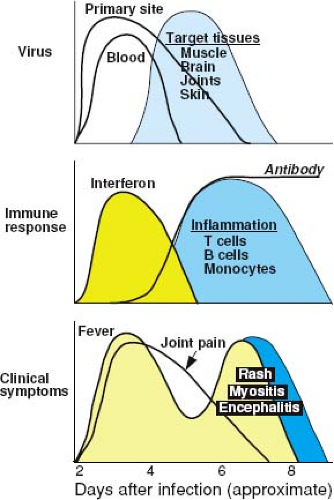 Figure 23.7. Schematic diagram of the pathogenesis of alphavirus-induced disease. Viremia may be accompanied by production of interferon, other proinflammatory cytokines, and fever. Virus then spreads through the blood to other target tissues. As the immune response is induced, the viremia is terminated; however, fever is renewed with appearance of a mononuclear inflammatory response in the infected tissue. In infections that lead to rash and arthritis, joint pain usually appears early after infection and prior to the appearance of the rash. |
The mechanism by which encephalitic alphaviruses enter the CNS is not entirely clear. Neuroinvasiveness is a component of virulence that varies between viruses and virus strains.188 Murine studies have shown infection or transport by cerebrovascular endothelial cells,396,602,748 infection of choroid plexus epithelial cells,466 infection of olfactory neurons,133,670,792,837 and transport by peripheral nerves.152 Once within the CNS, virus can spread cell to cell or through the cerebrospinal fluid (CSF).349,584,792 For most encephalitic alphaviruses, the targeted cell within the CNS is the neuron,350,466,584 where cellular protein synthesis is suppressed804 and damage can be severe and irreversible. In mice that recover from neuronal infection, infectious virus is cleared but viral RNA persists.223,446,521,821 SFV, RRV, and VEEV can cause persistent infection of microglial and oligodendroglial cells leading to demyelination.94,160,226,715
Immune Responses
Innate Responses
Early responses to alphavirus infection include production of cytokines and chemokines and activation of NK cells.271 Type I (a/β) IFN is induced in vivo after many, but not all, alphavirus infections of experimental animals242,255,358,721,807 and humans328,677,702,853 (see Fig. 23.7). The amount of IFN produced by infected tissues is usually linked to the level of virus replication, and IFN production continues if virus is not cleared.91,224,294,328,358,702 IFN rapidly appears in serum, and levels are diminished by splenectomy, suggesting that lymphoid tissue is one important source.358,415,416
Several different cell systems have been used to determine the mechanisms by which alphaviruses induce and control synthesis of IFN in response to infection. Viruses vary in their ability to induce IFN production by different types of cells,623,702,721 and the cellular sources of IFN probably differ with time after infection. In tissue culture cell lines, IFN production, shutoff of host protein synthesis, and CPE are controlled either by nsP2 (SINV, SFV) or C (VEEV, EEEV).14,81,235,246,247 In these cells, shutoff of host gene expression suppresses antiviral responses.14,81,247,894
The primary source of early IFN in vivo may be plasmacytoid DCs for some alphaviruses730 but not for others.702 SFV induction of IFN by myeloid DCs requires fusion but not replication, and it is independent of MyD88.322 For SINV and RRV, N-linked glycans on E2 are important determinants of IFN production by DCs.719
Induction of IFN requires viral entry and RNA synthesis.57,322,881 Data from ts mutants suggest that formation of dsRNA is the necessary step in replication for IFN induction. Viruses with mutations in the protease domain of nsP2, which cannot process the nonstructural polyprotein and thus cannot initiate plus-strand RNA synthesis, do not induce IFN.298,497 In infected fibroblasts and epithelial cells, dsRNA and higher-ordered RNA structures activate MDA5 and PKR, which are important intracellular sensors of alphavirus infection,49 and stimulate phosphorylation of interferon regulatory factor (IRF) 3610,706 and formation of the IRF3/CBP/p300 transcriptional activation complex for immediate early IFNs.57 This process is independent of mTOR pathway activation.175 NsP1/nsP2 cleavage affects IRF3 activation and IFN induction by SINV without affecting shutoff of host transcription or translation.158 Virulent strains of EEEV do not induce IFN after fibroblast infection, whereas attenuated strains do, which is potentially associated with decreased binding to HS and increased infection of lymphoid tissue.242,243
Production of IFN follows the initial release of virus from infected cells by 2 to 3 hours327 and for some, but not all, alphavirus strains is regulated by nuclear importation of nsP2 and by whether host protein synthesis is shut off before IFN can be synthesized.81,92,881 The ability of CHIKV to induce IFN mRNA and protein is cell type dependent.702 Primary human monocytes infected by CHIKV produce IFN-a, interleukin (IL)-6, and IL-12319; however, similarly infected primary human fibroblasts produce IFN-β mRNA but no protein, because the mRNA is not translated.881
IFN is an important part of the host response to alphavirus infection, and virus replication is sensitive to its effects.12,155,172,441,680,718,753,787,905 Animals can be protected from lethal infection if treated with IFN or IFN inducers before or soon after infection.12,358,375,477,714 Animals unable to respond to IFN owing to deletions of the IFN receptor or crucial IFN signaling molecules develop more severe disease than wild-type mice.12,95,155,157,210,224,279,679,702,756,880 Furthermore, absence
