

Since the last WHO classification of 2004, there have been many additions to our knowledge about renal neoplasms (23). A number of entities that were included among other subtypes earlier have been recognized as distinct clinicopathologic entities now. Thus, clear cell papillary RCC, many cases of microphthalmia-associated transcription factor (MiTF) family translocation–associated RCC, tuberous sclerosis–associated RCC, and multilocular cystic RCC tumors, all extracted from clear cell RCC group, are now considered as tumors distinct from it. Similarly, hereditary leiomyomatosis RCC (HLRCC) tumors, some cases of MiTF family translocation–associated RCC, clear cell papillary RCC, collecting duct carcinoma, and acquired cystic disease–associated RCC are some examples of tumors many of which were considered to be papillary RCC in the past. Succinate dehydrogenase B (SDHB) and Birt-Hogg-Dubé (BHD) syndrome–associated tumors are some examples of tumors separated from oncocytoma/chromophobe/oncocytic unclassified RCC group. Most of these extracted entities have biologic behaviors quite different from their originators. Therefore, recognition and proper classification of these “new” entities is pathologically and clinically essential.
With the publication of results on molecular investigations by the National Institutes of Health (NIH)–sponsored “The Cancer Genome Atlas” (TCGA) on clear cell RCC (24) and those of similar studies from Japan (25) and other parts of the world (26,27) in the past 2 to 3 years, it has become obvious that even the post-2004 clear cell RCCs are not a uniform group of renal tumors (28). Many tumors that were regarded as clear cell RCC in fact do not belong to this category, and others, even that are regarded as clear cell RCC as of now, show significant differences from the bulk of cases within clear cell RCC group at the molecular and morphologic levels. It is potentially quite likely that ongoing TCGA studies on papillary and chromophobe RCC will yield somewhat corresponding results, adding to our understanding, as well as requiring further modifications in the classification of renal tumors in the near future.
EPIDEMIOLOGY
RCC accounts for approximately 2% of all cancers (20,29). Its incidence varies among countries, with the highest rates being in North America and Scandinavia (30). They occur twice as frequently in men as in women, and in the United States, the incidence is equal among whites and blacks (29). At the same time, the proportion of different subtypes of RCC tends to be biased towards papillary phenotype among African Americans, compared to clear cell RCCs among Caucasians (31). In 2013, approximately 51,200 new cases of kidney and renal pelvic cancers were diagnosed in the United States, with close to 13,000 deaths reported (32). The disease resulted in more than 100,000 yearly deaths worldwide.
RCC may occur at any age, with peak incidence in the sixth and seventh decades of life. The incidence has increased substantially over the last two decades, accompanied by an improved 5-year survival. Undoubtedly, both trends are at least in part due to improved diagnostic techniques and early detection. Over the last 15 to 20 years, we have seen a very significant decrease in tumor size and pathologic stage among resected renal cancers at our institution (33,34). In spite of these facts, up to 30% of patients with RCC present with metastatic disease, and recurrence develops in approximately 40% of patients treated for a localized tumor. The median overall survival in patients with metastatic disease is approximately 12 months (35).
The better understanding of molecular genetics of renal tumors in the recent past has resulted in progress in the management of renal epithelial neoplasms. Until recently, interleukin-2 and interferon-α were considered the most reliable therapeutic options for metastatic clear cell RCC, with disappointing outcomes at the best (35). Molecular studies have now shown that normal von Hippel-Lindau (VHL) gene product, pVHL, is required to target and degrade hydroxylated hypoxia inducible factor (HIF) in normoxemic states. In cells that are hypoxic, or show absence of/abnormal pVHL (as in virtually all cases of VHL syndrome and most cases of sporadic clear cell RCC), HIF-1α escapes degradation, is overexpressed, and activates many downstream molecules, including vascular endothelial growth factor, glucose transporter 1 (GLUT1), carbonic anhydrase IX (CA-IX), and epidermal- and platelet-derived growth factors (EDGF and PDGF), among others. Similarly, mammalian target of rapamycin (mTOR) pathway activation has been shown to interact with the HIF pathway to produce excess of HIF, resulting in its overexpression. Some of the aforementioned downstream molecules are believed to have a role in tumor initiation and progression. Targeted therapies against these molecules in recent years have shown promising and much better results than previous best available therapeutic options in metastatic RCC (35). At the same time, the diffuse and strong expression of some of these downstream molecules at immunohistochemical level is now being used as diagnostic marker for clear cell RCC, including its sarcomatoid variants (36–38). More recently, a significant proportion of clear cell RCCs have been shown to harbor mutations in epigenetic regulators located close to the VHL gene (39–41), with some clinical and morphologic implications. How these findings will affect the management of clear cell RCC remains to be determined at this time. Modern immunotherapy promises to improve disease free survival even further.
Environmental factors play a significant role in the development of renal cancer (42–44). Cigarette smoking doubles the risk of RCC (45,46). At the same time, in a more recent study, although smoking history of 20 pack years or greater was associated with a significantly increased risk of advanced disease, it did not achieve an independent association after adjusting for age and gender (47). Obesity, particularly in women, is also a risk factor (42,48,49). Other risk factors include hypertension; estrogen therapy; and occupational exposure to petroleum products, heavy metals, and asbestos (42,46,49,50). A case control study based on statistics from the New Zealand Cancer Registry demonstrated an increased relative risk for development of renal cancer in firefighters and painters, both occupational groups likely to be exposed to known carcinogens (51).
HEREDITARY RENAL TUMORS
The majority of renal neoplasms are sporadic, although a small percentage may be hereditary, such as those clear cell (conventional) carcinomas associated with VHL syndrome (52–66). In almost all forms of inherited renal neoplasms, tumors are diagnosed at an earlier age and are more likely to be multifocal and bilateral (67). HLRCC syndrome, as of now, appears to be the only exception with most, although not all, cases reported to have solitary, unilateral tumors. VHL disease is an autosomal dominant syndrome characterized by retinal hemangiomas, clear cell (conventional) RCCs, multiple renal cysts, cerebellar and spinal hemangioblastomas, pheochromocytomas, endocrine pancreatic tumors, and epididymal clear cell cystadenomas. The tumor suppressor VHL gene is located at chromosome 3p25 and may be inactivated by various mutations, loss of heterozygosity, promoter hypermethylations, or alterations in VHL modifier genes. Some of the gene products related to lack of pVHL are shown to play a role in tumorigenesis. Recently, histone-modifying and chromatin-remodeling genes located at chromosome 3p21—PBRM1, SETD2, BAP1, and KDM5C—have been found to be mutated in clear cell RCC. These genes are located in close proximity to VHL and along with VHL are commonly lost (in ~90% of sporadic clear cell RCC with 3p losses). A recent addition to familial RCC syndromes is the germline mutations in BRCA1-associated protein-1 (BAP1) gene. These families are characterized by the presence of uveal melanoma, cutaneous melanoma, and mesothelioma and have been reported to have early-onset clear cell RCC in some members (68,69). Other familial non-VHL clear cell RCCs have been reported, most of which involve translocations of chromosome 3 including 3p14 (fragile histidine triad [FHIT]), 3q13.3, and 3q21 genes, whereas a few others do not involve chromosome 3 (67,70).
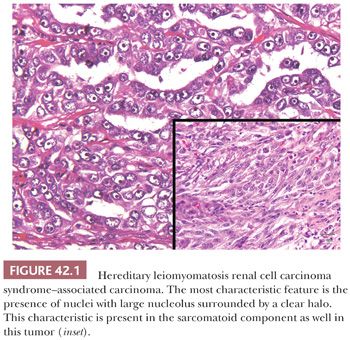
Hereditary forms of RCCs showing papillary architecture are sometimes associated with tumors of the breast, pancreas, lung, skin, and stomach. One of these syndromes is associated with activating mutations of c-MET proto-oncogene at chromosome 7q34 (71). Upregulation of the gene results in the activation of a heterodimeric membrane-spanning MET tyrosine kinase, which is a receptor for hepatocyte growth factor/scatter factor, and is involved in angiogenesis, cellular motility, growth, invasion, and cellular differentiation. The renal tumors with trisomy 7 in this syndrome have been shown to harbor c-MET mutants in two of the chromosome copies (67). The papillary RCCs associated with syndromic c-MET mutations are all described to be of type 1. The kidneys in this syndrome show numerous, bilateral type 1 papillary tumors, often manifesting at relatively older age (most in 50 to 70 years range). There seems to be no other systemic manifestations of this syndrome at present. HLRCC syndrome is an autosomal dominant familial syndrome with mutation in the fumarate hydratase (1q42.3-q43) gene and is characterized by the development of cutaneous and uterine leiomyomas and less commonly renal tumors. Fumarate hydratase deficiency prevents conversion of fumarate to malate in the Krebs cycle, increasing the intracellular levels of fumarate. The increased levels of fumarate act as competitive inhibitor of the prolyl hydroxylases required for hydroxylation of HIF. Nonhydroxylated HIF does not interact with pVHL and is overexpressed, leading to the consequences similar to that in clear cell RCC genesis (72). HLRCC renal tumors are clinically very aggressive and often show metastases at presentation, even when quite small in size. Many renal tumors in this syndrome show a high-grade papillary phenotype and were regarded as type 2 papillary RCC in the past. However, other architectural patterns, including tubular, solid, cribriform, and admixture of variable patterns are often present (73,74). The most diagnostic and characteristic feature of the tumors, irrespective of the architectural pattern, is the presence of large nuclei with very prominent orangeophilic or eosinophilic nucleoli, surrounded by a clear halo (Fig. 42.1). The designation of HLRCC-associated RCC rather than papillary type 2 RCC or collecting duct carcinoma appears more appropriate (23).
