TISSUE SAMPLING AND PREPARATION
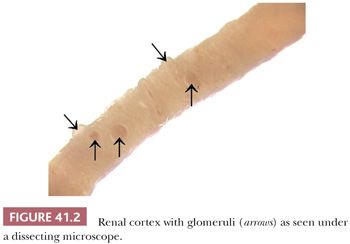
Renal tissue samples are usually obtained by percutaneous or open renal biopsy or by nephrectomy. The biopsy is divided for LM, IF, and EM. The specimen should be handled very carefully to avoid squeezing and compression, preferably by wooden sticks instead of forceps. A needle biopsy specimen should be divided under a dissecting microscope. This microscope allows for visualization and differentiation of the renal cortex and medulla. The medulla shows parallel structures (vasa recta and collecting ducts); on the other hand, the cortex appears irregular, and its glomeruli usually are visible as small red capillary conglomerates (Fig. 41.2). Even anemic glomeruli can be seen as vague shadows of “small balls” if one adjusts the micrometer screw.
We recommend cutting away two small (2-mm) pieces with glomeruli for EM, a small (~5 mm) piece with glomeruli for IF, and the remaining tissue for paraffin sections for LM. Of course, the size of these tissue pieces depends on the individual situation, primarily on the size of the core biopsy specimen. If no dissecting microscope is available, the best method is to cut 1- to 2-mm pieces from both ends of the biopsy specimen for EM and two 2- to 3-mm pieces for IF and submit the rest for routine paraffin processing for LM. When an excised kidney is received, it is important to bisect the kidney properly to make the entirety of the pyelocalyceal system and renal papillae visible. This is done by orienting the kidney longitudinally in one hand and carefully and slowly cutting into the renal cortex with a long knife. When one just enters a portion of the renal collecting system, it should then be opened with scissors along the pyelocalyceal system. The accompanying renal artery, renal vein, and ureter should always be examined in terms of gross and microscopic features. After obtaining kidney tissues that are as fresh as possible, it is necessary to process the tissue quickly and appropriately for IF and EM, if it is deemed necessary. One should remember that it is better to have appropriately processed tissue and not need it than to need it and not have it.
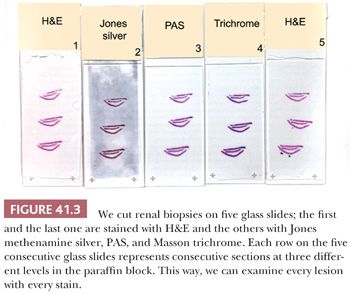
Tissue for LM should be placed in buffered formalin. The fixed tissue can be embedded in paraffin. For diagnosis of medical diseases of the kidney, multiple sections are routinely stained for LM with hematoxylin and eosin (H&E), periodic acid-Schiff (PAS), methenamine silver, and Masson trichrome. We cut three levels from the renal biopsies, and at each level, we obtain five consecutive sections on five separate glass slides (Fig. 41.3). The first slide is stained with H&E, the second with methenamine silver, the third with PAS, the fourth with Masson trichrome, and the fifth with H&E again. That way, we can examine every lesion on consecutive sections with the different stains. Thin (2- to 3-µm) sections are extremely important. Special additional stains, such as Congo red for amyloid (the only exception to the need for thin sections), may be useful.
Ideally, the tissue for IF should be rapidly snap frozen, usually in isopentane or methylbutane, and cooled in liquid nitrogen, after having been surrounded by optimal cutting temperature frozen section material. EM material is best diced into 1-mm cubes with a sharp razor blade (after cleaning off the oil on the blade with alcohol or xylene) and then placed in cold 3% glutaraldehyde or Carson formalin as soon as possible. In our experience, 10% neutral-buffered formalin is also adequate for diagnostic purposes if glutaraldehyde is not available. It is imperative to transfer this tissue carefully with a small wooden stick or forceps by capillary action (do not squeeze or compress the tissue). If necessary, tissue with glomeruli for EM can be retrieved from the paraffin blocks, although the ultrastructure is somewhat suboptimal.
A question that is frequently asked concerns the size of an adequate biopsy specimen and how many glomeruli it should contain. Obviously, the more the better; however, in certain cases, a single glomerulus may be enough to make a diagnosis (e.g., membranous GN). In focal glomerular diseases (e.g., early-stage focal segmental glomerular sclerosis), however, a sampling error cannot be excluded even if many glomeruli are examined. Usually, taking two cores of renal cortex is recommended; one core may not be representative of the kidney, particularly of a transplant kidney.
IMMUNOFLUORESCENCE
Tissue snap frozen for direct IF should be cut thin (3 to 4 µm) and stained with antisera known to be monospecific for immunoglobulin (Ig) G, IgM, IgA, κ and λ light chains, C3, C1q, fibrinogen-related antigens (FRA), and albumin. The antibody to albumin is used as a control to exclude nonspecific staining, particularly with IgG (in case of nonspecific staining, albumin is usually also present in similar intensity; see Point 4 below). Other antisera, such as antibodies to properdin, C4, and IgE, are applied by some laboratories. The following IF characteristics should be evaluated in every renal biopsy:
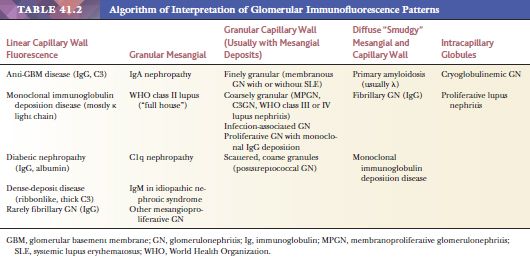
1. Is the deposition granular (probably immune complex) or intensely linear (classic antiglomerular basement membrane [anti-GBM] antibodies) (Table 41.2)? It should be noted, however, that linear staining for IgG may be nonspecific and should always be compared to the intensity of albumin staining.
2. What region of the glomerulus is staining—capillary wall or mesangium? Is this in an area of segmental sclerosis (Table 41.2)? If so, it would raise the possibility of nonspecific nonimmunologic trapping, especially if one found only IgM, C3, and FRA.
3. What class of Ig and fraction of complement is being deposited? Are both types of light chains positive or just one? It is important to determine the class of Ig because the Ig classes may correlate with specific diseases. For example, IgA can be seen in systemic lupus erythematosus (SLE), often associated with a “full house” of Igs, whereas IgA alone might suggest either Berger disease (IgA nephropathy) or anaphylactoid purpura (Henoch-Schönlein purpura [HSP]). Recently, we introduced IF with antibodies to the four IgG subclasses (IgG1, IgG2, IgG3, and IgG4). We find this helpful in certain glomerular diseases, such as in membranous GN (where IgG4 is the dominant IgG in the idiopathic form) and in glomerular diseases, which are associated with monoclonal Ig deposition (7,8). Finding C3 without C1q would suggest that the alternate complement pathway is involved.
4. Is the deposition of purported immune material associated with nonimmunologic material, such as albumin? In areas of plasmatic insudation, all plasma proteins flow into the region—not only Igs and complement but also albumin and FRA. If all are found, one is less certain of a primary immunologic phenomenon, and nonspecific staining should be a consideration.
5. How intense is the fluorescence? Grading the image/background signal may be quite subjective, but a skilled eye can easily distinguish a mild, moderate, or strong reaction. This is important in certain diseases, such as IgA nephropathy, where, for example, predominance or codominance IgA in the glomerular deposits is required for the diagnosis. Examination of the extraglomerular structures (tubules, interstitium, and vessels) is also important, although relevant deposits in these compartments are less common.
ELECTRON MICROSCOPY
EM is especially useful in the diagnosis of medical diseases of the kidney; indeed, one of the first uses of EM in medicine was in renal disease. The most important role of EM in the context of a diseased glomerulus is the identification of discrete electron-dense immune-type deposits (Tables 41.3 and 41.4) (9,10). In immune-mediated glomerular disease, EM is useful in determining the presence and the exact site of the deposits (which is often difficult to determine by IF, especially if deposits are present along the glomerular capillary wall [subendothelial vs. subepithelial]). In certain diseases, the presence of glomerular subendothelial deposits (especially if large and in the peripheral glomerular capillary walls) indicates more active disease (as in SLE). Glomerular mesangial deposits are present in many diseases, such as SLE and IgA nephropathy (Berger disease). A subtype of mesangial deposit is the so-called paramesangial deposit, located immediately beneath the GBM covering the mesangium. From a practical point of view, paramesangial deposits do not differ from other mesangial deposits.


Occasionally, EM can be very helpful in detecting structures other than immune complex deposits, such as the fibrillary deposits in amyloidosis or fibrillary glomerulonephritis, microtubular deposits in cryoglobulinemia, the GBM changes in thin basement membrane nephropathy or Alport syndrome, mitochondrial abnormalities, inclusion bodies (such as tubuloreticular inclusion in lupus nephritis or HIV infection), zebra bodies in Fabry disease, crystalline deposits in tubules or glomeruli in monoclonal gammopathies, and so on.
GENERALIZATIONS ABOUT GLOMERULAR DISEASES
The following points are important in understanding glomerular diseases:
1. A variety of renal morphologic patterns can lead to the same clinical syndrome; for example, nephritic syndrome and hematuria can be caused by hereditary nephropathy, membranoproliferative GN (MPGN), IgA nephropathy, acute proliferative GN, and so on. Nephrotic syndrome can be provoked by a variety of disease patterns, such as minimal change nephrotic syndrome (MCNS), focal segmental sclerosis, membranous glomerulonephropathy, diabetic nephropathy, or amyloid (11).