of an IFN response allows virus replication in cells previously resistant to infection.224,678,679,810 IFN appears to act primarily to limit virus replication early, during the time the specific immune response is being induced (see Fig. 23.7). Treatment of cells with IFN inhibits alphavirus replication90,172,358,540,551,681; however, the mechanism by which this occurs, and therefore the IFN-induced host responses important for control of replication, are not known. Attachment and entry are not affected,172,641 although later replication steps, including formation of replication complexes, structural protein synthesis, and morphogenesis, are inhibited.551,641,787
of an IFN response allows virus replication in cells previously resistant to infection.224,678,679,810 IFN appears to act primarily to limit virus replication early, during the time the specific immune response is being induced (see Fig. 23.7). Treatment of cells with IFN inhibits alphavirus replication90,172,358,540,551,681; however, the mechanism by which this occurs, and therefore the IFN-induced host responses important for control of replication, are not known. Attachment and entry are not affected,172,641 although later replication steps, including formation of replication complexes, structural protein synthesis, and morphogenesis, are inhibited.551,641,787
In addition to inhibiting host cell protein synthesis, alphaviruses can also interfere with IFN signaling by decreasing Janus kinase (JAK) activation and STAT phosphorylation and nuclear translocation.236,733,894 This is a property of either nsP1 or nsP2.236,734
Antiviral proteins PKR and RNase L have a limited role in the IFN-induced antiviral response in vitro and in vivo.681 However, activation of PKR improves the stability of IFN mRNA.49,706 Interestingly, infected ribonuclease (RNase) L–deficient fibroblasts fail to shut off minus-strand RNA synthesis or form stable replication complexes and establish persistent infection, suggesting a role for RNase L in virus replication.698
IFN-induced proteins that can inhibit alphavirus replication include human MxA,432 zinc-finger antiviral protein (ZAP),63,292,392,435,488,905 viperin,905 the large form of 2′,5′-oligoadenylate synthetase,82 interferon-stimulated gene (ISG) 20,905 and ISG15.440,441 Transgenic expression of MxA, a large cytoplasmic guanosine triphosphatase (GTPase), in IFN-α/β receptor-deficient mice results in decreased SFV replication by preventing accumulation of genomic and subgenomic RNA and provides some protection against fatal disease.311 ZAP is an RNA-binding protein that prevents accumulation of viral RNA by blocking translation of incoming viral genomic RNA.63,291 This is accomplished by binding to specific viral mRNA sequences and interaction with the host DEAD box helicase p72 and RNA-processing exosome.135,292 ISG15 is an ubiquitin-like molecule that exerts its antiviral effect by conjugating proteins, although the mechanism for suppression of virus replication remains unknown.251
Viruses and virus strains vary in their sensitivity to the antiviral activities of IFN, and this may or may not correlate with virulence.24,68,174,756,894 Mutations associated with altered sensitivity to IFN have been mapped to the 5′ nontranslated region (NTR), nsP1, and nsP2.235,667,756,880
IFN may also contribute to alphavirus-induced disease. Fever during the viremic phase of infection, as is seen with CHIKV and RRV, is probably a response to the IFN induced early after infection (see Fig. 23.7). It has been postulated that the rapidly fatal disease induced by alphaviruses in newborn mice may be owing to the production of large amounts of IFN and pro-inflammatory cytokines.807 Acute-phase responses induced by alphaviruses prior to the virus-specific immune response include up-regulation of toll-like receptor expression and increases in tumor necrosis factor (TNF), IL-1, and IL-6, and levels generally correlate with the extent of virus replication.278,512,702,807,874 Adult mice deficient in IL-1β have reduced mortality after CNS infection with a neurovirulent strain of SINV, again suggesting that cytokine effects may contribute to mortality.457
Virus-Specific Adaptive Responses
Both cellular and humoral immune responses are induced by infection (see Fig. 23.7). In experimentally infected adult mice, antiviral antibody is usually detected in serum within 3 to 4 days after infection.280,592,713 The cellular immune response, manifested by the presence of virus-reactive lymphocytes in draining lymph nodes and blood and the infiltration of mononuclear cells into infected tissues, also appears within 3 to 4 days after infection.283,509 These responses appear later (7–10 days after infection) in neonatal mice.721 Both appear to play a role in recovery from infection and protection against reinfection.