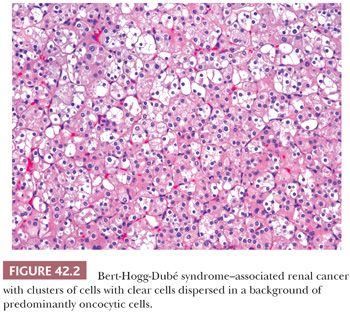
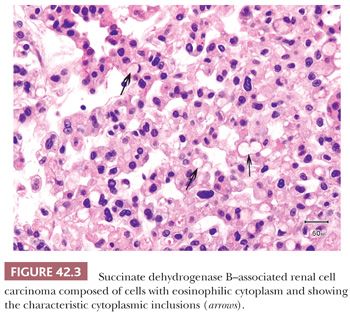
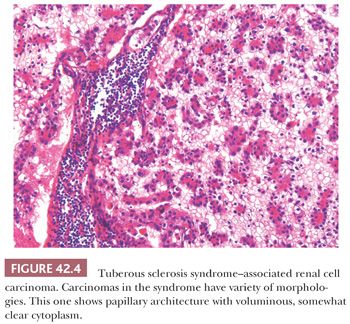
BHD is another syndrome associated to multifocal and bilateral renal tumors. This autosomal dominant syndrome is characterized by cutaneous lesions (fibrofolliculomas, trichodiscomas, and acrochordons), spontaneous pneumothorax, bronchiectasis and bronchospasm, colonic neoplasms, and lipomas, which occur in conjunction with the renal tumors (75,76). The renal tumors have variably been reported as renal oncocytomas or chromophobe RCC. However, in our experience, these tumors are predominantly oncocytic neoplasms displaying hybrid features of both types or tumors with some unusual morphologic features (Fig. 42.2). A study on a large number of renal tumors from patients with BHD syndrome revealed similar findings (77). The BHD gene has been mapped to chromosome 17p12-q11.2 and it codes for the protein folliculin (76). Folliculin is known to form a complex with its binding proteins, folliculin-interacting protein 1 (FNIP1) and FNIP2, which activates 5-adenosine monophosphate-activated protein kinase (AMPK). AMPK is normally known to downregulate mammalian target of rapamycin complex 1 (mTORC1) and mTORC2 proteins. Mutations in BHD gene remove this downregulatory effect on mTOR pathway, ultimately resulting in overexpression of HIF-1α (68). Of the families identified with familial renal oncocytoma, a number of these have subsequently been found to have BHD syndrome (67,70). Rarely, a constitutional reciprocal translocation between 8q24.1 and 9q34.3 has been reported in cases of bilateral, multifocal renal oncocytoma (78). Renal tumors arising in another familial condition, SDH gene mutations, particularly of SDHB, are also predominantly oncocytic and are characterized by large clear or eosinophilic cytoplasmic inclusions (Fig. 42.3) (79). Although initially believed to be a less aggressive form of familial renal cancer, recently, a number of tumors developing metastases have been reported (80). SDH deficiency prevents conversion of succinate to fumarate in the Krebs cycle, increasing the intracellular levels of succinate. The increased levels of succinate cause competitive inhibition of prolyl hydroxylases, similar to that due to fumarate excess in HLRCC syndrome, resulting in overexpression of HIF (68). Tuberous sclerosis complex (TSC) syndrome is characterized by inactivating mutations in the tumor suppressor genes TSC1 or TSC2, which encode the proteins hamartin and tuberin, respectively. The protein products are essential parts of the mTOR pathway and both genes act as tumor suppressors. Tuberin and hamartin proteins form a heterodimer, which inhibits mTOR activity. Lack of mTOR inhibition by these proteins ultimately results in accumulation of HIF-1α and increased expression of HIF-responsive genes, including vascular endothelial growth factor (68). Renal tumors arising in TSC are mostly angiomyolipomas, but rarely, RCCs may be also seen. Although the reported histology of TSC-related renal epithelial tumors has been varied, including clear cell, chromophobe, and unclassified RCC and oncocytoma, in our experience, they seem to have varying but characteristic morphologic features, which only superficially resemble the more common tumor types (81). These morphologic features include often an admixture of architectural patterns including nests, tubules, solid sheets, and variable papillary areas. The cytology ranges from exclusively clear cells with voluminous cytoplasm traversed by cobweb-like cytoplasmic strands to purely eosinophilic cytoplasm with variably sized pink granules (Fig. 42.4). However, most tumors show an admixture of clear and eosinophilic cells in varying proportions (81).
Recently, RCCs with ALK mutations have also been described. Whether they have a familial basis still needs to be determined (82).
GRADING AND STAGING OF RENAL TUMORS
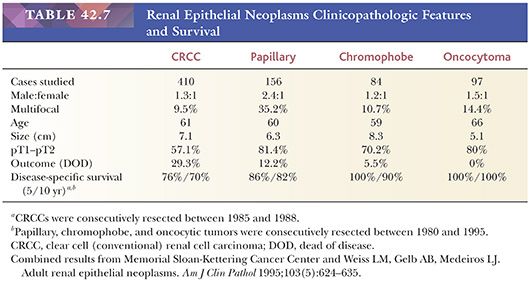
Several systems over the years have been proposed for the grading of renal tumors. Of these, the one proposed by Fuhrman and colleagues (83–88) has been the most widely used. This system uses nuclear grades based on nuclear size, irregularity of the nuclear membrane, and nucleolar prominence. Although we and others have demonstrated good correlation between Fuhrman nuclear grade and survival in clear cell carcinomas, application may be cumbersome because it requires the pathologist to measure nuclear size (87). Even though the identification of nucleoli at 400× and 100× is frequently used to differentiate between grades 2 and 3, the overall interobserver variability has been significant (87,88). The use of the Fuhrman or any other grading scheme in papillary and chromophobe cell carcinomas remains controversial. Whereas Amin and colleagues (89) report use of this scheme as a strong predictor of survival in papillary RCC on univariate analysis, we and others have not found any significant correlation between the nuclear grade and survival (90,91) nor did Amin and colleagues (89) on multivariate analysis. Similarly, Fuhrman nuclear grading appears to be inappropriate for chromophobe RCC (92), as it very frequently shows marked nuclear irregularities (grade 3 nuclei) in spite of the overall excellent survivals. Although a modified Fuhrman grading was proposed and reported to be useful by Paner et al. (93), we and others did not find any predictive significance of this or similar grading scheme on multivariate analyses in three large cohorts of chromophobe RCC (94–96). There is no reason to grade renal oncocytomas because they are benign tumors.
More recently, a different “nucleolar grading” scheme has been proposed to represent a better grading system for renal tumors, particularly clear cell and papillary RCC (97,98), although its use in papillary RCC was questioned in one study (99). In a more recent study on a large number of cases, taking both the microscopic tumor necrosis and nucleolar grading into account, a modified grading scheme has been reported to be a superior predictor of clinical outcome in clear cell RCC but not papillary or chromophobe RCCs (97).
In 1969, Robson and colleagues (100) published a staging scheme, which was adopted by most clinicians and pathologists; it was shown to correlate well with survival. During the ensuing years, the AJCC in collaboration with the UICC published the tumor-node-metastasis (TNM) classification in which stage I and II tumors were confined to the kidney but the former measured 2.5 cm or less (101). This classification was modified in 1997, and even further modifications were made in 2002 and 2010 (102) (Table 42.4). Undoubtedly, any of these staging schemes offer valuable prognostic information but only within specific tumor types. For example, large size and extracapsular extension does not have the same prognostic significance in cases of oncocytoma or even chromophobe carcinomas that it has in clear cell RCC. Up to 20% of renal oncocytomas may present with pT3 disease even though they are a benign tumor. The mean tumor size of chromophobe carcinomas is possibly the largest of all renal cortical neoplasms, and yet the 5-year disease-free survival in virtually all published series is greater than 90% (94,103). We recommend that tumors be classified using the latest TNM classification in an attempt to establish uniformity in reporting, although future editions will surely correct deficiencies in the present system (102,104).
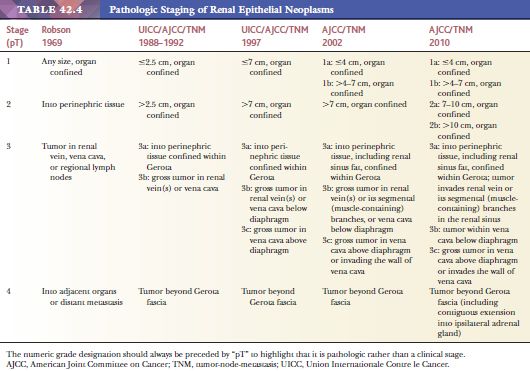
After a series of publications highlighted the significance of the renal sinus and sinus vein invasion (105,106), these were for the first time included in the AJCC staging system in 2002; involvement of muscular branches of renal vein in the renal sinus was assigned the same pT stage as the renal vein invasion, that is, pT3b. Some studies performed thereafter support the rationale for equating sinus vein invasion with renal vein involvement (107). In the 2010 AJCC staging scheme, similar pathologic stage for the sinus vein invasion and renal vein invasion has been retained, but both were downstaged to pT3a (102). Thus, a careful gross examination and adequate sampling of the renal sinus–tumor interface, especially in tumors 4 cm or more in maximum diameter, is strongly recommended in both the radical and partial nephrectomy specimens (Fig. 42.5). This is primarily based on the observations after meticulous dissection of the renal sinus by Dr. Bonsib (106), who showed that 67% of clear cell RCCs between 4 and 7 cm in size and 97% of those above 7 cm in diameter show sinus vein and/or sinus fat invasion.