2. One disease may produce a variety of distinct patterns of renal injury. SLE is one example; in this instance, disparate patterns of injury might each have a different prognosis and may vary in their etiology and treatment.
3. A distinctive type of pathologic pattern or process may be caused by or associated with many different diseases (e.g., the pattern of intracapillary proliferative GN can be seen in SLE, hepatitis C infection, and various other infections).
4. Renal biopsy is thought to be specific to, and diagnostic of, only a few conditions, such as fibrillary GN. Thus, diagnostic renal pathology is an integrated “gestalt” or holistic process, wherein one must analyze and collate all the clinical and laboratory data and results of LM, EM, and IF studies to arrive at the best overall diagnosis.
5. One renal syndrome may be associated with different patterns of diseases and yet have a common pathogenetic mechanism of action (e.g., crescentic GN can be seen in a variety of diseases, such as vasculitis, SLE, anaphylactoid purpura, and acute GN; in all of these conditions, the crescents are probably related to FRAs and other proteins that reach Bowman space through breaks or “gaps” in the GBM).
THE HYPERCELLULAR OR TRUE OR “PROLIFERATIVE” GLOMERULONEPHRITIDES (GLOMERULAR DISEASES ASSOCIATED WITH NEPHRITIC SYNDROME)
ACUTE POSTINFECTIOUS GLOMERULONEPHRITIS (USUALLY POSTSTREPTOCOCCAL GLOMERULONEPHRITIS)
Epidemiology
This condition has become less prevalent after the advent of antibiotics. It is more frequently seen in warm climates. Epidemic outbreaks may occur among people living in crowded, highly populated communities in which poor hygiene and malnutrition are common. It occurs at any age, but it is primarily a disease of children.
Clinical Symptoms
Patients usually have nephritic syndrome that develops 1 to 4 weeks after an episode of pharyngitis or skin infection caused most often by certain nephritogenic Streptococcus types 12, 4, 1, and 49. Serum complement is temporarily depressed in more than 90% of patients. Antibodies to certain streptococcal antigens, such as antistreptolysin O (ASO), are elevated.
Light Microscopic Features
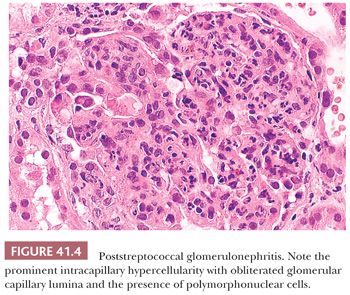
In the acute phase of postinfectious GN, all glomeruli are enlarged, and all glomerular tufts are extremely hypercellular (as a result of either proliferation of native cells or exudation of polymorphonuclear leukocytes and other inflammatory cells) (Fig. 41.4). Severe intracapillary hypercellularity leads to closure of the glomerular capillary lumens (12) and sometimes accentuation of the normal lobules of the glomeruli. There may be a slight increase in the amount of mesangial matrix. In some cases, crescents may be present; occasionally, they are abundant. Erythrocytes, red blood cell casts, and a few polymorphonuclear leukocytes may be seen in the tubular lumens. Small collections of interstitial inflammatory cells can be noted. Vessels are usually normal (12).
Immunofluorescence Characteristics
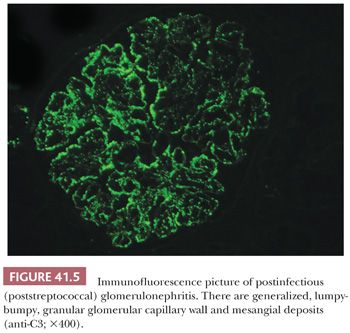
There are granular, “lumpy-bumpy,” glomerular capillary wall deposits of mainly C3 and less commonly IgG (and sometimes IgM and other immunoreactants) in a diffuse and global distribution (Fig. 41.5). From time to time, C3 may be the only immunoreactant (12–14).
Electron Microscopic Features
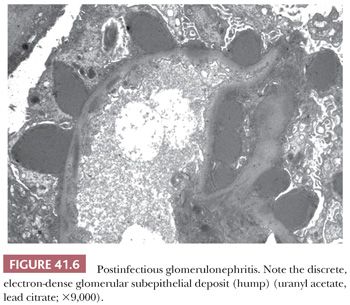
The presence of discrete, large, electron-dense, glomerular, subepithelial deposits (“humps”) is the most characteristic finding (Fig. 41.6). These humps are semilunar and vary in shape, size, and number. Small, discrete, immune-type electron-dense deposits are usually also present in the glomerular mesangium and occasionally in subendothelial regions as well.
Differential Diagnosis
Other diseases that may show the light microscopic pattern of intracapillary proliferative GN are C3GN, MPGN, cryoglobulinemic GN, and acute GN associated with Staphylococcus infection, endocarditis, deep-seated visceral abscesses, line abscess, or infected atrioventricular shunts (“shunt nephritis”). In these conditions, the GN may show a focal pattern by LM, but the immune deposits are present in every glomerulus (12). Diffuse proliferative lupus nephritis may also show a pattern similar to postinfectious GN.
Outcome
The prognosis (especially in children) is very good; patients usually spontaneously recover from this “one-shot serum sickness” over weeks to months. Subepithelial humps and neutrophils disappear from the glomeruli in 6 to 8 weeks. A slightly increased amount of mesangial matrix and/or more numerous glomerular cells can be detected for 1 to 2 years; proteinuria and/or hematuria may also persist for some time. Transition to a slowly progressive GN has not been well documented, and many such reported cases may represent postinfectious GN occurring in a patient who has an underlying alternative complement pathway activation abnormality and the Streptococcus infection evoked a protracted or chronic course of the disease, in fact, a form of C3GN (see the following text). The prognosis in adults is more variable and is the subject of controversy, but it appears to be good.
Etiologic Factors and Pathogenesis
Glomerular deposition of immune complexes (circulating or in situ) formed by antibodies, coupled with streptococcal antigens and complement, appears to be the most important pathogenetic factor. The exact streptococcal antigens involved are still uncertain.
GLOMERULONEPHRITIS ASSOCIATED WITH ONGOING INFECTIONS, INCLUDING ENDOCARDITIS, DEEP-SEATED VISCERAL ABSCESSES, AND INFECTED VENTRICULOATRIAL SHUNTS
These glomerulonephritides are rare, but their correct diagnosis is important because immunosuppressive medications should be avoided. For successful outcome, treatment of the underlying infection is necessary. LM usually shows a proliferative GN, frequently with a membranoproliferative pattern. Occasionally, particularly in GN associated with endocarditis, the proliferative GN may show a focal pattern (several glomeruli may appear light microscopically unremarkable). Segmental glomerular necrosis and crescents may occur, particularly in endocarditis-associated GN. IF usually shows C3 and IgG-dominant mesangial and granular glomerular capillary deposits. EM reveals electron-dense immune-type deposits in the mesangium and along the glomerular capillary loops. The glomerular capillary deposits are frequently intramembranous, particularly in endocarditis-related GN, but subendothelial deposits are common. Subepithelial deposits, including “humps,” may also occur. A number of underlying etiologic agents can be identified, including Staphylococcus aureus. Staphylococcus infection–associated GN is discussed more in detail in the following section.
STAPHYLOCOCCUS INFECTION–ASSOCIATED GLOMERULONEPHRITIS
In recent years, we have been experiencing a worsening Staphylococcus infection epidemic, particularly infection with methicillin-resistant S. aureus (MRSA). The incidence of poststreptococcal GN is declining in well-developed countries; however, in the Western world, including the United States, we see a rapid increase in the incidence of S. aureus infection–associated GN. It is important to emphasize that Staphylococcus infection–associated GN is usually not a postinfectious GN because many times, the active Staphylococcus infection is still ongoing when the symptoms of GN emerge (15,16).
Epidemiology
Staphylococcus infection–associated GN usually affects people with underlying predisposing conditions, such as diabetes, morbid obesity, malignancies, and the elderly. However, occasionally, the disease can emerge in previously apparently healthy individuals as well (12,17–19).
Clinical Symptoms
Most patients present with acute kidney injury, heavy proteinuria, and hematuria. The proteinuria is frequently nephrotic. The underlying Staphylococcus infection is not always obvious, and many times, a careful search has to be undertaken to find the source of infection. Other patients are overtly septic. Some patients have a clinical presentation resembling HSP (20). As opposed to poststreptococcal GN, the serum complement levels are usually normal, but in 20% to 30% of patients, there may be slightly low C3 levels.