Humoral Immunity
Virus-specific immunoglobulin M (IgM) antibody is detectable very early in human disease, often provides a means for rapid diagnosis of infection, and can persist for many months after recovery.69,99,103,112,115,288,424,495,571 Virus-specific immunoglobulin A (IgA) also appears early in infection but declines rapidly.114 Immunoglobulin G (IgG) antibody is present in serum after 7 to 14 days and is maintained at relatively high levels for years.101,183,396 Many lines of evidence support the hypothesis that recovery from alphavirus infection depends in large part on the antibody response.91,284,906 Rapidity of antibody synthesis is predictive of outcome from encephalitis because patients without evidence of antibody at the time of illness onset are most likely to die.99 Antibody can neutralize virus infectivity and promote virus clearance by the reticuloendothelial system in conjunction with complement.356 Appearance of antibody correlates with cessation of viremia (see Fig. 23.7).
The most extensive experimental studies to define the antibody specificity and the mechanisms of antibody-mediated recovery and protection have been done using VEEV, SFV, and SINV infection of mice. Passive transfer of antibody before or after infection is protective. Both neutralizing and nonneutralizing MAbs against multiple epitopes on the E1, E2, and E3 glycoproteins can protect against alphavirus challenge and promote recovery.65,66,289,343,502,519,597,757,892 There is a correlation of protection with the ability of the MAb to bind to the surface of infected cells, although this is not absolute.757 Protection requires intact bivalent antibody but does not require complement.323,503 However, virus clearance from blood is delayed in complement-deficient mice.324
Treatment of immune deficient mice persistently infected with SINV or SFV with antiviral antibody clears infectious virus from the CNS without causing neurologic damage.21,448 Clearance of infectious virus is rapid, whereas the decline in viral RNA occurs more slowly.448,521 E2-specific MAbs can down-regulate intracellular virus replication in vivo and in vitro by a nonlytic mechanism.448 Antibody against an N-terminal peptide of VEEV E2 that is not neutralizing can limit virus replication in vivo,342 and a nonneutralizing MAb to SFV E2 can limit virus replication in vitro.66 Anti-E3 MAbs inhibit production of VEEV.597 Anti-E1 MAbs may also be able to alter intracellular virus replication.128
In vitro studies show that the process by which antibody alters intracellular virus replication requires bivalent antibody but does not require the Fc portion of the MAb, complement, or other cells448,824; however, the effects of antibody are amplified by treatment of infected cells with IFN-α.172 Soon after antibody binding, virion budding from the plasma membrane is inhibited.173 In vivo studies also show that IFN and antibody act
synergistically to promote recovery, although the mechanisms by which these systems interact have not been identified.95,154
synergistically to promote recovery, although the mechanisms by which these systems interact have not been identified.95,154
After recovery from encephalitis, viral RNA remains detectable in the CNS for life. Therefore, one consequence of a nonlytic mechanism for clearance of virus from tissue is that the virus genome is not completely eliminated if the originally infected cells survive.446,447 This leads to a need for long-term control of virus replication that is accomplished in part by infiltration of antibody-producing B cells into the CNS.281,286,521,821
Cellular Immunity
Alphavirus infection induces virus-specific lymphoproliferative, cytokine, and cytotoxic T-cell responses.4,283,385,498,531 Cytokines increased in plasma during acute disease include IL-4, IL-6, IL-10, IL-12, IL-13, and IFN-γ.328,853 After epidermal virus inoculation, Langerhans cells increase expression of MHC class II antigens and accessory and costimulatory molecules that enhance activation of naive T cells.370 The mononuclear inflammatory process in alphavirus encephalitis is immunologically specific509 and includes infiltration of NK cells, CD4+ and CD8+ T lymphocytes, B cells, and macrophages.346,521,529,541 Relative proportions of these mononuclear cells vary with the time after infection.346,521,529 T cells play a role in virus clearance and in protection from challenge.200,897 Viral RNA levels in the CNS of SINV-infected mice decrease more rapidly when CD8+ T cells are present.400 Mice lacking the ability to produce antibody can clear infectious virus from some populations of neurons through production of IFN-γ,64,84 and IFN-γ down-regulates SINV replication in mature neurons in vitro through a JAK/STAT-dependent mechanism.89,90 In animals infected with virulent strains of virus, cellular immune responses contribute to tissue damage and fatal disease.273,399,563,669