IMMUNOHISTOCHEMISTRY
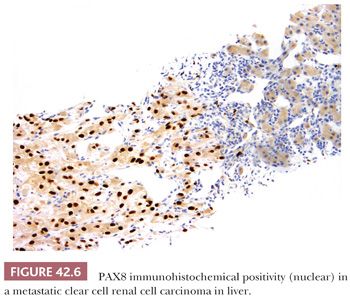
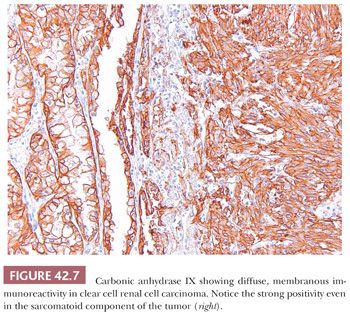
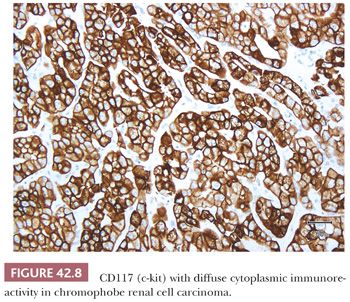
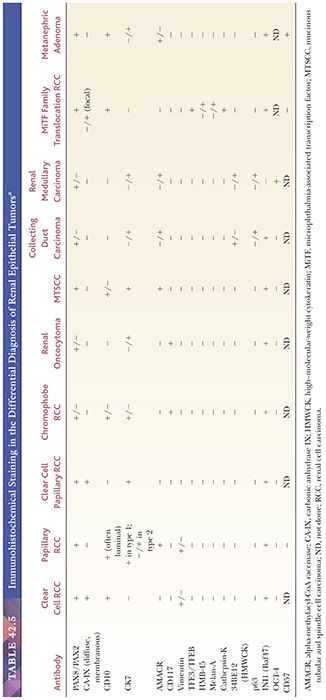
Immunohistochemistry has the potential for use to differentiate between different subtypes of primary renal epithelial neoplasms or to confirm the renal origin in a metastatic tumor. Numerous antibodies have been used and reported to be of use in both these situations (108–117a) (Table 42.5). In our experience, many of these antibodies are able to discriminate between better differentiated tumor subtypes. However, such tumors are also easily classifiable on light microscopic evaluation. In situations where the antibodies are in fact really required, that is, in high-grade or poorly differentiated tumors, the results tend to be inconsistent and unreliable. Among the more recent antibodies, PAX8/PAX2 and CA-IX appear to be most useful in determining the renal origin and in the differential diagnosis of renal cell tumors (36,37,38). PAX8 and PAX2 show diffuse nuclear immunoreactivity in most—but not all—primary and metastatic renal cell tumors (Fig. 42.6) (117a–123). Differentiation from female genital tumors should always be considered from a clinical perspective. CA-IX shows diffuse and strong membranous reactivity in more than 90% of clear cell RCC, compared to absent or at the most focal, usually perinecrotic, positivity in other common subtypes of renal cell tumors (36,38). Interestingly, this diffuse reactivity is retained even in the sarcomatoid components of most cases of clear cell RCC (Fig. 42.7) (37). Other antibodies that we have found of practical use in the differential diagnosis of renal cell tumors include c-kit (CD117), which consistently stains chromophobe RCC (Fig. 42.8) and renal oncocytoma in a diffuse manner, whereas most other tumors, even those with eosinophilic cytoplasm, are either negative or only focally positive (38,124). We also find diffuse positivity for cytokeratin (CK) 7, as in a large proportion of cases of chromophobe RCC, quite useful in their distinction from renal oncocytoma.
Depending on the subtype and the stage of the renal tumor, as well as its expected biologic behavior, clinical comorbidities, etc., multiple therapeutic options tailored to an individual patient are now being offered. Among others, such options include in situ ablation of the tumor, targeted therapies in a neoadjuvant setting, or even watchful waiting in selected cases. In view of these developments, availability of a robust and dependable panel of immunohistochemical stains has become even more important, as pathologists are frequently asked to render diagnosis on limited material. Using a select panel of five antibodies—CA-IX, CK7, c-kit, racemase, and CD10—we have reported greater than 90% diagnostic accuracy on in vivo needle core biopsies of renal cell tumors. This fact indicates that if pathologists are provided with adequate biopsy samples, close attention to cytomorphologic features, together with a judicious use of immunohistochemistry, will allow for an accurate diagnosis in the overwhelming majority of the cases (124,125).
CLEAR CELL (CONVENTIONAL) RENAL CELL CARCINOMA
Clear cell carcinomas comprise approximately 60% of renal tumors (126) (Table 42.6). Although they are still the most common of all renal cancers, over the past few years, clear cell RCC as a proportion of all renal cell tumors has shown a declining trend. This is mainly the result of identification and separation of multiple subtypes of RCC, which were previously included within the clear cell RCC category. As a consequence of TCGA and other recent extensive genomic studies (24,25), it is potentially likely that some additional tumors may be extracted out of the clear cell RCC category in near future.
Clear cell RCCs are characterized by the loss of genetic material of the short arm of chromosome 3 (3p) and mutations in VHL gene (Table 42.2) (4,14,24,25,64,65,127,128). In patients with VHL syndrome, such losses and mutations are described in virtually all cases. Losses of 3p/somatic mutations/hypermethylations in the same region are found in an overwhelming majority of the more common sporadic tumors as well (24,25,129). Histone-modifying and chromatin remodeling genes located at chromosomes 3p21, PBRM1, SETD2, BAP1, and KDM5C have also been commonly found to be mutated in sporadic clear cell RCC. These genes are located in close proximity to VHL and together with VHL are mutated or lost in greater than 90% of sporadic clear cell RCCs (24–28). Sato et al. (25) recently reported another mechanism of VHL inactivation in clear cell RCC through mutations in the TCEB1 gene that encodes for elongin C; the gene is located at 8q21.11. Mutations in the gene abolish elongin C–pVHL binding, which in turn prevents the binding of pVHL to HIF, therefore hampering the proteasomic degradation and consequential overexpression of HIF.
CLINICAL FEATURES
Males are affected more commonly than women in a ratio of 1.7 to 2.0:1 with a peak incidence during the seventh decade of life. In our series of 155 patients, age at the time of presentation ranged from 34 to 90 years with a mean of 61 years. There are several case reports of these tumors arising in children and young adults, although this is a rare occurrence. Hereditary clear cell RCCs tend to arise at an earlier age, usually in the fourth or fifth decade of life, and are more likely to be bilateral and multifocal. In sporadic disease, both kidneys are affected equally and most tumors are solitary. Bilateral disease was documented in only 3.2% of the cases in our series. Multifocality, defined as a collection of neoplastic clear cells regardless of size, is present in approximately 11% of the cases.
The most common symptoms at the time of presentation are hematuria, abdominal pain, and a palpable mass, seen in 50% to 60%, 40%, and 30% to 40% of cases, respectively (130). A combination of all three components of the “classical triad” occurs in less than 10% of cases. Other signs and symptoms are fairly nonspecific and include fever, night sweats, general malaise, and weight loss. Rarely, clear cell RCC may present with features of erythrocytosis due to the erythropoietin gene expression and production of erythropoietin by the tumor (131). At present, greater than two-thirds of the cases are diagnosed after the detection of an asymptomatic, incidental renal mass using modern imaging techniques (132–135).
GROSS FEATURES
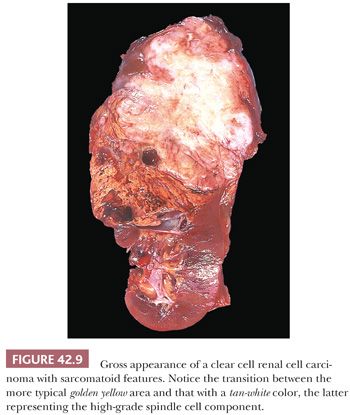
Grossly, the classic case has a golden yellow color due to abundant intracytoplasmic lipid (Fig. 42.9). Higher grade tumors contain less lipid and glycogen and show a more variegated appearance including areas of hemorrhage and necrosis (Fig. 42.9). In our series, the main tumor mass ranged in size from 1.8 to 21 cm with a mean size of 8 cm. Approximately 12% showed marked cystic degenerative changes and renal vein invasion was grossly identified in 30%. Slightly more than half of the cases presented with disease extending outside the kidney (pT3 or higher). It is important to note that modern imaging techniques and early detection has brought about a significant stage migration in renal cancer. Clear cell RCC resected at our institution during the years of 1995 to 1997 had a mean tumor size of 5.5 cm, 2.5 cm less than the series quoted earlier, which included tumors resected between the years 1983 and 1985. At present, approximately 70% of the resected tumors at our institution are 4 cm or less in size (136). We documented significant clinical and pathologic downstaging as well (33,34).
MICROSCOPIC FEATURES
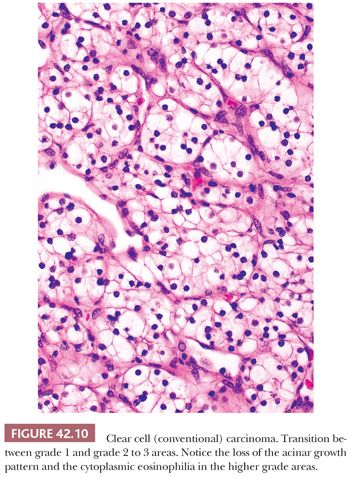
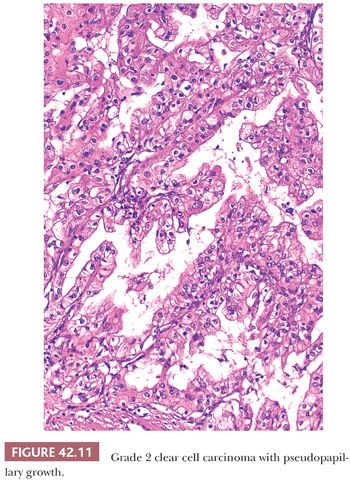
Microscopically, approximately 50% of cases exhibit exclusive acinar growth pattern (Fig. 42.10). They are characterized by solid acinar structures or nests of tumor cells, separated by delicate, intricately branching fibrovascular septa. In some cases, the acinar pattern is less well defined, yet the prominent vascularity is retained. In the remaining cases, these tumors contain a mixture of growth patterns including solid sheetlike, cystic, papillary/pseudopapillary, tubular, and sarcomatoid. A rich capillary vascular network is prominent in all but the solid sheetlike and sarcomatoid areas. Growth pattern is loosely associated with grade because most low-grade lesions have an acinar growth, whereas higher grade areas are more likely to exhibit solid, pseudopapillary, rhabdoid, or sarcomatoid features (Fig. 42.11). It is imperative to prosect tumors properly, making sure that all visually disparate areas are sampled, as well as sampling areas of transition and areas that are golden yellow. Tumors exhibiting a pseudopapillary growth pattern, which must not be mistaken for the true papillary growth pattern, are characterized by pseudopapillae lacking a true fibrovascular core. They are often due to degenerative changes in areas of solid and/or acinar growth. True papillations, when present in clear cell RCC, are usually very focal. Presence of more than focal papillary architecture in association with clear cell cytology should raise other differential diagnostic possibilities, including clear cell papillary RCC and MiTF family translocation–associated RCC, among others, irrespective of immunohistochemical positivity for CA-IX. Focal fibrosis or hyalinization due to degenerative changes is seen in more than 75% of the cases; however, true desmoplasia is absent or minimal. Geographic necrosis and focal hemorrhage are common histologic findings as well. These tumors may contain an inflammatory infiltrate, which is predominantly lymphocytic. Calcifications and osseous metaplasia are rare findings, seen in less than 10% and 4%, respectively.