Light Microscopic Features
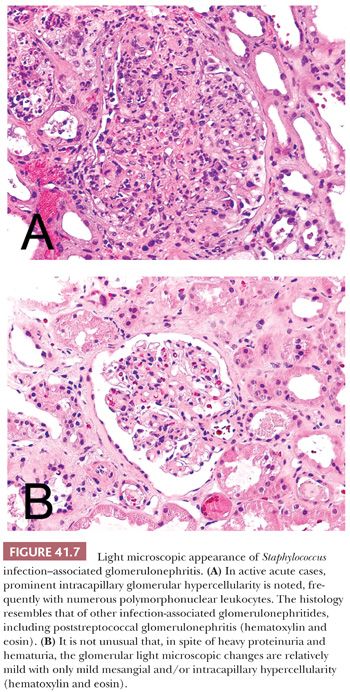
Glomerular hypercellularity is present in almost every biopsy; however, the degree of it can be quite variable (Fig. 41.7A,B). In some patients, only surprisingly mild glomerular hypercellularity is seen in spite of the heavy proteinuria and hematuria. In other patients, there are prominent intracapillary proliferative glomerular changes, but intracapillary polymorphonuclear leukocytes are usually less prominent than in poststreptococcal GN. Crescents may occur, but they are relatively rare. Patients with Staphylococcus infection–associated GN usually have acute tubular injury and patchy active interstitial inflammatory cell infiltrate as well. The vasculature is not involved.
Immunofluorescence Characteristics
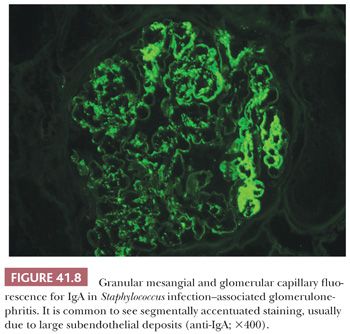
In almost all biopsies, variable degree of mesangial and also segmental glomerular capillary IgA deposits is noted. This is usually associated with C3 deposits and frequently also with some degree of IgG. However, in most biopsies, the IgA deposits are either dominant or codominant (Fig. 41.8). In a few biopsies, the glomerular IgA and immune complex deposition appears surprisingly mild relative to the severe clinical presentation.
Electron Microscopic Features
Mesangial electron-dense immune-type deposits are almost always evident. In several biopsies, glomerular capillary deposits are also seen. Subepithelial deposits, particularly large subepithelial “humps,” may be present (21) but not always (in our experience, in <50% of biopsies are typical “humps” noted).
Differential Diagnosis
The most important differential diagnosis is IgA nephropathy, which can be sometimes quite difficult. If a patient with IgA-dominant/codominant GN has evidence of underlying infection or the GN presents with acute kidney injury and heavy proteinuria, and if the patient has underlying comorbidities such as diabetes, a Staphylococcus infection should be considered. If an older patient presents with HSP-like clinical symptoms, Staphylococcus infection should always be excluded because, in adults and elderly patients, HSP is less common than Staphylococcus infection–associated HSP-like presentation, in our experience (20). If subepithelial “humps” are noted in association with IgA deposits, idiopathic IgA nephropathy is unlikely. Low serum complement levels are uncommon in Staphylococcus infection–associated GN, but if serum C3 is low, Staphylococcus infection should definitely be considered because C3 levels are normal in idiopathic IgA nephropathy. It is crucially important to differentiate Staphylococcus infection–associated GN from IgA nephropathy because, in many of these patients, the Staphylococcus infection is still active and administering immunosuppressive medications can result in sepsis. Other infection-associated glomerulonephritides are only exceptionally associated with IgA deposits, in our experience.
Outcome
The outcome depends on the elimination of the infection, the degree of underlying chronic renal injury, and the general condition of the patient. In our experience, roughly 50% of patients will recover good renal function.
Etiologic Factors and Pathogenesis
Staphylococcal antigens, in particular staphylococcal superantigens, probably play an important role.
C3 GLOMERULOPATHY (C3 GLOMERULONEPHRITIS) AND MEMBRANOPROLIFERATIVE GLOMERULONEPHRITIS
During the recent years, there were major developments in our understanding of proliferative GN with C3-dominant immune complex deposits. After careful genetic and proteomic studies, it turned out that a large proportion of patients with C3-dominant proliferative GN have congenital or acquired deficiencies in regulatory factors of the alternate complement activation pathway. Therefore, MPGN and GN with C3-dominant immune complex deposition has recently been reclassified. C3 glomerulopathy now includes glomerulonephritides with C3-dominant immune complex deposits in the absence of Ig deposition and in the presence of detectable congenital or acquired or congenital dysregulation of the alternate complement pathway activation. MPGN is now diagnosed if, in addition to glomerular C3 deposition, IgG deposition is also evident (22–25). However, the distinction between the disease groups is not always clear because patients with alternate pathway complement activation abnormalities may have some degree of Ig deposition (26).
C3 Glomerulopathy
C3GN first as a separate diagnostic entity was proposed by Servais et al. (27). The currently accepted classification was proposed by Fakhouri et al. (22). According to this, C3 glomerulopathy encompasses the following:
1. Dense-deposit disease (formerly known as type II MPGN) where dense glomerular intramembranous deposits are present in the glomerular capillary loops, which stain for C3 only by IF.
2. C3GN, a proliferative GN resembling MPGN type I without Ig deposits.
3. Rare diseases such as familial MPGN type III and complement factor H–related protein 5 abnormality–associated familial GN cases.
Epidemiology. C3 glomerulopathy is most frequently seen in pediatric patients or in young adults. It is less common in adults because most of the congenital abnormalities of alternate pathway complement regulatory proteins surface early during life. The disease may be familial.
Clinical Symptoms. Most patients present with worsening proteinuria and microscopic hematuria. Gross hematuria is rare. Initially, particularly if the disease is diagnosed during early childhood, the renal function is usually normal; however, if the disease starts insidiously, some degree of renal insufficiency may already be evident at the time of diagnosis. Hypertension is common. Serologies and laboratory findings are usually negative except for serum C3 levels, which are almost always persistently low.
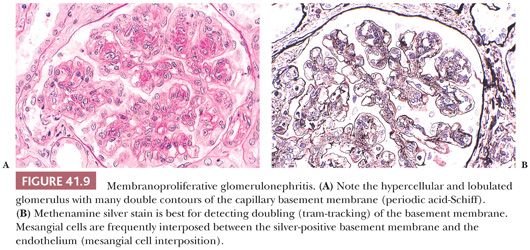
Light Microscopic Features. Most patients have variable degree of intracapillary proliferative GN, frequently with an MPGN pattern. In an MPGN pattern, the glomeruli are diffusely enlarged with marked diffuse and global intracapillary hypercellularity, lobular accentuation, and thickening of the glomerular capillary walls by abundant, sometimes large subendothelial deposits (Fig. 41.9A). There is “tram-tracking” or “reduplication” of the glomerular capillary wall caused by circumferential mesangial cell interposition (mesangial cells spreading into the contiguous space between the adjacent glomerular endothelium and the GBM) with creation of a new inner GBM-like material, which gives rise to the double or the “reduplicated” GBM (Fig. 41.9B). Occasionally, there may be a focal and even segmental MPGN pattern. This MPGN pattern of glomerular injury, however, is not always seen, and in some cases, the hypercellularity is quite mild and mesangial only. Crescents and tubulointerstitial disease may be present, the latter of which typically correlates with the severity and duration of the GN.
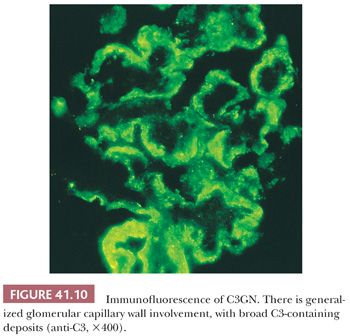
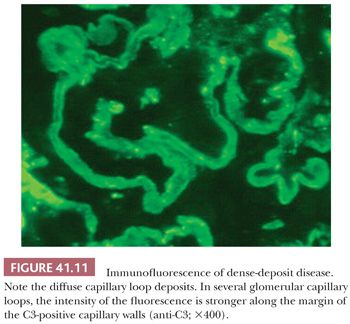
Immunofluorescence Characteristics. Large, intense granular, sometimes confluent, glomerular capillary wall and mesangial deposits of C3 (Fig. 41.10) are evident. IgG deposition should not be present (if significant IgG deposits are seen, the diagnosis of MPGN type I should be made). In dense-deposit disease, strong, granular, glomerular, mesangial, and linear, sometimes discontinuous, capillary loop deposits of C3 are noted. It is frequently apparent that C3 is present along the margins of, but not within, the dense intramembranous deposits (Fig. 41.11). This contributes to a double-linear appearance of the glomerular capillary wall and a ring appearance around the mesangial deposits (mesangial rings) on IF (28).