Pathologic Changes
Encephalomyelitis
Pathologic changes in the CNS of humans with fatal neurologic disease and mice with experimentally induced encephalomyelitis begin with perivascular infiltration of mononuclear and polymorphonuclear inflammatory cells.509,529,577 Adhesion molecules (e.g., ICAM-1, VCAM-1) are up-regulated on endothelial cells and integrins LFA-1 and VLA-4 are important mediators of mononuclear cell entry.347,741,750 This phase of infection may include extravasation of red blood cells and endothelial cell swelling and hyperplasia.577 Lymphocytes and monocytes move from the perivascular regions to areas of the parenchyma with virus-infected neurons. This inflammatory process is accompanied by gliosis and evidence of inflammatory and glial cell apoptosis.245
Neonatal mice and human infants may die with widespread virus-induced neuronal cell death before the inflammatory process—a manifestation of the cellular immune response—can be initiated.555 Immature neurons die by an apoptotic process,452 whereas death of mature neurons may be characterized by cytoplasmic swelling, vacuolation, membrane breakdown, and cellular degeneration suggesting necrosis.245,307,555 Demyelination has been described as a consequence of EEEV and WEEV infection in humans52,576,577 and of WEEV, RRV, and SFV infection of mice, probably as a result of infection of oligodendrocytes.94,532,715
Reticuloendothelial Infection
The pathology of VEE in horses includes cellular depletion of bone marrow, spleen, and lymph node tissue, and pancreatic necrosis.405 Small mammals also develop widespread infection of reticuloendothelial system tissues and may develop ileal necrosis.40,266,845 Leukopenia is commonly observed during human infection.534
Arthritis
In CHIKV- and RRV-induced arthritis, there is hyperplasia of the synovial lining, vascular proliferation, and mononuclear cell infiltration.328,747 Synovial fluid contains increased protein, CD4+ T lymphocytes, activated NK cells and macrophages, and increased levels of monocyte chemotactic protein (MCP)-1/CCL2, IL-6, and IL-8.145,229,328 Persistent infection is suggested by the presence of RRV RNA 5 weeks after onset of symptoms747 and CHIKV antigen and RNA in synovial macrophages 18 months after acute disease in a patient with chronic arthritis.328
Rash
Skin biopsies taken from patients with RRV-induced rash show perivascular infiltration of lymphocytes (primarily CD8+ T cells) and monocytes without evidence of immune complex deposition.230
Release and Transmission
A common feature of alphaviruses is their transmission by insects and maintenance in a natural cycle of replication in vertebrate and invertebrate hosts. Arthropod vectors become infected by feeding on a viremic host, are able to transmit the virus 4 to 10 days later (external incubation), and remain persistently infected. Maintenance of this cycle requires an amplifying host that develops a viremia of sufficient magnitude to infect feeding mosquitoes. For many alphaviruses, humans are dead-end hosts unable to infect mosquitoes efficiently. However, human-mosquito-human transmission has been important in epidemics of RRV, o’nyong nyong virus (ONNV), and CHIKV-induced polyarthritis,478,689,790 and horse-mosquito-horse transmission is important in epizootics of VEE.659,771
Other modes of transmission are occasionally important. Horses infected with VEEV may shed virus in nasal, eye, and mouth secretions, as well as in urine and milk, resulting in the potential for transmission by the respiratory route.118,405 Aerosol transmission of VEEV, CHIKV, and Mayaro virus has occurred in laboratory settings,118,378,439,788,799 and aerosolized VEEV has been developed as an agent of biological warfare.728 EEEV persists in the feather follicles of infected pheasants, and transmission among penned pheasants can occur through feather picking and cannibalism.697 Person-to-person transmission has not been documented.439,659
Veterinary Correlates and Animal Models
WEEV, EEEV, and VEEV—the first alphaviruses to be cultured—came to the attention of virologists because they caused fatal disease in horses; these viruses remain important equine pathogens.182,522,786 EEEV and WEEV cause encephalitis
in horses, whereas VEEV causes severe respiratory disease associated with leukopenia; encephalitis may or may not be present. Getah virus causes an urticarial rash and hind leg edema in horses.717 WEEV, EEEV, and Highlands J virus (HJV) cause disease in domesticated birds such as chickens, pigeons, pheasants, turkeys, and emus.42,167,213,221,476,822,868 The alphaviruses associated with arthritis in humans have not been recognized as important causes of disease in domestic animals.