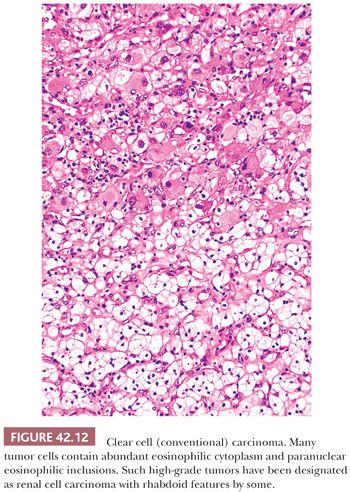
In most cases, clear cell RCC tumor cells are either polygonal or cuboidal in shape. Cell shape is more variable in higher grade lesions. Fuhrman grade 4 lesions contain bizarre, pleomorphic cells, some of which mimic the giant cells seen in some chromophobe cell tumors and angiomyolipomas. The spindle cells seen in sarcomatoid areas are an evidence of high-grade disease. The majority of tumors show a mixture of cytoplasmic features, namely clear or granular eosinophilic cytoplasm. Again, the cytoplasmic features can be correlated with tumor grade because low-grade lesions invariably contain clear cytoplasm, whereas the cytoplasm of higher grade lesions is more likely to be variably eosinophilic and granular (Fig. 42.12). Some grade 3 and 4 lesions may lack clear cytoplasm altogether.
Fuhrman nuclear grading system correlates with tumor stage and reliably stratifies patients into prognostic groups (83,85,137). However, as discussed before, measurements of nuclear size renders it “user-unfriendly.” In addition, grading schemes that group cases into four rather than two or even three categories are usually hampered by marked interobserver variability (87). In our experience, slightly less than 80% of cases are classified as grade 2 or 3 tumors with the remainder split between grades 1 and 4. As discussed earlier, the recently proposed nucleolar grade may be a more objective and reproducible grading system for these tumors (97,98). However, additional studies on a larger number of cases with input from a wider range of pathologists is required to replace the current, popular Fuhrman grading scheme. Mitotic activity is not a prominent feature of these tumors with very few cases exhibiting more than five mitoses per 10 high-power fields. The majority of tumors are not encapsulated, although 10% to 15% may be partially or completely surrounded by a fibrous pseudocapsule. The number of cases with renal vein invasion, extension into perinephric fat, and regional lymph node involvement has decreased dramatically over the last 15 years due to stage migration secondary to early detection. At the same time, in our experience, we are recognizing many more tumors with invasion of large branches of renal vein in the renal sinus (pT3a), mostly but not always in tumors greater than 4 cm in diameter. This finding becomes more apparent particularly if more attention is paid to the renal sinus by careful gross examination and submitting abundant sections from the parenchymal–sinus fat junction, more so in larger tumors even when these appear cortical in location (106). Tumors, particularly those that are large and high grade, may exhibit areas of either macroscopic or microscopic necrosis. Some authors have suggested that the presence of necrosis is a feature of poor prognosis (138,139). As a matter of fact, microscopic tumor necrosis has been proposed as an additive prognostication factor to nucleolar grade in a recent study on a large number of tumors that included 3017 clear cell RCCs (98).
Clear cell RCCs are usually immunoreactive with CK CAM 5.2 as well as epithelial membrane antigen (EMA) and often show diffuse membranous positivity with CA-IX. We have found EMA to be more sensitive and to stain a larger proportion of cells particularly in higher grade tumors. CD10 is positive in a membranous distribution, whereas CK7 is generally negative except in occasional cells. But CK7 may be positive in the clear cell lining cystic areas as well as in tumor cells entrapped in areas of sclerosis (140,141). α-Methylacyl-coenzyme A racemase (AMACR) is usually negative, as in HMB-45, A-103, and synaptophysin.
DIFFERENTIAL DIAGNOSIS
Some tumors may be confused with papillary or chromophobe RCC, although microscopic examination of all available areas should reveal areas with classic histologic features. In most cases of clear cell RCC with papillary growth, this growth pattern occurs as a result of cell drop-off in which the tumor cells situated away from the blood supply die and those close to the vessels are preserved, thus exhibiting a pseudopapillary growth pattern. Histiocytes are unlikely to be present within the fibrovascular “stalk” in these pseudopapillary areas. Intracellular hemosiderin deposition is absent as well. Papillary carcinomas often show diffuse expression of CK7, whereas conventional tumors do not. Among the exceptions to such expression used to be clear cell papillary RCC; however, it is now regarded as a separate distinct entity (23). Chromophobe RCCs have characteristic nuclear and cytoplasmic features, which are rarely seen in conventional tumors and then only focally. Diffuse cytoplasmic positivity for Hale colloidal iron is a hallmark of chromophobe tumors, although this stain may be extremely difficult to perform and interpret. Electron microscopy is a more reliable means to resolve the differential diagnosis but not in every case. Ultimately, cytogenetics and molecular genetics can resolve problems in classification in virtually all cases because all three lesions are associated with distinct genetic abnormalities (Table 42.2). Epithelioid variants of angiomyolipomas can be readily misclassified as clear cell RCC and are usually assigned a high nuclear grade. The problem is compounded by the fact that some angiomyolipomas lack the morphologic diversity seen in classic cases. Close attention to morphologic features, immunohistochemistry, electron microscopy, and even cytogenetic properties can help establish the correct diagnosis. The usual immunoreactivity with CK CAM 5.2 as well as EMA, and often the diffuse membranous positivity with CA-IX (Fig. 42.7), as seen in clear cell RCC, is not seen in angiomyolipoma. On the other hand, the latter stain for HMB-45 and smooth muscle actin. Some clear cell RCCs are so poorly differentiated that metastatic disease may enter into the differential diagnosis. Knowledge of the patient’s prior medical history will help in this regard. Another entity to be considered in the differential diagnosis is adrenal cortical carcinoma, particularly in the case of upper pole tumors. Adrenal cortical tumors are not immunoreactive with EMA and rarely stain with CKs; they express inhibin and A-103 (MART-1), which are negative in renal carcinomas.
OUTCOME
Disease-free and overall survival correlates with grade and stage (84,86,136). In our consecutive series of 155 cases accrued between 1983 and 1985 and with a median follow-up of 62.5 months, 38% of patients died of disease. Similar outcomes have been reported by other investigators (142,143). Although clear cell RCCs are the most virulent of all common types of renal cortical carcinomas, we would expect the survival statistics to improve as tumors are detected earlier (Table 42.7). Effective systemic therapy has remained elusive, making surgical resection the best chance for cure. However, targeted therapies against the molecules of VHL/HIF and more recently mTOR pathways have shown promising results. Although durations of survival have increased with these targeted therapies, improvements in overall survivals have been disappointing.
MULTILOCULAR CYSTIC RENAL CELL CARCINOMA
Multilocular cystic RCC represents a rare variant of clear cell (conventional) renal carcinoma (144). They constitute between 3% and 6% of clear cell RCC (145–149). The mean age at diagnosis is 51 years. None of the reported cases have progressed (23,150–152). Because of its indolent biologic behavior, 65% of the participants at the International Society of Urological Pathology (ISUP) renal consensus conference at Vancouver favored changing the name to multilocular cystic renal cell neoplasm of low malignant potential (23). Recently, it has been shown that these tumors are associated with 3p deletions and the silencing of the VHL gene similar to that in conventional clear cell carcinomas (144). The gross appearance is that of a well-circumscribed, multicystic mass which is separated from the adjacent renal parenchyma by a fibrous pseudocapsule (Fig. 42.13). Size can be quite variable but may reach 13 cm in greatest dimension. The cysts are variable in size, may contain serous or bloody fluid or clot, and are separated by thin fibrous septa. No solid or expansile masses of tumor are present anywhere.
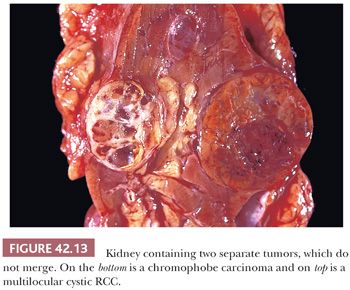
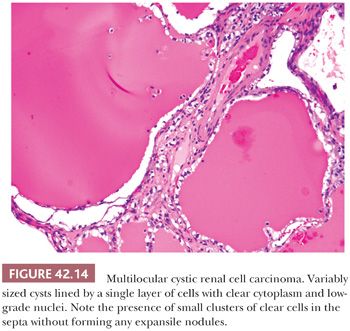
Microscopically, the thin fibrous septa are lined by one or several layers of cells, although in many areas, the epithelial lining may be absent. Foamy macrophages may also line the cyst wall. Occasionally, the tumor cells exhibit papillary tufting into the cyst lumen. The tumor cells contain clear cytoplasm and Fuhrman grade 1 or 2 nuclei. Small collections of tumor cells are present within the fibrous septa (Fig. 42.14) or in the adjacent pseudocapsule, but no expansile or solid masses of tumor are evident. In cases where the epithelial lining is not readily apparent, immunohistochemical stains for EMA, pankeratins, or CA-IX should help in its identification. One must be aware that conventional clear cell RCCs may be partially cystic and contain thin fibrous septa virtually identical to what is seen in multilocular cystic RCC. The key to the correct diagnosis is the presence of expansile masses of clear cells either adjacent to or within the cystic component. There is no agreement as to the size of the expansile mass required to classify a tumor as conventional clear cell carcinoma. However, in order to maintain the correlation to benign clinical outcome, we do not accept any expansile mass in tumors classified as multilocular cystic RCC, irrespective of its size. Occasionally, a clear cell RCC may be almost entirely composed of cystic epithelial component, with hyalinization and scarring in the broad septa in between. We and others do not regard such tumors as multilocular cystic RCC, as these most likely represent clear cell RCCs with partial regression and cystic change (153).
The tumor cells are diffusely positive for CA-IX in a membranous distribution, similar to conventional clear cell RCC (140,141). CD10 is often negative or focally positive, with diffuse staining in only one-fourth of the tumors. Although AMACR is negative, it is common to see focal to diffuse staining for CK7. CK 34βE12 is generally negative although an occasional cell may be immunoreactive (140). HMB-45, A-103, and synaptophysin are negative. CA-IX and CK7 positivity can raise the differential diagnostic possibility of predominantly cystic clear cell papillary RCC (see the following section). However, the CA-IX staining pattern (boxlike) is different from that in clear cell papillary RCC (cuplike). Clear cell papillary RCC also shows the characteristic linear arrangement of nuclei (see the following section), which is not present in multilocular cystic RCC.