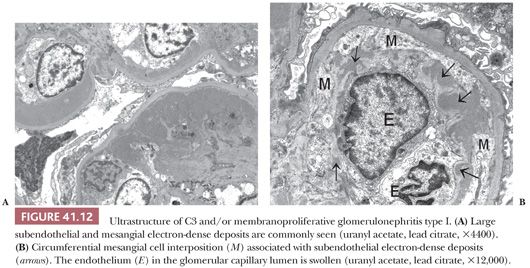
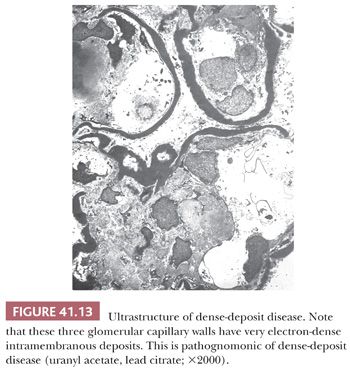
Electron Microscopic Features. There are widespread electron-dense, glomerular subendothelial deposits of varying sizes (Fig. 41.12A); sometimes, they are large and confluent, resembling the “wire loop” lesions in severe proliferative lupus nephritis. Circumferential mesangial cell interposition is frequently seen (Fig. 41.12B). Subepithelial deposits (even a few large “humps”) may be present. In some cases, mesangial deposits dominate with no or few glomerular capillary deposits. In such cases with mainly mesangial deposits, the intracapillary hypercellularity may be mild or absent. In dense-deposit disease, there are large, dense, glomerular, intramembranous deposits on EM, often involving long segments of the GBM, which are unique and pathognomonic (Fig. 41.13). The deposits may be discontinuous. There may be similar deposits in the nearby mesangial regions. Glomerular subepithelial deposits (“humps”) may also be present (28). Deposits similar to the GBM deposits often develop in Bowman capsule and in tubular basement membranes (TBMs) and have even been noted in the basement membranes of other organs, such as the liver, spleen, and eye.
Differential Diagnosis. Glomerular intracapillary hypercellularity with a membranoproliferative pattern may be associated with a variety of glomerular diseases (29), with or without the glomerular deposition of immunoreactants. The term membranoproliferative glomerulonephritis should be applied to conditions in which the characteristic glomerular immune deposits can be identified. Diseases other than C3 glomerulopathy and MPGN in which a membranoproliferative pattern can be noted include the following:
1. Postinfectious (intracapillary proliferative) GN.
2. Other infection-associated glomerulonephritides (e.g., endocarditis, deep-seated abscesses, infected shunts, schistosomiasis, and hepatitis C infection). GN associated with hepatitis C is usually indistinguishable from MPGN (30,31); in fact, many cases previously diagnosed as MPGN are probably hepatitis C–associated GNs. Because patients with hepatitis C virus–associated GN also have cryoglobulinemia, there is an overlap with cryoglobulinemic GN. Not all cases of hepatitis C–associated GN, however, have glomerular capillary cryoglobulin thrombi.
3. Cryoglobulinemic GN (with or without hepatitis C virus infection). The distinctive feature is the presence of glomerular, intracapillary, “hyaline” (cryoglobulin) thrombi.
4. Idiopathic lobular GN without immune complex deposition may represent an advanced, “burned-out” form of C3 glomerulopathy or MPGN, but it may also be a distinct diagnostic entity (see also the differential diagnosis of diabetic glomerulosclerosis) (32).
5. Monoclonal Ig (light chain) deposition disease.
6. Proliferative GN with monoclonal immune complex deposition.
7. Diabetic glomerulosclerosis (in early stages, there may be some glomerular hypercellularity).
8. Later stages of glomerular thrombotic microangiopathy.
9. Transplant glomerulopathy.
10. Lecithin-cholesterol acyltransferase deficiency, a rare hereditary disease of young persons. Glomerular mesangial and subendothelial lipid deposition is characteristic (33).
The differential diagnosis in C3 glomerulopathy relies now primarily on the IF (C3-positive glomerular deposits only) and the detection of alternate pathway complement regulatory protein abnormality.
Outcome. C3 glomerulopathy is generally slowly progressive, and the overall long-term renal prognosis is poor. The recurrence rate after renal transplantation is not very well established, but we know that there is a very high recurrence rate in dense-deposit disease (34,35). It is likely that other forms of C3 glomerulopathy also frequently recur in renal allografts.
Etiologic Factors and Pathogenesis. As mentioned earlier, the disease is secondary to acquired or congenital abnormalities in the factors regulating the alternate complement pathway activation (22,23,36,37). The described abnormalities are growing, and they include mutation in genes for factor H, factor H regulatory proteins, factor I, factor B, and C3. Some patients develop autoantibodies to the C3 convertase (such as C3 nephritic factors), factor H, and to other complement regulatory proteins.
Membranoproliferative Glomerulonephritis
MPGN in the past was subdivided into type I, type II, and type III. Type II is now classified under C3 glomerulopathy as dense-deposit disease and so are most forms, particularly the genetic forms of type III MPGN. Forms of type I MPGN with C3 deposits only are also classified now as C3 glomerulopathy. Therefore, one has to reevaluate what really idiopathic MPGN is (38).
Epidemiology and Clinical Symptoms. Very similar to that described under C3 glomerulopathy. Serum C3 levels are frequently low because many patients with type I MPGN also have abnormal complement pathway activation.
Light Microscopic Features. By definition, glomeruli in MPGN have a membranoproliferative pattern; they are enlarged, hypercellular, and lobulated with frequent double contours along the glomerular capillaries on methenamine silver stain. Type I and type III MPGN are indistinguishable under the light microscope (type II MPGN is not considered a form of MPGN; it is discussed as dense-deposit disease under C3 glomerulopathy).
Immunofluorescence Characteristics. The dominant finding is glomerular C3 deposition in the mesangium and along the glomerular capillary loops. In MPGN type I and type III, unlike in C3 glomerulopathy, IgG deposits and sometimes other immune reactants may also be present. C1q and C4 are usually not seen in the glomerular deposits.
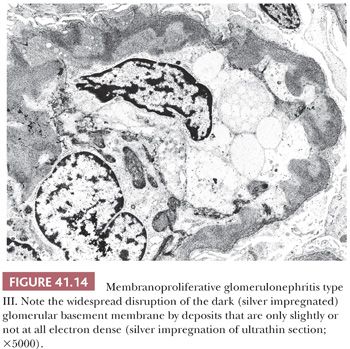
Electron Microscopic Features. In type I MPGN, in addition to mesangial deposits, numerous subendothelial deposits can be seen just like in C3GN (Fig. 41.12). Occasional intramembranous and subepithelial deposits, even large subepithelial “humps,” may occur. In type III MPGN, extensive glomerular subendothelial, subepithelial, and transmembranous deposits are seen along the glomerular capillaries (39,40). These deposits are frequently less electron dense than in type I MPGN, and they may blend into the irregularly thickened GBM. Sometimes, only silver impregnation of the ultrathin sections can reveal the deposits, which do not pick up the silver granules (Fig. 41.14).
Differential Diagnosis. The differential diagnosis is not different from that of C3 glomerulopathy. In MPGN, in addition to C3 deposits, IgG deposits are also present according to current criteria.
Outcome. Slowly progressive disease similar to C3 glomerulopathy. Recurrence in renal allografts occur in approximately 27% to 65% of patients according to different series (35).
Etiologic Factors and Pathogenesis. In the opinion of authors, it is likely that many forms of type I and type III MPGN are related pathogenetically to C3 glomerulopathy. In our and others’ experience, it is somewhat arbitrary to separate otherwise two very similar glomerular diseases (C3GN and MPGN) based on the absence or presence of IgG staining. Some degree of IgG staining is not unusual in renal biopsies otherwise entirely consistent with C3 glomerulopathy (26). The reclassification of MPGN/C3 glomerulopathy is clearly just evolving.
EXTRACAPILLARY PROLIFERATIVE CRESCENTIC GLOMERULONEPHRITIS
General Considerations
Crescentic GN is a pattern caused by, or associated with, a large number of different disease entities (Table 41.5). Crescents are defined as two or more cell layers between the parietal and visceral epithelia of the glomerulus. The definition of diffuse crescentic GN requires at least 50% of glomeruli to contain crescents.
Crescents are often associated with gaps, or “focal disruptive lesions,” of the GBMs through which large proteins (such as fibrin[ogen] and thrombin) and cells are thought to pass (41). It is this passage of fibrin(ogen) and cells (and possibly other growth factors) that is thought to be the stimulus for crescent formation. The cellular crescents consist of large polyhedral cells most likely derived from glomerular parietal epithelial cells and macrophages as well as other inflammatory cells (Fig. 41.15A). In rare instances, giant cells give the crescent a granulomatous appearance. Glomerular tuft necrosis, which is usually segmental, may occur in severe forms (Fig. 41.15B). The presence of fibrin is a common finding in cellular crescents.