in horses, whereas VEEV causes severe respiratory disease associated with leukopenia; encephalitis may or may not be present. Getah virus causes an urticarial rash and hind leg edema in horses.717 WEEV, EEEV, and Highlands J virus (HJV) cause disease in domesticated birds such as chickens, pigeons, pheasants, turkeys, and emus.42,167,213,221,476,822,868 The alphaviruses associated with arthritis in humans have not been recognized as important causes of disease in domestic animals.
Good small animal models exist for the encephalitogenic alphaviruses but are less satisfactory for study of the arthritogenic alphaviruses. In mice, alphaviruses generally infect lymphatic tissue, muscle, brown fat, brain, and spinal cord; however, the extent and relative importance of infection at these sites differs among these viruses. For instance, RRV and Getah virus cause primarily myositis, VEEV causes reticuloendothelial infection, and WEEV and EEEV cause encephalitis with neurons as the main target cells.9,526,554 In mice infected with relatively avirulent strains of SFV, RRV, and VEEV, the acute encephalitic phase is accompanied by infection of oligodendroglial cells and demyelination.94,715 For all alphaviruses, fatal disease in mice is usually associated with CNS infection even if encephalitis is not a manifestation of the human infection. For instance, SINV infection of mice is studied as a model for acute viral encephalitis, although SINV causes arthritis and rash, not encephalitis, in humans.349,489,820 Specifics of these animal model systems are discussed with the individual viruses.
Virulence
Virulence is a measure of the ability of the virus to cause fatal disease; for alphaviruses, this usually reflects the severity of neurologic disease. Outcome is influenced by characteristics of both the host and the virus. An early virus determinant of virulence is induction of IFN and susceptibility to IFN-mediated inhibition of replication. Viruses that induce IFN and are susceptible to IFN are generally attenuated.12,92,158,235,243,734,756,880 Most alphaviruses show an age-dependent susceptibility to disease.9,75,294,554,584,713 Resistance increases with maturation and is associated with decreased virus replication in tissues at the site of virus inoculation and in target tissues (e.g., brain), not with changes in induction of IFN or the ability of infected mice to mount a virus-specific immune response.280,287,554 The ability of a virus strain to cause fatal disease or a particular complication of infection also often depends on the genetic background of the host186,759,792,814; however, the specific genetic determinants of susceptibility are just beginning to be identified.793 Avirulent alphavirus strains may replicate poorly even in newborn animals, whereas virulent strains can usually replicate well and cause disease in adult and newborn animals. The role of the response to IFN is unclear, although older mice increase ISG12 whereas young mice do not.429
For encephalitic alphaviruses, another viral determinant of virulence is their ability to enter the CNS efficiently (neuroinvasiveness). Many alphavirus strains can cause fatal disease after intracerebral or intranasal inoculation but not after subcutaneous or intraperitoneal inoculation. The duration of viremia often correlates with virulence with virulent strains sustaining longer viremias than avirulent strains.355,357,360 Peripheral replication, viremia, neuroinvasiveness, and neurotropism (ability to replicate in CNS cells) all contribute to virulence.
The viral determinants of virulence have been most extensively studied in murine models for SINV, SFV, RRV, and VEEV infections. Viruses with altered virulence have been selected after chemical mutagenesis,48,85 by passage in tissue culture,58,166,395,783 by passage in mice,284,517,783 by isolation of MAb escape mutants842 or plaque variants,354 and by manipulation of complementary DNA (cDNA) virus clones.166,485,779,815 Nucleotide and amino acid changes affecting virulence have been mapped to the 5′ NTR, nsP1, nsP2, nsP3, E1, and E2. Virulence determinants in the glycoproteins map to receptor-binding regions of the E2 A and B domains840 (see Fig. 23.4). Specifics are covered in the sections dealing with each of these viruses.