CLEAR CELL PAPILLARY RENAL CELL CARCINOMA
Clear cell papillary RCC was initially described by Tickoo et al. (154) in the setting of end-stage kidneys, although we now know that they do occur without evidence of impaired renal function (155–158). A recent publication claims it to be the fourth most common subtype of RCC in nephrectomy specimens (158). At the recent Vancouver ISUP renal consensus conference, it was recognized as one of the five new distinct epithelial tumors within the renal classification system (23). Although originally considered to be a morphologic variant of conventional clear cell carcinoma, the tumor shows no evidence of 3p25.3 losses, VHL gene mutations, or promoter hypermethylations, supporting its being a distinct entity (155–157). Additionally, polysomy of chromosome 7 is not seen, as would be expected in many papillary RCCs. These tumors are usually unifocal, unilateral, and small; however, the largest described tumor measured 6 cm in diameter. Multifocality and bilaterality may be present in some cases, particularly in end-stage kidneys (154). Multifocality and bilaterality could also raise the differential diagnostic possibility of VHL syndrome, in which clear cell papillary–like tumors may be seen in association with usual clear cell RCCs. Williamson et al. (159) recently report that such tumors in VHL syndrome are different from the nonsyndromic tumors at the level of immunohistochemistry, as well as the molecular profile.
GROSS FEATURES
Grossly, they are well circumscribed and usually encapsulated, although the capsule may be of variable thickness. It is common for the lesion to be cystic or partially cystic and partially solid. Areas of apparent hyalinization are common.
MICROSCOPIC FEATURES
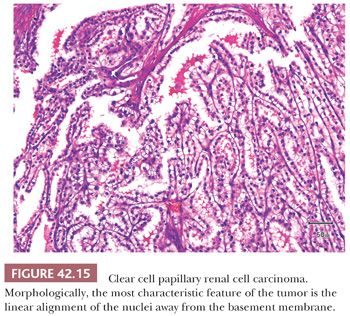
Microscopically, the tumor cells have uniformly clear cytoplasm and low-grade nuclei. The cells are cuboidal and arranged in tight tubular/acinar structures, particularly in the tumor-bearing solid areas. If the tumor cells have minimal cytoplasm (collapsed acini), these areas can have a solid, sheetlike appearance. Invariably clear cells also line small, tightly packed papillary structures. In other cases, the tubules show branching and infoldings, imparting an apparent papillary architecture. Sometimes these papillary structures are tufting into cystic spaces, which are also lined by clear cells. Characteristically, the tumor nuclei are arranged in a linear fashion, away from the basal aspect of the cell, either in the center of the cell or close to the apical aspect (Fig. 42.15). Stromal hyalinization and myoid metaplasia may be evident within the lesion. Tumor necrosis and vascular invasion are not the features of these tumors; rare cases extending into the renal sinus have been described. Although the number of cases in the literature with extended clinical follow-up information is limited, no cases with metastasis have been described, overall suggesting that these are most likely indolent tumors.
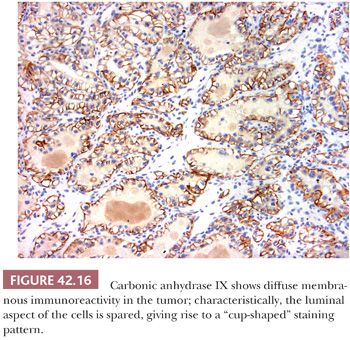
The immunohistochemical features on this tumor are quite characteristic. Tumor cells express CA-IX diffusely in a membranous distribution. However, it is common to see absence of staining along the luminal border of the tumor cells (cup-shaped distribution) (Fig. 42.16) (156,160). There is diffuse positivity with CK7, in almost 100% of the tumor cells, but racemase is negative. CD10 is negative in most cases, whereas it is common to see patchy of even diffuse immunoreactivity for 34βE12. HMB-45, A-103, and synaptophysin are negative.
The differential diagnosis includes cystic nephroma/mixed epithelial stromal tumor (MEST), multilocular cystic RCC, and benign multilocular cyst of the kidney. Close attention to the epithelial lining and the fibrous cyst wall should lead to the correct diagnosis. Cysts in cystic nephromas/MEST are usually lined by a single layer of flattened or hobnail epithelium, which only occasionally contains clear cytoplasm. The fibrous stroma may be more cellular, resembling ovarian stroma. The septa may contain small tubules lined by bland epithelial cells, reminiscent of renal tubules. Occasional clear cell RCC may also contain foci with nuclear arrangement similar to that in clear cell papillary RCC. However, in addition to such histology being only focal, these tumors show CA-IX and CD10 positivity of classical clear cell RCC. CK7 is also negative or at the most only focally positive.
PAPILLARY RENAL CELL CARCINOMA
Papillary renal cell carcinoma (PRCC) is the second most common subtype of RCC after clear cell RCC (8,21,142,143,161,162). Detailed morphologic features of the entity were first provided in a study of 34 cases published in 1976 by Mancilla-Jimenez and colleagues (163), although earlier clinical studies had suggested specific clinical characteristics for these tumors (164). The term chromophil renal carcinoma has been used to describe these tumors, but papillary renal cell carcinoma is the most widely accepted term, primarily based on the growth pattern in the majority of tumors.
Cytogenetic and molecular studies have revealed distinct findings in PRCC that distinguish them from other renal epithelial tumors (10,13). The majority of sporadic PRCCs are characterized by trisomy of chromosomes 7 and 17 as well as loss of chromosome Y (3,7,21,155,156,165,166,167). Some investigators have suggested that tumors exhibiting trisomy 7/17 only are likely to be benign, whereas those tumors exhibiting additional genetic abnormalities will behave aggressively, a hypothesis that has not been substantiated in the literature. Approximately 10% of the sporadic PRCC have also been reported to show somatic mutations in c-MET gene, a genetic abnormality that is commonly seen as a germline mutation in familial cases (168).
Although PRCC have distinct clinical and cytogenetic features, its histomorphologic features are still a subject of controversy. Although true papillae with a fibrovascular core lined by neoplastic epithelial cells is the hallmark of this tumor, the amount of papillary component needed to classify a tumor as a PRCC is not uniformly agreed upon.
CLINICAL FEATURES
PRCCs are reported to comprise 11% to 20% of primary epithelial renal neoplasms (7,169,170). In our institution, they represented 15% of renal epithelial neoplasms resected over a 10-year period. Age at presentation ranges from the third to eighth decade of life with peak incidence in the sixth and seventh decades, similar to other tumor types. An increasing number of cases present as incidental masses discovered during workup for unrelated causes; like other common renal cell tumors, more than 50% of cases present as incidental masses, often detected on radiologic investigation for unrelated conditions (171,172). Metastases at presentation are uncommon, unlike some other primary renal tumors that also may show prominent papillary architecture (173). The male-to-female ratio ranges from 2:1 to 4:1 (165,169,170,174,175). The majority of patients present with unilateral tumors, although PRCC quite often is bilateral as well as multifocal; multifocality has been reported in up to 49% of cases in the older literature (165,169). However, such studies likely included the smaller than 5 mm lesions (which are currently regarded as papillary adenomas) for determination of multifocality. Size of the dominant tumor mass can range from 1.0 cm to 23.0 cm (163,165,169,175). In the series from Memorial Hospital, the median size was 6.4 cm (90). Grossly, visible areas of necrosis and hemorrhage are common, being present in 32% to 70.5% of the tumors (163,170). Of all renal epithelial tumors, PRCCs are the most likely to be surrounded by a fibrous pseudocapsule. The reported incidence of gross cystic change is quite variable, ranging from 9% to 70% (163,169).
GROSS FEATURES
Tumors appear as a discrete mass within the renal cortex. Greater than a third of tumors are surrounded by a fibrous pseudocapsule on gross evaluation. Most tumors exhibit a variegated cut surface (Fig. 42.17). The color of the tumor in great part is related to the microscopic findings; tumors containing abundant foamy macrophages are tan to yellow, whereas those with intratumoral hemorrhage are dark tan to brown. Some lesions may be frankly necrotic.
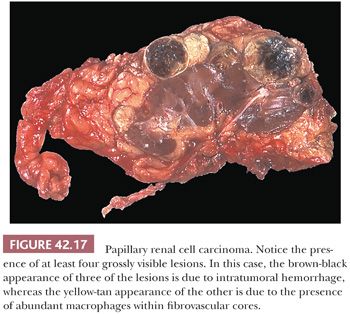
MICROSCOPIC FEATURES
Conforming to the gross circumscription, a majority of PRCCs show well-defined fibrous pseudocapsule on light microscopic evaluation. Even when the capsule is incomplete, most tumors tend to be well circumscribed. Although focal infiltration beyond the level of circumscription is not uncommon (172–175), the presence of extensively infiltrative edges into the surrounding renal parenchyma should raise other differential diagnostic possibilities (173).
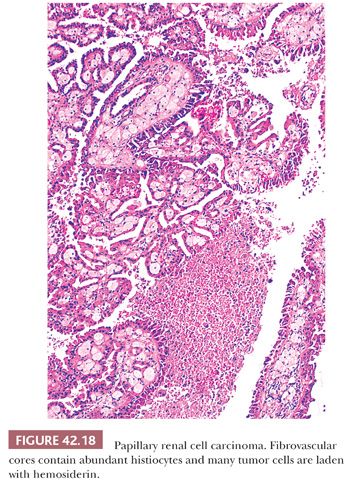
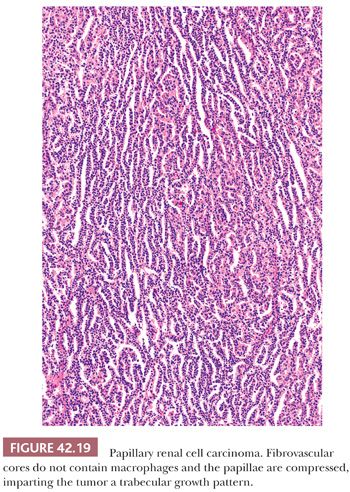
The majority of PRCC exhibit a broad morphologic spectrum, including papillary, tubulopapillary, papillary-trabecular, and papillary-solid areas (91,169,171,173,176–179). The classic papillary pattern is characterized by discrete papillary fronds lined by neoplastic epithelial cells and containing a central fibrovascular core, easily recognized on low magnification (Fig. 42.18). The papillary-trabecular areas exhibit papillations, which are delicate, elongated, and arranged in a parallel fashion (Fig. 42.19). In the papillary-solid areas, the papillae are closely packed, masking their true growth pattern. The fibrovascular cores and consequently the papillary nature of the lesion are more difficult to discern, requiring examination under medium-to-high magnification. In cases exhibiting delicate fibrovascular cores devoid of foamy macrophages, these tumors may be mistaken to have a solid growth pattern (176,178).