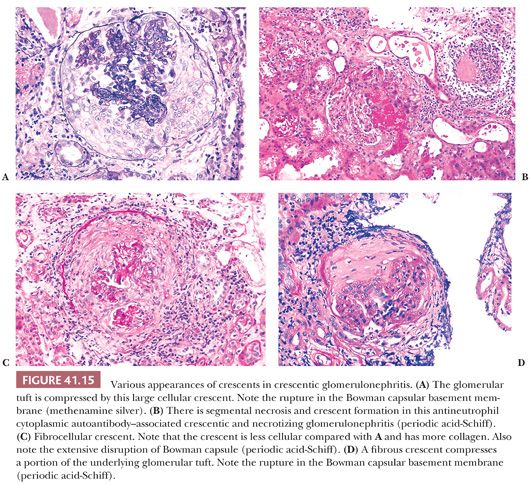
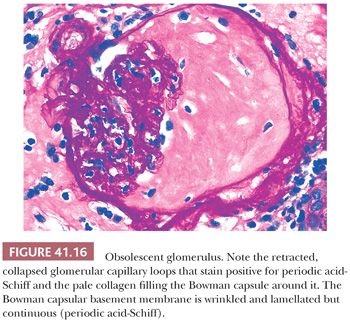
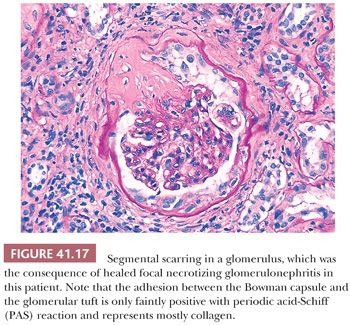
These cellular crescents quickly transform into fibrocellular crescents with the appearance of myofibroblasts and deposition of extracellular matrix, including interstitial collagen (Fig. 41.15C). Eventually, fibrous crescents develop, consisting mainly of collagen and a few fibroblasts (Fig. 41.15D). It is possible that myofibroblasts/fibroblasts migrate into the Bowman capsule through gaps in the Bowman capsular basement membrane. It may be difficult to differentiate an old fibrous crescent (Fig. 41.15D) from Bowman space fibrosis in obsolescent glomeruli secondary to ischemia or nephrosclerosis (Fig. 41.16). A good way to make the distinction is to perform a PAS reaction, which stains the basal lamina of Bowman capsule. If the Bowman capsular basement membrane is disrupted, the lesion probably represents an old crescent (Fig. 41.15D); if not, the glomerulus may simply be obsolescent from a noncrescentic cause (Fig. 41.16). Also, in obsolescent glomeruli, the PAS-positive wrinkled glomerular capillaries are retracted towards the vascular pole and are surrounded by faintly PAS-positive interstitial-type collagen. In typical fibrous crescents, interstitial-type collagen bands frequently separate remnants of PAS-positive glomerular matrix.
Inspecting glomeruli with the least amount of morphologic injury (i.e., those glomeruli without advanced crescents) by light IF and EM is always important because, if crescentic GN stems from a well-defined underlying immune complex glomerular disease (such as SLE, HSP, acute postinfectious GN, and others), the noncrescentic, or least involved, glomeruli may show diagnostic changes of the underlying disease.
Crescentic GN can be classified based on IF and EM into three types (Table 41.5): (a) pauci-immune, in which no or only insignificant deposits are noted; (b) anti-GBM disease, with intense linear deposits of IgG along the GBM (Fig. 41.14); and (c) immune complex crescentic GN, with diffuse granular IF and discrete immune-type, electron-dense deposits visible on EM. In idiopathic (or primary) immune complex crescentic GN, the pattern of immune complex deposition is not diagnostic of any well-defined immune complex GN (such as MPGN, SLE, postinfectious GN, etc.). In the secondary forms, the pattern of an underlying well-defined immune complex GN can be recognized in better preserved or noncrescentic glomeruli.
Pauci-Immune Crescentic and Necrotizing Glomerulonephritis
Epidemiology. The incidence of this form of crescentic GN in renal biopsy material appears to be increasing. This is the most common form of crescentic GN seen today (particularly if we disregard immune complex glomerulonephritides with some focal secondary crescent formation). It develops most often in the elderly population, and it rarely occurs in children.
Clinical Symptoms. Acute renal failure (often rapidly progressive), usually with nephritic syndrome (rapidly progressive GN), is the rule. The disease can be renal limited or associated with systemic vasculitis, such as granulomatosis with polyangiitis (formerly known as Wegener granulomatosis), microscopic polyangiitis, or rarely with eosinophilic granulomatosis with polyangiitis (Churg-Strauss syndrome) (42). Granulomatosis with polyangiitis usually involves the upper and lower respiratory tract and the kidney with necrotizing vasculitis affecting primarily small and medium-sized vessels. Microscopic polyangiitis also involves small and medium-sized arteries, usually without granulomatous reaction, and can involve any organ in the body. Churg-Strauss syndrome can involve not only medium-sized arteries but also small vessels. In Churg-Strauss syndrome, renal involvement is less common but may occur (42). Antineutrophil cytoplasmic antibodies (ANCAs) are detected in approximately 90% of patients, irrespective of whether the disease is limited to the kidney or is systemic (42,43). ANCAs originally were detected with IF, and two subtypes, based on fluorescence pattern, were described. So-called perinuclear antineutrophil cytoplasmic antibody (p-ANCA) shows perinuclear fluorescence in alcohol-fixed neutrophil granulocytes, whereas cytoplasmic antineutrophil cytoplasmic antibody (c-ANCA) shows a diffuse cytoplasmic fluorescence. Enzyme-linked immunoassay is a more sensitive and specific method to detect ANCA. It has become clear that p-ANCAs are usually antibodies to myeloperoxidase, and c-ANCAs are antibodies against proteinase-3, a serine protease in the azurophilic granules of neutrophils. Patients with granulomatosis with polyangiitis (Wegener granulomatosis) usually have c-ANCA, whereas patients with polyarteritis and kidney-limited disease have predominantly p-ANCA (41). However, the finding of ANCA (particularly p-ANCA) is not specific to crescentic GN. It is interesting that in approximately 5% of patients with ANCA-positive crescentic GN, anti-GBM antibodies also can be detected (44).
Light Microscopic Features. Pauci-immune crescentic and necrotizing GN can be diffuse or focal. In addition to crescents, segmental glomerular tuft necrosis and fibrin exudate in the crescents are typical findings (Fig. 41.15A,B) (41,45). Noncrescentic glomeruli and glomeruli with small crescents usually show no or little intracapillary hypercellularity. In the glomeruli with large crescents, the compressed glomerular tuft may appear hypercellular because of nuclear crowding. Secondary interstitial nephritis, composed mainly of lymphocytes and macrophages, is almost always present. Berden et al. (46) proposed that T-cell tubulitis and tubular atrophy are independent predictors of impaired renal function 12 months after the biopsy. Eosinophils and plasma cells frequently are found in the interstitial infiltrate. Vascular necrosis is rarely seen in renal biopsy material, even when GN is associated with systemic vasculitis, because of the focally involved vessels and the limited tissue sample. Recently, classification of ANCA-associated pauci-immune crescentic GN has been developed by an international working group of renal pathologists (47), creating four classes of ANCA-associated GN: (a) focal ANCA-associated GN: Over 50% of glomeruli are normal (uninvolved by crescent formation or necrosis); (b) crescentic: Over 50% of the glomeruli are involved by cellular crescents and/or necrosis; (c) mixed: Less than 50% of glomeruli are normal (uninvolved by crescent formation), less than 50% of glomeruli show cellular crescents and/or necrosis, and less than 50% of glomeruli are globally sclerotic (usually with fibrous crescents); (d) sclerotic: Over 50% of glomeruli are globally sclerotic (usually with fibrous crescents).
Immunofluorescence Characteristics. Using the routine panel of antibodies, IF portrays negative or nonspecific findings. Some glomerular tuft C3 is commonly found. Fibrinogen is frequently found in crescents or in necrotic glomerular segments. While evaluating IF, one has to be careful evaluating the glomeruli with crescent formation and necrosis because necrotic glomerular segments frequently show nonspecific staining because of entrapped serum proteins.
Electron Microscopic Features. No, or only few, small, immune-type, electron-dense deposits are visible. Electron-dense fibrin strands and wisps, with their typical periodicity, ruptures or gaps in the GBM, and other features of severe glomerular injury are commonly seen.
Differential Diagnosis. IF and EM are necessary to exclude immune complex GN or anti-GBM disease. Certain cases with ANCAs may also have anti-GBM antibodies (see discussion of anti-GBM disease). It is worth noting that ANCAs can be detected in certain diseases that are not usually associated with crescentic GN, such as “collagen vascular diseases,” inflammatory bowel disease, and some infectious diseases to name just a few.
Outcome. In untreated cases, there is rapid development of irreversible glomerulosclerosis and scarring. With aggressive treatment (cyclophosphamide/rituximab plus steroids), however, the 2-year patient and renal survival rates are greater than 70% in ANCA-associated cases (44,45,48,49). It appears likely that small early crescents, causing little glomerular tuft destruction, may heal with segmental Bowman capsule fibrosis and disruption with or without glomerular capillary loop adhesions if the injury is not continuous. If the glomerular tuft is affected as well, the healing process will result in segmental or global glomerular sclerosis and scarring (Fig. 41.17). The percentage of normal unaffected glomeruli appears to be the best histologic predictor of outcome (46,50). The recurrence rate of pauci-immune crescentic GN in renal transplantations seems to be relatively low (17%), probably because of the continuous immunosuppressive treatment of transplantation recipients (51).
Etiologic Factors and Pathogenesis. The pathogenetic role of ANCAs is debated; some argue that ANCAs merely represent an epiphenomenon. However, increasing number of clinical and experimental studies suggest the pathogenic role of ANCA (52). It appears that the alternate complement pathway activation also plays an important role in the glomerular/microvascular damage (53–55).
Antiglomerular Basement Membrane Disease
Epidemiology. This is the least common form of the three subtypes of crescentic glomerulonephritides. In our experience, it is found in less than 0.5% of all native renal biopsy specimens. It is a disease mainly affecting adults and is rare in children.
Clinical Symptoms. The renal-limited form manifests as acute renal failure, rapidly progressive GN, and nephritic syndrome. If GN is associated with pulmonary hemorrhage, the condition is classified as Goodpasture syndrome (56).