Persistence
There is substantial evidence that alphaviruses can persist after appearance of an immune response and clearance of infectious virus from the circulation and from tissue.447 Pathologic examination of CNS tissue from human cases of progressive WEEV months to years after resolution of acute encephalitis has shown an active inflammatory process.575,577 Viral RNA and proteins can be detected in the nervous system long after recovery of mice from SINV or SFV-induced encephalitis and for several weeks in the joints of humans with RRV-induced arthritis.186,285,396,447,521,747,821 It is postulated that this persistence of RNA is attributable to failure of the virus or the immune system to eliminate the infected cells. Interestingly, passive antibody protection predisposes to persistent infection and the late onset of progressive disease.399,712
Congenital Infection
Alphaviruses can be transmitted transplacentally. This has been documented in mice for RRV, SFV, VEEV, and Getah virus3,30,35,524,755 and in humans for RRV, WEEV, VEEV, and CHIKV.6,231,380,723 In mice, the virus infects the placenta, where it is able to persist and spread to the fetus despite the development of maternal antibody. The outcome of fetal infection depends on the timing of infection relative to transfer of maternal antiviral IgG to the fetus. Fetuses are protected if transfer occurs prior to infection; however, transfer of antibody after fetal infection does not mediate recovery.524 In monkeys, congenital infection with VEEV induces malformations of the brain and eye.471 In humans, no abnormalities were observed in infants infected with RRV at 11 to 19 weeks gestation; however, earlier infection may lead to fetal death.6 Epidemics of VEE are associated with increases in spontaneous abortion.659,862 No effect on pregnancy outcome was identified during the CHIKV outbreak on Reunion Island.231
Pathogenesis and Pathology in Mosquitoes
The ability of alphaviruses to infect mosquitoes efficiently with spread to and replication in the salivary glands is essential for maintaining the natural cycle of transmission. Not all mosquitoes taking a blood meal from a viremic host will become infected, and not all infected mosquitoes develop the ability to transmit virus. Many alphaviruses preferentially infect a narrow range of mosquito species, and this host specificity plays an important role in determining the geographic distribution of the virus. Even within a species, strains of mosquitoes may vary in
susceptibility to infection. Ae. albopictus collected from different geographic regions show differences in susceptibility to infection with CHIKV and in the amount of virus produced after infection.789 Field and laboratory populations of Culex tarsalis differ in susceptibility to WEEV.334 Strains of virus also differ in their abilities to infect mosquitoes, and laboratory-adapted strains may establish infection relatively inefficiently.557,710,711,831,833
susceptibility to infection. Ae. albopictus collected from different geographic regions show differences in susceptibility to infection with CHIKV and in the amount of virus produced after infection.789 Field and laboratory populations of Culex tarsalis differ in susceptibility to WEEV.334 Strains of virus also differ in their abilities to infect mosquitoes, and laboratory-adapted strains may establish infection relatively inefficiently.557,710,711,831,833
The extrinsic incubation period—or the time between taking an infected blood meal and ability to transmit infection—depends on the rapidity of virus replication and dissemination to the salivary glands. This period is relatively short (2–7 days) for alphaviruses compared to other arboviruses.710
Entry and Sites of Primary Replication
Posterior midgut epithelial cells are the initial sites of infection,77,558,612,708,864 and infection is facilitated when virus in the serum is concentrated next to these cells as the blood meal clots866 (Fig. 23.8). Susceptibility of mosquitoes to alphavirus infection is determined in large part by the ability of the virus to infect midgut epithelial cells.334,397,710 Changes both in the virus and vector can affect this interaction.889 WEEV rapidly fuses with microvillar membrane preparations from Cx. tarsalis mosquitoes, and both WEEV and CHIKV bind better to membranes from susceptible mosquitoes than to membranes from refractory mosquitoes,334,546 which is consistent with a role for viral structural proteins as determinants of vector specificity, midgut infection, and dissemination.612,813,832,833 The high-affinity laminin receptor A is a receptor for VEEV and SINV on the surface of C6/36 larval Ae. albopictus cells,474,850 and proteins of 60 and 38 kDa have been identified as putative receptors for CHIKV on brush-border membranes from Ae. aegypti.546
Host cell membrane cholesterol levels affect the efficiency of alphavirus entry.473 Mosquitoes, like other insects, do not synthesize cholesterol and obtain sterols needed for reproduction and development from dietary blood. Cholesterol-independent mutants of SFV replicate better than parental SFV in cholesterol-depleted C6/36 cells and in adult Ae. albopictus mosquitoes.16
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