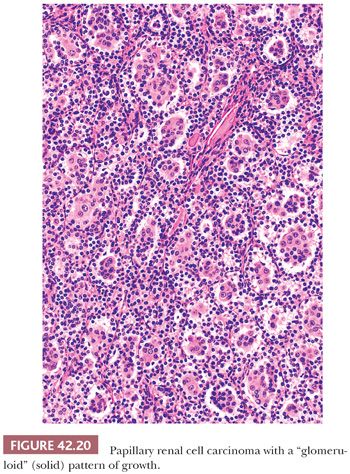
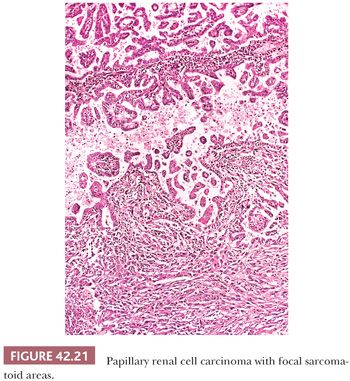
Areas containing classic papillary, papillary-trabecular, and/or papillary-solid patterns of growth are seen in virtually all cases. However, more than half the cases can exhibit other morphologies including truly solid, tubular, and/or glomeruloid growth patterns. Areas of true solid growth are characterized by closely juxtaposed polygonal tumor cells, often with cytoplasmic clearing, and lacking a fibrovascular core. These areas are almost always microscopic and are present adjacent to areas of more conventional PRCC. The tubular growth pattern is characterized by small to medium–sized tubules lined by cuboidal or columnar cells. The intervening stroma between the tubular structures is usually sparse. Tumors exhibiting a glomeruloid growth pattern are composed of closely juxtaposed, small tubule-like structures with intraluminal tufting of tumor cells (Fig. 42.20). In most cases, it is possible to identify two populations of tumor cells. The cells lining the tubular-like structures are cuboidal and contain scant to moderate amounts of mostly amphophilic cytoplasm, whereas the cells tufting into the lumen are polygonal; contain more abundant, often eosinophilic, cytoplasm; and often have larger nuclei with easily identifiable nucleoli. In rare cases, the glomeruloid growth pattern predominates or comprises virtually the entire tumor. Sarcomatoid growth may be seen rarely and is a predictor of aggressive clinical behavior (Fig. 42.21).
Although a papillary growth pattern predominates in the majority of cases, 20% to 25% of cases exhibit less than 50% papillary growth. In isolated cases, papillary growth comprises a very minor component of the tumor. For this reason, the percentage of papillary growth should not be used to determine whether or not a tumor should be classified as a PRCC. Solid variants of PRCC contain the same genetic alterations as other classic papillary tumors (176).
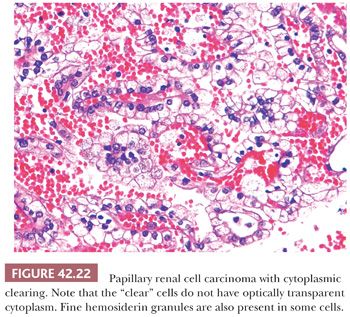
Cytologic features in PRCC are quite variable. The cytoplasm may be scant to moderate and amphophilic, or abundant and eosinophilic (89,163,169,175,179–181). The cells in some tumors with eosinophilic cytoplasm resemble those observed in renal oncocytoma. The low-grade nuclei in such tumors are often aligned towards the lumen. Such tumors have been called oncocytic PRCC (182). Occasional PRCCs contain variable, or sometimes prominent, “clear cell” areas. These clear-appearing cells, unlike the optically transparent cells in clear cell RCC, have fine cytoplasmic reticulations and granularity (Fig. 42.22) (171,173). The granularity is often due to the presence of fine particles of hemosiderin in the cytoplasm. Tumor cells with clear cytoplasm may be seen in association with a solid growth pattern or in tumor cells in close association with necrosis and degenerative changes (89,163,169–172). The majority of cases contain tumor cells exhibiting different cytoplasmic features, although the predominant tumor cells are either amphophilic or eosinophilic (Figs. 42.12 to 42.16). Nuclear features can be quite variable, ranging from small, round nuclei with inconspicuous nucleoli to large, moderately irregular nuclei containing coarse chromatin and prominent nucleoli.
Geographic necrosis is a common finding, as is the presence of psammoma bodies and foamy macrophages within the fibrovascular stalk. At times, the fibrovascular stalk becomes hyalinized or fibrotic, masking the papillary nature of the lesion. Occasionally, the stalks show very prominent edema, with the papillae superficially resembling microcysts. Hemosiderin pigment may be present in the cytoplasm of the tumor cells in a patchy distribution (Fig. 42.12); this finding is more common, although not exclusive, in cells with eosinophilic cytoplasm.
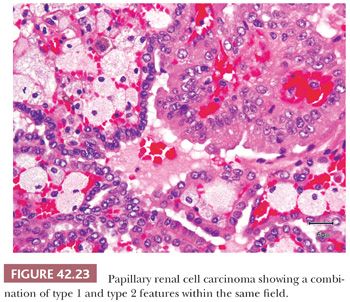
WHO divides PRCC into two types: type 1 with papillae covered by smaller cells with scant to moderate amphophilic cytoplasm and type 2 with larger cells with eosinophilic cytoplasm, nuclear pseudostratification, and often with higher nuclear grade (180). However, there are some tumors that show a combination of type 1 and 2 features (Fig. 42.23). WHO system does not address the issue of classification of such PRCCs with combination of cytologic features.
OUTCOME
The prognosis of PRCC is more favorable than clear cell RCC and less so than chromophobe RCC (Table 42.7) The reported 5-year disease-free survival varies from 79% to 92% (163,179). In a more recent large study from Mayo Clinic, the estimated cancer-specific survival rates at 1, 5, and 10 years after surgery were 98%, 92%, and 88%, respectively (181). Grading remains controversial. Amin and colleagues (89) found that it was a strong predictor of survival on univariate analysis but not on multivariate analysis. Delahunt and Eble (162) divided their cases into type 1 and type 2 lesions, based on architectural and cytologic features (Fig. 42.16), with some genetic differences reported between the type 1 and type 2 tumors (129,183). Subsequently, Delahunt and colleagues (91) have reported significant differences in overall survival between the two groups. Based on a study of 103 cases of PRCC, we were not able to confirm these results (90) nor were Warrick et al. (174) in another more recent study. One of the reasons for these differences in results may be the small number of events that occur in PRCC (e.g., progression or deaths due to disease) as reported in our series and by others (89,90,174,181). However, in the recent largest series to date of surgically resected 395 cases, tumor grade and TNM stage were significantly associated with death from RCC on multivariate analysis (181). On the other hand, the type of PRCC (type 1 or type 2) showed significant association only on univariate, and not on multivariate, analysis. Surprisingly, and contrary to the known behavior of PRCC, Delahunt and colleagues (91) have reported a very high rate of death in their series (27 deaths in 66 patients). Sika-Paotonu and colleagues (184) showed that nucleolar prominence, and not the overall Fuhrman nuclear grade, is a better indicator of the biologic behavior in PRCC. Using a modified Fuhrman grading scheme, based only on nucleolar prominence other than grade 4, grade was also found to be associated with deaths from RCC on multivariate analysis in the recent Mayo Clinic study (181). Yang and colleagues (185) reported that tumors with eosinophilic cytoplasm but low nuclear grade, as well as those with combination of type 1 and such low-grade eosinophilic areas, tumors that otherwise would be considered type 2 RCC by Delahunt and Eble criteria, showed molecular signature and biologic behavior similar to type 1 PRCC. More recently, based on a series of 220 PRCCs containing any type 1 area, we showed that the presence or the extent of type 2 morphology, Fuhrman nuclear grade, or nucleolar grade had no association with the disease-specific or overall survivals over a median follow-up duration of 90 months (175). Thus, in spite of the consensus view of the recent ISUP renal consensus conference (98), the use of grading of PRCC and the system by which they should be graded still needs to be validated clinically on an even larger number of cases likely across different medical institutions.