Light Microscopic Features. There is no way to distinguish anti-GBM disease from other forms of crescentic GN by LM alone (Fig. 41.15). The crescent formation may be focal or diffuse. Glomerular necrosis is typical. The noncrescentic glomeruli usually do not show intracapillary hypercellularity. An interstitial inflammatory cell infiltrate, just like that found in other forms of crescentic GN, is often present.
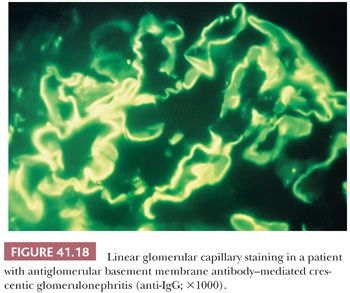
Immunofluorescence Characteristics. Intense linear staining for IgG and C3 along the glomerular capillary walls is characteristic (Fig. 41.18). C3 is often weaker than IgG, and in certain cases, only IgG is detected. When there is intense linear immunofluorescent staining, a search for anti-GBM antibodies in the circulation is in order. Indirect IF, using the patient’s serum placed on normal kidneys, is the easiest method, but it is neither specific nor sensitive. Radioimmunoassay for circulating anti-GBM antibodies is the benchmark diagnostic procedure.
Electron Microscopic Features. EM is not helpful in differentiating anti-GBM disease from pauci-immune crescentic and necrotizing GN because no, or only very few, vague, small, electron-dense deposits can be detected in either case.
Differential Diagnosis. Linear staining for IgG (in our experience, IgG1 dominant) by IF is not sufficient for the diagnosis of anti-GBM antibody–induced GN. The diagnosis must be confirmed by the presence of circulating anti-GBM antibodies in patients’ serum samples (56). False-positive, mild, linear staining by IF may be noted most commonly in patients with diabetes mellitus but even in 10% of supposedly normal kidneys from autopsy. A similar, mild, linear glomerular capillary fluorescence for albumin indicates the nonspecific nature of the IgG fluorescence. In monoclonal Ig deposition disease (mostly light chain, rarely heavy chain, deposition disease), only one of the two light chains (κ or λ) or a single subclass of heavy chain is deposited. In fibrillary GN, linear glomerular capillary IgG (usually IgG4 with or without IgG1) fluorescence can be noted, but it is frequently interrupted and segmental. In all of these conditions, mesangial staining with the same immunoreactants is also seen, and EM and laboratory data will help in establishing the correct diagnosis.
Finally, we have to reiterate that there is an overlap between ANCA-associated crescentic GN and anti-GBM disease. Approximately 30% of patients with anti-GBM antibodies also have ANCAs (38,44). It is possible that in these cases, anti-GBM antibodies appear following severe destruction of the GBM and subsequent release of GBM material into the circulation.
Outcome. The prognosis is frequently poor. End-stage renal disease (ESRD) develops in many patients within a short time. Aggressive therapy may somewhat improve the outcome, but results are worse than in ANCA-associated forms. Plasmapheresis may be helpful in the elimination of circulating anti-GBM antibodies. Poor prognostic indicators are oligoanuria at onset and diffuse involvement of glomeruli by crescents and necrosis (56,57). The recurrence rate in renal transplants is low, probably caused by the associated continuous immunosuppressive therapy. Delaying transplantation also decreases the chance of recurrence. In 50% of patients with transplantations who have had anti-GBM disease, linear GBM staining may be noted, but this is rarely accompanied by crescent formation and nephritic syndrome (34).
Etiologic Factors and Pathogenesis. Binding of anti-GBM antibodies to normal (native) GBM epitopes and subsequent complement activation initiate the inflammatory response and tissue injury. The Goodpasture epitope is on the alpha 3 chain of type IV collagen. Why anti-GBM antibodies appear is unknown. Cross-reaction with environmental antigens, including infectious agents, is one possibility. The best “experimental” model has been established in humans. Classic anti-GBM disease will develop in some patients with Alport hereditary nephropathy who lack or have an aberrant Goodpasture epitope when they undergo transplantation with normal kidneys (which have the normal Goodpasture epitope) (58).
Idiopathic (or Primary) Immune Complex Crescentic Glomerulonephritis
Immune deposits are present in the glomeruli, but the pattern is often not consistent with any of the known immune complex glomerulonephritides, such as MPGN, lupus nephritis, postinfectious GN, IgA nephropathy, and others. If one of the well-defined patterns of immune complex GN is identified, crescentic GN is considered to be secondary and is best defined as a crescentic MPGN, a crescentic postinfectious GN, or another form based on the underlying immune complex disease of the glomerular tuft.
Epidemiology. Idiopathic immune complex crescentic GN is rare. The secondary forms (crescentic GN superimposed on a well-defined immune complex GN) are more common.
Clinical Symptoms. The clinical picture is not different from that of other forms of crescentic GN (see relevant earlier sections), except for negative ANCA and anti-GBM antibodies. Rarely, ANCA may be positive.
Light Microscopic Features. In essence, the light microscopic features are similar to those of other forms of crescentic GN. Glomerular intracapillary hypercellularity is more frequently evident than in other forms, although this is not always the case. In contrast, glomerular necrosis is probably less frequent.
Immunofluorescence Characteristics. There is obvious granular staining for IgG and/or C3 and sometimes IgM in the mesangium and frequently along the glomerular capillary loops as well. If strong IgA or C1q staining is noted, the possibility of IgA nephropathy, HSP, or lupus nephritis should be considered, respectively.
Electron Microscopic Features. Discrete, immune-type, electron-dense deposits are usually noted in the glomerular mesangium and scattered in or along the GBM. The distribution of these deposits in idiopathic immune complex crescentic GN does not follow a well-defined pattern (such as in membranous GN or postinfectious GN).
Differential Diagnosis. An underlying pattern of a well-known primary immune complex GN (such as SLE, HSP, IgA nephropathy, etc.) should be considered. Careful examination of the LM, IF, and EM patterns in the most well-preserved glomeruli is the best method to differentiate idiopathic (primary immune complex) crescentic GN from the secondary forms. The possibility of infection should always be considered in the background. Endocarditis may be associated with immune complex crescentic GN, and sometimes, in such cases, even the ANCA test may be positive. If crescentic GN is IgA dominant, the possibility of an underlying Staphylococcus infection should be considered, particularly in an adult. In children and younger patients, HSP should be in the differential diagnosis in such cases.
Outcome. The prognosis of idiopathic (primary) immune complex crescentic GN is worse than that of other forms of crescentic GN. The secondary forms may have somewhat better outcomes, particularly crescentic poststreptococcal GN. There is not enough information regarding the recurrence rate in renal transplants, but in general, the recurrence rate of crescentic GN is low.
Etiologic Factors and Pathogenesis. Immune complex deposition obviously plays an important role in this disease process.
The Issue of Pulmonary-Renal Syndrome
Pulmonary hemorrhage and/or infiltrate associated with GN can be seen in many conditions other than anti-GBM antibody–induced GN. Such cases are frequently designated “pulmonary-renal syndrome” (59). In fact, less than one-third (much less, according to some) of patients with pulmonary hemorrhage and/or infiltrate and glomerular disease actually have demonstrable anti-GBM disease. Pulmonary hemorrhage and infiltrate in association with GN can be seen in anti-GBM antibody–induced disease; vasculitides, including granulomatosis with polyangiitis; cryoglobulinemia; SLE; penicillamine therapy; HSP; and cases of GN combined with pulmonary edema or pneumonia.
FOCAL GLOMERULONEPHRITIS
Focal GN is a pattern of reaction to renal injury in which some, but not all, of the glomeruli are seen on histologic examination to have areas of severe hypercellularity, with closure of capillary loops, necrosis, and/or sclerosis (Figs. 41.17 and 41.19) (60,61). If the term focal is used in a diagnostic report, the exact percentage of affected glomeruli should be stated. Most investigators consider a glomerular disease focal if less than 50% of glomeruli are involved. The remaining glomeruli appear relatively normal on LM. Frequently, focal glomerular involvement is segmental, affecting only a portion of a glomerulus. The pattern of focal segmental glomerular hypercellularity may be caused by, or associated with, several disease processes, such as SLE (see detailed description in relevant section), infective endocarditis, deep-seated visceral abscess, infected atrioventricular shunts (shunt nephritis), polyarteritis (usually the microscopic form), Granulomatosis with polyangiitis and other vasculitides, IgA nephropathy/HSP (anaphylactoid purpura), Goodpasture syndrome, the cellular lesion in focal segmental glomerular sclerosis (FSGS), the edge of renal infarcts, and from time to time, many other renal diseases.
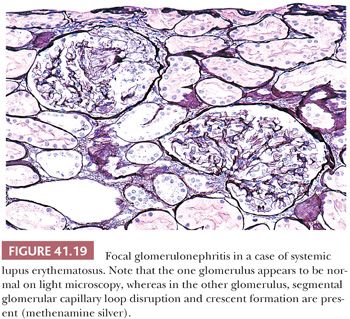
Many glomerular processes termed focal are not truly focal; they are focal only as seen by LM. For example, in patients with focal GN in Berger disease (IgA nephropathy), HSP, SLE, or the early stages of anti-GBM–induced GN, there are widespread, diffuse, often global glomerular immune deposits noted on IF and/or EM. What are focal are the manifestations of diffuse deposition of immune and immune-like materials on LM. However, in diseases where no immune complex deposition can be detected, such as in many vasculitides and ANCA-associated glomerulonephritides, the process truly appears to be focal. Focal GN can heal with FSGS and scarring (Fig. 41.17). The distinction between healed focal GN (as in a treated SLE or polyarteritis) and idiopathic FSGS may occasionally be quite difficult (61).