DIFFERENTIAL DIAGNOSIS
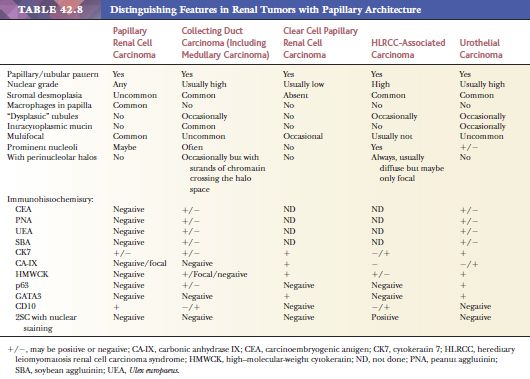
Papillary architecture may be seen in a number of renal cell tumors other than PRCC (Table 42.8). Separation of such tumors from PRCC is essential, as their biologic behavior is often quite distinct from PRCC, and the clinical management may also be quite different. PRCCs may contain tumor cells with clear cytoplasm focally, especially in areas with a solid growth pattern. Clear cell RCC may also be focally papillary, although this is usually due to cell drop-off in areas away from feeding vessels that creates a pseudopapillary appearance. Adequate sampling should clarify the issue. Psammoma bodies, hemosiderin deposition within tumor cells, and fibrovascular cores containing foamy macrophages are more likely to be seen in PRCC. CK7 immunoreactivity is present at least focally in many PRCCs; however, it is negative in most clear cell RCCs. Diffuse, membranous positivity for CA-IX is diagnostic of clear cell RCC in this differential. If needed, molecular genetics can be used to solve difficult cases. Collecting duct carcinomas may have a papillary growth pattern and resemble PRCC. Nevertheless, the former are centered in the medulla and are virtually always high grade, with associated desmoplastic stroma. Collecting duct carcinoma also shows prominent multinodular growth pattern. In our opinion, tumors with prominent papillary architecture but more than focal desmoplasia, particularly when associated with solid, cribriform, or cystic areas, most likely represent collecting duct carcinoma/carcinomas with distal nephron differentiation, rather than PRCC. Intracytoplasmic and luminal mucin is a common feature in collecting duct carcinoma, as is reactivity for carcinoembryogenic antigen (CEA), peanut and soybean agglutinins, Ulex europaeus, and high–molecular-weight CK 34BE12. CK7 may be expressed in both tumors. Again, cytogenetic studies can be used to solve difficult diagnostic problems. MiTF family translocation–associated RCC may have a prominent papillary growth pattern (186,187). However, they are more common in young patients, are rarely multifocal, and usually have a high nuclear grade and abundant (sometimes voluminous) cytoplasm. In general, they are either negative or only focally positive for CKs and EMA. Characteristically, they will exhibit nuclear immunoreactivity for TFE3 or TFEB, depending on the translocation. They are, in addition, often immunoreactive for cathepsin K, unlike PRCC. HLRCC-associated renal carcinomas show variable and often prominent papillary architecture (171,173). As a matter of fact, these were initially considered to be PRCC, type 2. HLRCC tumors often show a combination of architectural patterns within the same tumor. The most characteristic morphologic feature in these is the presence of a very large nucleolus surrounded by a perinucleolar halo. Genetic testing shows mutations in fumarate hydratase gene. Metanephric adenoma may be mistaken as a type 1 PRCC with prominent tubular features (171,173). Unlike type 1 PRCC, metanephric adenoma mostly lacks a pseudocapsule and shows a uniform cell population with no significant atypia and absence of, or at the most very inconspicuous, nucleoli. CK7 and AMACR are usually negative in metanephric adenoma, whereas CD57 is usually positive. A difficult differential diagnosis is separating mucinous tubular and spindle cell carcinoma (MTSCC) from the rare type 1 PRCC with low-grade spindle cell areas (171). Unlike such a PRCC, the former often shows prominent mucinous/myxoid stroma. The tubules are often elongated and branching, and the tubular luminal contours are smooth in MTSCC. In PRCC with low-grade spindle cell areas, the tubular lumina often show an irregular contour. CK7 and AMACR stains do not distinguish these tumors, but CD10 is often negative or only very focally positive in MTSCC.
PAPILLARY (CHROMOPHIL) “ADENOMA”
Although size often correlates well with clinical outcome, characterizing a tumor as either carcinoma or adenoma based solely on size is scientifically unsound. Despite the fact that these lesions are architecturally and cytologically identical to larger lesions, it is a common practice among pathologists to use the term papillary adenoma for small (<5 mm) papillary lesions in which common sense tells us that the likelihood of metastasis is minimal. Empirical data available has taught us that metastatic potential for lesions measuring even up to 1 cm and containing small, regular nuclei with compact chromatin and small or absent nucleoli is also virtually nil (Fig. 42.24).

CHROMOPHOBE RENAL CELL CARCINOMA
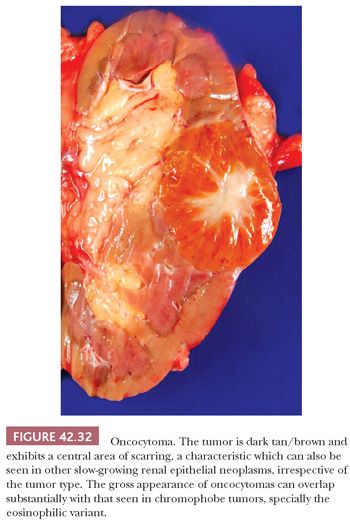
Chromophobe renal cell carcinomas (CRCC) are thought to arise from intercalated cells of the renal cortex, similar to oncocytomas. Bannasch and colleagues were first to report the occurrence of CRCC in kidneys of animals exposed to nitrosomorpholine. In 1985, Thoenes and colleagues (188) recognized this variant in human renal epithelial tumors and illustrated its morphologic, histochemical, and ultrastructural findings. During the ensuing years, our awareness of CRCC has increased dramatically, as evidenced by the increasing number of studies published on this subject (103,189–194). In the more recent years, a number of publications have presented the clinicopathologic features of CRCC based on large numbers of cases. Compared to the earlier ones, these recent publications have the added advantage of using the knowledge accumulated over more than 25 years since the initial recognition of CRCC, as well as long clinical follow-ups (93,94,143,195,196). All these studies make it amply clear that these tumors have distinct morphologic, histochemical, and ultrastructural features, as well as unique molecular genetic and clinical characteristics. It was in fact the discovery of the existence of human CRCCs by Thoenes, Kovacs, Storkel, and their collaborators (17–19) that led these investigators to initiate a reassessment of the morphologic classification of renal tumors, which has served as the foundation for the classification in use today. It is important to recognize this subtype of RCC for two main reasons. First, stage-for-stage, CRCC has a significantly better prognosis than clear cell (conventional) RCC with which it has some overlapping cytologic and growth pattern characteristics (Table 42.7). This morphologic overlap was further exemplified recently in the recent TCGA clear cell RCC cohort; initial data evaluation of the “clear cell RCCs” showed more than 10 cases with chromophobe-like copy number alterations. On review by a panel of expert genitourinary pathologists, all these tumors in fact turned out to be CRCC (personal observations); these cases, therefore, were excluded from the final analysis (24). Secondly, the morphologic features of the eosinophilic variant of CRCC may overlap with and be confused for oncocytoma, a benign tumor.
CRCCs are characterized genetically by loss of chromosomes 1 and Y as well as combined chromosomal losses, usually affecting chromosomes 1, 6, 10, 13, 17, and 21 (6,197–199). Loss of multiple chromosomes leads to hypodiploid tumor cells, a unique feature seen in many of these tumors (200). Abnormalities in mitochondrial DNA may be observed, but its specificity remains controversial (201). It is hoped that the extensive genetic analysis by TCGA that is under process for CRCC will answer many of these controversies.
CLINICAL FEATURES
CRCCs comprise 6% to 11% of renal epithelial tumors. The age and sex distribution is virtually identical to that seen in clear cell RCC (Table 42.7). In our series, the mean age at presentation was 60 years (94). They usually present as a unilateral renal mass. Of the cases resected at our institution, 58% were asymptomatic, 30% presented with a palpable mass, and 19% with hematuria. The tumor may involve the right and left kidneys equally, and rarely, patients present with bilateral disease. Angiographic examination usually reveals a hypervascular mass; however, some may be hypovascular. Radiographic examination may reveal a central scar, similar to that seen in oncocytomas and large, low-grade clear cell RCC. Thus, the presence of a radiographically detected central scar in a renal neoplasm offers little diagnostic information except for suggesting a slow-growing neoplasm.
GROSS FEATURES
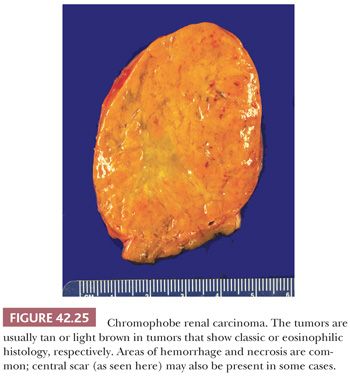
The gross appearance is very helpful in the diagnosis of CRCC. Characteristically, these tumors are well circumscribed but not encapsulated and classic cases show a homogeneous beige or pale-tan cut surface (Figs.42.13 and 42.25). Nonetheless, rare tumors are dark brown and mahogany; such tumors often turn out to be the eosinophilic variants. A central scar was present in approximately 15% of our cases. Tumors may be quite large; more than one-third of tumors in our study were greater than 7 cm in diameter (94). Tumors commonly have a lobulated appearance and roughly one-fifth of cases exhibit gross necrosis. Cystic areas are rare. Smaller tumors are centered in the renal cortex, a feature that may prove difficult to determine in larger neoplasms. Evidence of multifocality is rare and was seen in only 4% of our cases (94) and in 8% in the series by Amin and colleagues (195). Gross involvement of the renal vein may be seen in a small number of cases, whereas up to a third of patients may exhibit disease invading renal sinus or perirenal adipose tissue (22% in the recent Memorial Sloan-Kettering Cancer Center series) (94).
MICROSCOPIC FEATURES
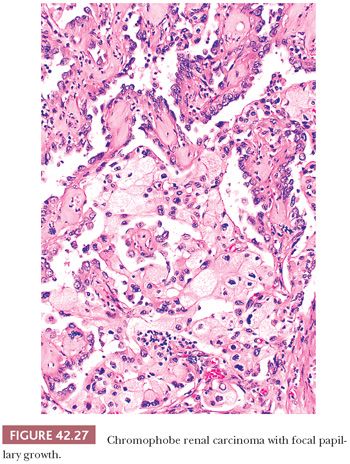
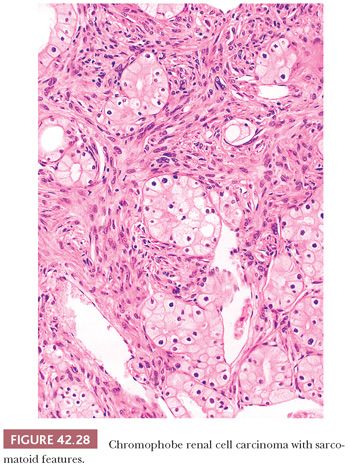
Greater than 80% of the tumors are nonencapsulated (94). The pattern of growth is predominantly solid sheetlike, separated by thin, incomplete fibrovascular septa (Fig. 42.26) but may be focally admixed with tubular, nested, broad alveolar, trabecular, solid, cystic, or papillary patterns (Fig. 42.27). A small percentage of cases may exhibit a sarcomatoid pattern of growth (Fig. 42.28). Akhtar and colleagues (202) state that CRCC may be the renal epithelial tumor most frequently associated with a sarcomatoid growth pattern. However, in the study of 203 tumors with primary resection at our institution, sarcomatoid features were noted in only 2% of the tumors (94), whereas the recent series by Cheville et al. (143) and Amin et al. (195) had 7% and 8% tumors with sarcomatoid areas, respectively. In most cases, islands of typical CRCC are embedded within expansive areas of spindle cell growth, and direct transition from epithelial to spindle cell components is difficult to discern. Geographic necrosis and calcification are present in 17% and 46% of cases, respectively (94). In our series, areas of hyalinized stroma intervening with tumor nests were present in 41% of cases and were associated with smooth muscle metaplasia in 26%.
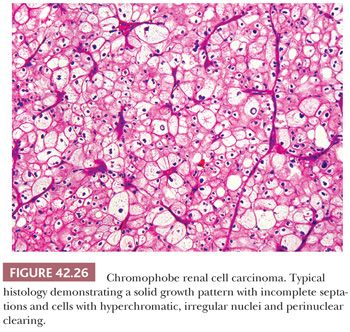
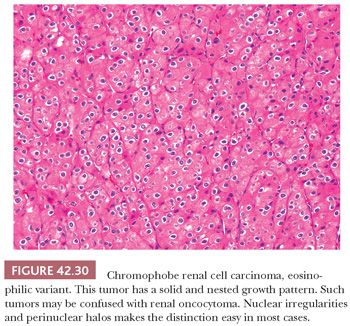
The typical histology of CRCC consists of large round-to-polygonal cells with well-defined cell borders and amphophilic to pale basophilic cytoplasm admixed with a smaller population of polygonal cells with eosinophilic cytoplasm (Figs. 42.26 and 42.29A,B). Unlike that in clear cell RCC, the cytoplasm is not entirely clear but rather translucent and finely reticulated. Up to 25% of cases contain larger cells with abundant clear to foamy (“hydropic”) cytoplasm (Fig. 42.29A). As stated earlier, in the past, and for that matter even now (see previous discussion), chromophobe carcinomas with typical histology were often classified as clear cell carcinomas. A predominance of tumor cells with densely eosinophilic “oncocytic” cytoplasm is seen in 20% to 41% of cases (94,195). This latter group of tumors constitutes what has been called the “eosinophilic variant” of CRCC. Although the distinction between the “classic” and the “eosinophilic” variants of CRCC is clinically not important, its introduction was necessary to alert pathologists to the wide spectrum of morphologic changes seen in CRCC and to avoid misinterpreting the tumor as an oncocytoma (Fig. 42.30) (189). Perinuclear halos are invariably seen but are more pronounced in the classic variant. Perinuclear clearing in CRCC is a consequence of perinuclear concentration of microvesicles (see the following texts); it results in the other cytoplasmic organelles being pushed towards the periphery of the cytoplasm, causing apparent accentuation of the cell membrane. At low- to medium-power magnification, this feature imparts the tumor with a “cobblestone” appearance, also reminiscent of the thick cell wall in plant cells as seen through the microscope (Fig. 42.27). The nuclei are typically hyperchromatic, with irregular nuclear membranes. This nuclear cytology predominates in the great majority of cases, although round nuclei with only focally wrinkled contours are prominent in many eosinophilic variants. Binucleate cells are present in virtually all cases. Focal cytologic atypia with bizarre nuclei are seen in half the cases but prominent in only a small minority of tumors. Mitotic activity is very low, except in sarcomatoid or frankly high-grade epithelial areas. In the classic cases, tumor cells are arranged in large nests/acini or solid sheets separated by incomplete septations. In general, the rich, arborizing vascular network seen in clear cell carcinomas is lacking, a useful clue to arriving at the proper diagnosis.
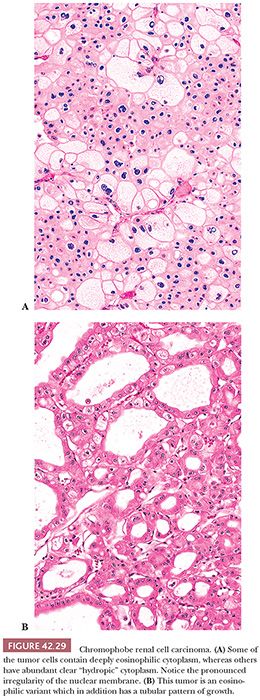
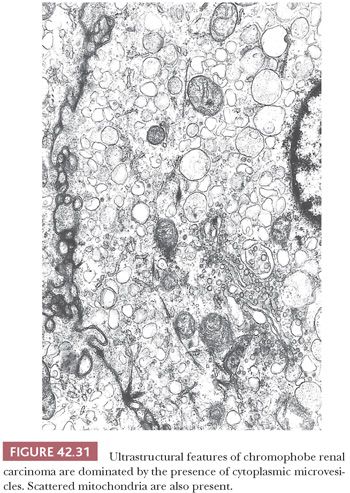
Variable granular or reticular and diffuse cytoplasmic staining with Hale colloidal iron is reported in the majority of cases (203), although focal, weak, and even absent staining may be seen in cases with a predominance of eosinophilic cells (188,204,205). A few cases exhibit staining of the luminal borders of acini. It is a difficult stain to perform, and we have found it to be of limited use in eosinophilic variants. Hale colloidal iron stain detects polyanionic compounds including acid mucopolysaccharides. A unique ultrastructural feature of CRCC is the presence of cytoplasmic microvesicles (206–209) (Fig. 42.31). Their origin is uncertain but believed to be related to defective development of mitochondria (209). The vesicles and their content lack affinity for hematoxylin and eosin dyes, imparting these cells with a “clear” cytoplasmic appearance. Bonsib and colleagues (207) have shown that tissue processing for paraffin embedding results in solubilization of these vesicles and cannot be employed for the ultrastructural confirmation of a histologic diagnosis of CRCC. The work by the same author confirms the relationship between the cytoplasmic vesicles and Hale colloidal stain in osmicated tissue, a process that preserves the integrity of these vesicles (205). Ultrastructural studies have shown that small eosinophilic cells have abundant mitochondria and a few microvesicles, explaining why these tumor cells show a different pattern of expression of Hale colloidal iron (210). Mitochondria often show tubulocystic cristae (209,210). Weak Hale colloidal iron positivity has been demonstrated in other renal tumors, including a focally diffuse distribution in oncocytoma and dropletlike pattern in conventional and PRCC (203). Luminal staining can also be seen in the neoplastic tubules of oncocytomas (203,205). For the reasons stated earlier, we rarely rely on Hale colloidal iron to make a diagnosis of CRCC.
Tumor cells show immunoreactivity for EMA as well as pankeratins such as CAM 5.2 and AE1/AE3. Expression of CK7 is usually diffuse and strong, as is c-kit (Fig. 42.8) (124). However, CK7 positivity may be very focal or even absent in the eosinophilic variants. Vimentin is positive only in the sarcomatoid areas. High–molecular-weight CK 34BE12 was negative in all cases we studied. AMACR and CD10 are negative, although the eosinophilic tumor cells may stain for the latter. Although these tumors have been reported to show a characteristic staining pattern with antimitochondrial antibody 113-1, the staining patterns may overlap between oncocytomas and the eosinophilic variant of CRCC (117). CA-IX, HMB-45, A-103, and synaptophysin are negative.
OUTCOME
The prognosis of CCRC is much better than other types of RCC (103,191,192) (Table 42.7). In our study of 203 tumors (200 patients), 5 had metastatic disease at the time of diagnosis. Of the other 195, 8 (4%) developed metastases/died of disease over a mean follow-up of 81 months (94). Although classic CRCC may metastasize and kill the patient, those with a sarcomatoid component are the most likely to behave in an aggressive fashion. In our series, the tumors developing events (metastasis/death due to disease) had microscopic tumor necrosis, small vessel angiolymphatic invasion, and/or sarcomatoid features (94). Microscopic tumor necrosis and sarcomatoid features were also associated with outcome in the series by Amin et al. (196). Interestingly, one study of metastatic non–clear cell RCC showed a longer survival in patients with metastatic CRCC, compared to those of other morphologic subtypes (211). These findings are in further support of the relatively indolent nature of these tumors, compared to clear cell RCC.
DIFFERENTIAL DIAGNOSIS
Many tumors called granular RCC in the past are actually CRCCs, whereas others are either clear cell RCCs, RCC unclassified, PRCC, epithelioid angiomyolipoma, or oncocytomas (Table 42.3). Given the differing clinical behavior among these tumors, distinction between them is mandatory and can be performed easily in most cases by adequate sampling and paying close attention to the growth pattern and cytologic characteristics of the tumor. Immunohistochemistry, electron microscopy, as well as cytogenetics may be useful in difficult cases.
RENAL ONCOCYTOMA
Oncocytoma is a benign renal epithelial neoplasm first described by Zippel in 1942 (212) and then largely ignored until the publication of 13 additional cases by Klein and Valensi (213). It is estimated that oncocytomas comprise from 3.2% to 7% of all primary renal neoplasms (214–217). Over a 10-year period, oncocytomas constituted 10% of 860 renal epithelial neoplasms resected at the Memorial Sloan-Kettering Cancer Center (Table 42.5). Most series show a wide age distribution at presentation with a peak incidence in the seventh decade of life. Males are affected nearly twice as often as females (215,217). The majority are asymptomatic at presentation and uncovered during the workup of other unrelated, nonurologic conditions. A minority of the patients present with hematuria, flank pain, or a palpable mass.
Unlike other renal epithelial neoplasms, no consistent unique genetic abnormality characterizes oncocytoma. However, the most frequent genetic change is loss of chromosomes 1 and Y (218). Less common are translocations involving chromosomes 9 and 11 [t(9;11)(p23;q13)] (219–221), chromosomes 5 and 11 [t(5;11)(q35;q13)] (222), and chromosomes 6 and 9 [t(6;9)(p21;p23)] (223). It is of interest that genes encoding mitochondrial DNA are found in the translocation region of chromosome 11. Also, the 11q13 region is in the vicinity of the cyclin D1 gene (224,225). Most oncocytomas with rearrangement involving 11q13 also demonstrate cyclin D1 overexpression (224,225). Familial cases of renal oncocytoma have been described but are uncommon (226).
GROSS FEATURES
Oncocytomas are well-circumscribed, nonencapsulated neoplasms that are classically mahogany brown and less often tan to pale yellow (Fig. 42.32). The presence of a central, stellate, radiating scar is seen in roughly 33% of cases and is related to size. For example, in one series, the mean size of tumors with a central scar was 6.5 cm, whereas that of tumors lacking a scar was 4.6 cm (217). One should bear in mind that low-grade clear cell RCC and CRCC may also contain a central scar. Gross hemorrhage is present in 20% of cases but necrosis is almost nonexistent (217). Cysts and gross extension into perinephric adipose tissue are rarely seen. Multifocal tumors are seen in up to 17% of cases and bilateral tumors in 4% (215,217).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