THE NONGLOMERULONEPHRITIC GLOMERULONEPHROPATHIES (GLOMERULAR CAPILLARY WALL LESIONS LEADING TO THE NEPHROTIC SYNDROME)
These diffuse, glomerular capillary wall lesions are characterized by predominant or exclusive involvement of the glomerular capillary walls (usually little change in the mesangial regions). The glomerular tufts are usually not hypercellular. The patterns described here are almost always associated with marked loss of the permselectivity of the glomerular capillary walls with subsequent heavy proteinuria and quite often with the nephrotic syndrome.
MINIMAL CHANGE NEPHROTIC SYNDROME (MINIMAL CHANGE DISEASE)
Epidemiology
Minimal change disease (MCNS) occurs primarily and not infrequently in children, although it may be seen in adults. It is the most common cause of idiopathic nephrotic-range proteinuria in children and adolescents (62).
Clinical Symptoms
Severe proteinuria, usually in conjunction with nephrotic syndrome (edema, hypoalbuminemia, hyperlipidemia, and lipiduria), is the leading symptom. Proteinuria is “selective” in the majority of cases (urine protein consists mainly of albumin and low–molecular-weight proteins with little IgG or larger proteins) compared with the findings in other glomerular diseases that feature the nephrotic syndrome, such as FSGS, in which proteinuria is frequently “nonselective” (high–molecular-weight proteins, such as Igs, are also present in the urine). Mild microscopic hematuria may occur, but it is not characteristic. Renal function remains normal for the most part, but acute renal failure may develop in adults, particularly elderly patients with major shifts in their hemodynamics. The blood pressure characteristically remains normal. Serum complement levels are also normal. In adults and elderly patients with acute-onset heavy nephrotic syndrome and the morphology of minimal change disease, we always recommend to test the patients for underlying lymphoma because lymphomas may be associated with minimal change disease (63).
Light Microscopic Features
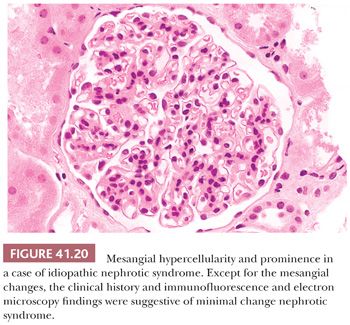
The renal parenchyma appears normal. In adults, and particularly in the elderly, there may be a few obsolescent glomeruli, with very mild focal interstitial fibrosis and tubular atrophy. If MCNS is associated with interstitial nephritis, nonsteroidal anti-inflammatory drug (NSAID)–induced renal disease should be suspected (see discussion of acute interstitial nephritis). Several minor LM changes have been found in what seems to be otherwise typical MCNS, such as mild mesangial prominence and hypercellularity (Fig. 41.20). The International Study of Kidney Disease in Children has shown that these minor variants do not seem to affect the patient’s response to steroids and that most of these patients appear to have a good prognosis with resolution of their severe proteinuria (64). Ectatic glomerular capillary lumina and slightly prominent podocytes may be seen.
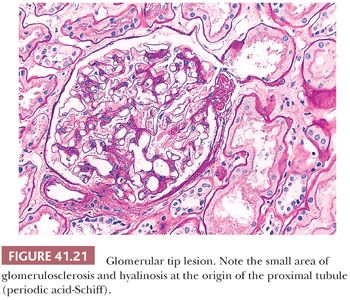
The adhesion and localized sclerosis of a glomerular capillary loop to Bowman capsule, adjacent to the origin of the proximal tubule, have been designated “glomerular tip lesion” (Fig. 41.21) (65). Tip lesions can develop in a variety of diseases with heavy proteinuria, including MCNS, but may not influence outcome (66). The Columbia classification of FSGS considers glomerular tip lesions as a variant of FSGS (see section on FSGS).
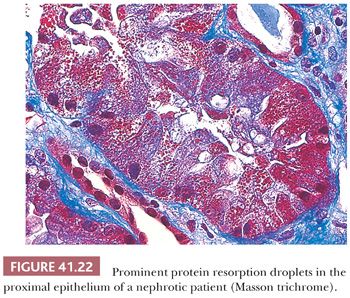
Tubular protein resorption droplets (also called hyaline droplets) are frequently found (Fig. 41.22). These are usually PAS positive, fuchsinophilic (dark red) with trichrome stain, and sometimes (but not always) methenamine silver positive. In elderly patients with MCNS, some degree of acute tubular necrosis may develop.
Immunofluorescence Characteristics
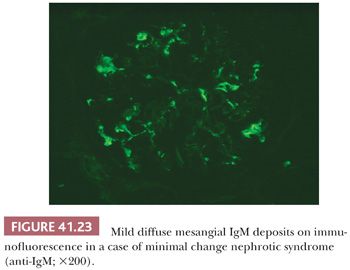
IF results are negative in most cases. Positive glomerular mesangial IF for IgM and C3 has been noted in some patients (Fig. 41.23). These are often cases with mild mesangial prominence.
Occasionally, immunoreactants other than IgM may also be present in otherwise typical MCNS, apparently without major clinical significance (67).
Electron Microscopic Features
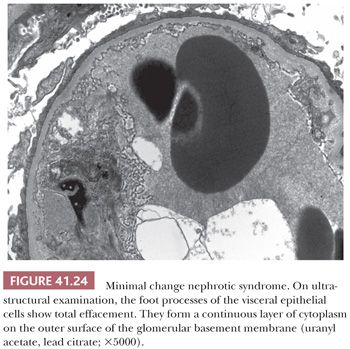
The most striking finding is the extensive loss of glomerular visceral epithelial cell foot processes (so-called fusion, which is not really fusion but rather effacement of visceral epithelial cell foot processes with swelling) (Fig. 41.24); no other prominent ultrastructural lesions of the glomerular capillary wall are usually noted. Immune-type, electron-dense deposits are usually absent. Many investigators believe that the effacement of the foot processes stems from enhanced permeability of the glomerular capillary wall. Foot process effacement may not be obvious in biopsy specimens taken after steroid treatment. Other nonspecific glomerular visceral epithelial cell changes, such as increased intracytoplasmic lipid and protein droplets, hypertrophy of the visceral epithelium with microvillous transformation, and basal condensation of intracytoplasmic microfilaments, are also frequently seen.
Differential Diagnosis
The main differential diagnostic problem is FSGS. A sampling error (absence of diagnostic segmentally sclerotic/hyalinized glomeruli) is difficult to exclude, particularly in biopsy specimens containing fewer than 10 glomeruli and in superficial specimens not containing the corticomedullary junction and juxtamedullary glomeruli (see later section on FSGS). The paraffin block of the biopsy material should be step-cut to make sure that every available glomerulus is examined. Although FSGS cannot be established in the absence of diagnostic segmental lesions, there are certain clinical and histologic features that favor FSGS, such as microscopic hematuria, nonselective proteinuria, enlarged glomeruli, and focal interstitial fibrosis with tubular atrophy. Recently, expression of certain markers by podocytes (such as decreased dystroglycan and CD44 expression in minimal change disease) was proposed as a possible tool to differentiate between early FSGS and minimal change disease (68,69). The practical usefulness of these methods remains to be confirmed (dystroglycan is probably not a sensitive method to differentiate between the two diseases). Recently, Hodgkin et al. (70) isolated mRNA from laser-captured glomeruli with biopsies showing FSGS and minimal change disease and described a distinct molecular profile in FSGS. In FSGS, transcription factors associated with developmental processes as well as genes known to be part of the slit diaphragm and genes associated with the so-called dysregulated podocyte phenotype were overexpressed.
In adults, particularly in elderly patients with severe proteinuria and glomeruli that appear normal on LM, early-stage membranous GN or early-stage amyloidosis should be considered. These conditions can easily be confirmed or excluded by IF, EM, and Congo red stain. A few patients with mesangial IgA deposition (IgA nephropathy) may have nephrotic syndrome and normal-appearing glomeruli; most respond to steroid treatment.
Outcome
Most patients respond to steroids with complete resolution of proteinuria. Some patients will become steroid dependent (proteinuria recurs after the tapering of steroids); others may be steroid resistant. Many of the steroid-resistant patients turn out to have FSGS according to follow-up biopsy results.
Etiologic Factors and Pathogenesis
MCNS is considered as podocytopathy, which is transient and responsive to steroids by most investigators. However, the exact underlying pathogenesis is unclear. On the basis of circumstantial evidence, some researchers have suggested that MCNS stems from an immune disorder. Both cellular and humoral immune responses have been implicated. The source of proteinuria is also unclear, but loss of the fixed glomerular capillary polyanionic layer(s), which normally lead to the negative charge of the glomerular capillary wall, may allow albumin and other negatively charged proteins to seep through in increasing amounts. Circulating serum factors, cytokines that amplify glomerular capillary permeability, may also be of importance (71,72). Recently, it has been found that the amount of nephrin or podocin in the podocytes is decreased and its distribution is altered in MCNS (73,74). Nephrin and podocin are molecules localized to the slit diaphragm between the foot processes. Therefore, it is reasonable to assume that changes in their expression may be involved in the pathogenesis of MCNS. Recently, it has been proposed that increased and persistent CD80 expression of podocytes is responsible for the foot process changes and proteinuria (75). MCNS is not always idiopathic; it may be associated with a variety of NSAIDs. In such cases of MCNS, interstitial nephritis also may develop. Symptoms usually regress on withdrawal of the drug (76). In the adult and elderly population, underlying lymphoproliferative disorder can also lead to acute-onset heavy proteinuria with minimal change disease (63).
DIFFUSE MESANGIAL HYPERCELLULARITY IN IDIOPATHIC NEPHROTIC SYNDROME
The association of diffuse mesangial hypercellularity (DMH) and idiopathic nephrotic syndrome (INS) was first described in 1970 (Fig. 41.20). Various studies have verified that DMH is present in 2% to 8% of children and adolescent patients with INS. This histopathologic form is thought by some investigators to be a variant of MCNS clinically associated with an increased incidence of microscopic hematuria; in some series, it has been reported to have a less favorable response to steroid therapy.
The response of children with DMH and nephrotic syndrome to treatment with steroids and/or cytotoxic agents is relatively poor compared with patients with MCNS, although the long-term renal prognosis appears to be good (at least in published short follow-up studies) (77). Some cases of nephrotic syndrome combined with DMH may represent undiagnosed FSGS; follow-up biopsies of these cases may show definite signs of FSGS. The majority of cases, however, probably represent a variant of MCNS with a greater than usual mesangial reaction. It has been shown that mesangial hypercellularity in MCNS may be a transient feature (67). In our opinion, DMH in INS should be considered an MCNS, particularly if the patient is steroid sensitive or dependent. If steroid resistance and, in particular, renal impairment develop, FSGS (i.e., undiagnosed FSGS) most likely lies in the background.
GLOMERULAR MESANGIAL IMMUNOGLOBULIN M DEPOSITION IN IDIOPATHIC NEPHROTIC SYNDROME (IMMUNOGLOBULIN M NEPHROPATHY)
In 1978, two groups independently described mesangial deposition of IgM in renal biopsy samples that otherwise appeared to indicate the presence of MCNS (Fig. 41.23) (78,79). Since that time, many reports of this so-called IgM nephropathy have been published, but the lesion continues to be the subject of controversy. In our opinion, the presence of mesangial IgM in the absence of other immunoreactants merely represents a disturbance in the mesangial transport of macromolecules and should not be considered an entity separate from MCNS.
Several observations support our hypothesis. First, reports that compare cases with and without mesangial IgM within a distinct glomerular morphologic pattern did not find a difference in outcome (62). Second, in cases where IgM is the only immunoreactant, EM usually does not show electron-dense, immune-type deposits in the mesangium, except for a few vague electron densities. If discrete electron-dense deposits are present, other immunoreactants, such as IgG, IgA, C3, and C1q, are almost always evident on IF. Third, mesangial IgM deposition may be present in normal kidneys (e.g., autopsies, donor kidney biopsies). Fourth, mesangial IgM may disappear on follow-up biopsy results (67). Fifth, we do not know the incidence of mesangial IgM in cases of steroid-sensitive MCNS because such cases are not subjected to biopsy.
Despite these arguments, we do not think that the presence of mesangial IgM is a totally innocent finding that should be disregarded. It probably represents mesangial dysfunction, and its presence, particularly in steroid-resistant cases, is a further argument in favor of the possibility that there is underlying early-stage FSGS. Rarely, we have seen cases with mesangial IgM and C3 in which small electron-dense deposits were evident on EM. These small deposits may truly represent IgM containing mesangial immune complexes, but their significance is unknown.
C1Q NEPHROPATHY
Epidemiology
This rare condition occurs mainly in children, adolescents, and young adults (80–83).
Clinical Symptoms
Severe proteinuria, often with nephrotic syndrome, is the predominant finding. Microscopic hematuria is common.
Light Microscopic Features
Renal biopsy either shows normal renal parenchyma or FSGS. Mesangial hypercellularity is a common finding (83).
Immunofluorescence Characteristics
Predominant mesangial C1q staining in the absence of SLE is a prerequisite for the diagnosis. In addition to C1q, IgG, IgA, IgM, and C3 may be seen; indeed, sometimes all are present.
Electron Microscopic Features
Electron-dense, immune-type deposits are always present in the mesangium. Glomerular subendothelial or subepithelial deposits are rarely seen. There is usually widespread foot process effacement.
Differential Diagnosis
SLE should be excluded before making the diagnosis. Patients with lupus with histologically normal glomeruli and mesangial deposits on IF and EM (WHO class II lupus nephritis) may have prominent mesangial C1q deposits but do not have nephrotic-range proteinuria. Endothelial tubuloreticular inclusions (frequently seen in lupus) are usually absent in C1q nephropathy. Minimal change disease and FSGS are easy to exclude on IF and EM.
Outcome
It appears that patients who have light microscopically normal–appearing glomeruli respond well to steroid treatment. In contrast, patients who present with the light microscopic pattern of FSGS show a poor response to steroids (83). ESRD has reportedly developed in some patients, whereas others did not progress to this stage.
Etiology and Pathogenesis
The origins of this condition are unclear.
FOCAL SEGMENTAL GLOMERULAR SCLEROSIS
It is important to emphasize that the morphologic pattern of FSGS and hyalinosis is nonspecific and that this pattern is associated with a variety of diseases. FSGS can be primary (idiopathic) or secondary. The secondary forms are by definition associated with an identified underlying primary disorder (84,85). The diagnosis of idiopathic FSGS should be made only after careful clinicopathologic correlation and exclusion of other possible underlying conditions (secondary forms).
Epidemiology
FSGS is one of the glomerular lesions that most frequently leads to progressive renal insufficiency in children. In major renal pathology laboratories in the United States, the proportion of biopsy samples with FSGS is on the rise; it represents the most frequently seen glomerular pattern, not only in children but also in adults, in whom it reaches or surpasses the incidence of membranous GN (85–88). This is not the case in Europe and Asia, where IgA nephropathy is the most common glomerular disease and where membranous GN is the typical cause of INS in adults. It is interesting that in the United States, African Americans are at considerably higher risk of FSGS than whites (85–88). There is a male predominance in most studies. Although the incidence of various forms of FSGS is rising, FSGS associated with intravenous drug abuse (heroin-associated nephropathy) is almost disappearing in the New York City area despite the continuing high incidence of heroin abuse (89).
Clinical Symptoms
Idiopathic FSGS is characterized by severe proteinuria; 70% to 80% of patients have overt nephrotic syndrome. There is often associated microhematuria, hypertension, and poorly selective proteinuria (as opposed to MCNS with “highly selective” proteinuria/albuminuria but no passage of larger proteins into the urine). In secondary forms of FSGS, the proteinuria is frequently nonnephrotic and may develop gradually through many years. In secondary forms of FSGS, it is not unusual to have nephrotic-range proteinuria in the absence of nephrotic syndrome.
Light Microscopic Features
Some, but not all, glomeruli are affected (focal involvement) (Fig. 41.25). Glomerular sclerosis means abnormal increase in methenamine silver and PAS-positive glomerular extracellular material (GBM material and mesangial matrix). Typically, in the affected glomeruli, only a portion of the glomerular tuft (segmental involvement) shows sclerosis, collapse, and closure of glomerular capillary lumens. Lining up of prominent visceral epithelium (“visceral epithelial cell caps”) along the sclerotic segments and adhesions or synechiae are common (Fig. 41.25B,C) (90). In the early stages, the segmental lesion may appear hypercellular primarily because of the hypertrophic podocytes overlying the sclerosing collapsing glomerular capillary loops. There may also be regions of superimposed hyalinosis (plasma protein insudation in sclerotic regions [Fig. 41.25C and Table 41.6], between the GBM and endothelium) and intracapillary foam cell accumulation. Because of the frequent presence of hyalinosis, the disease is also called focal segmental glomerular sclerosis and hyalinosis, although hyalinosis is not invariably present.
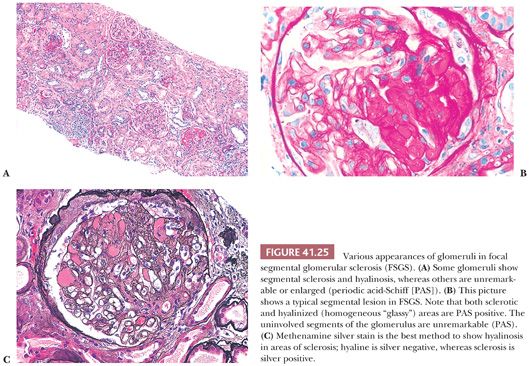
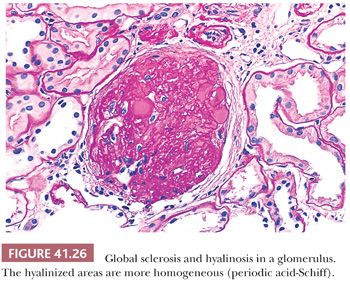
The segments of the glomerulus uninvolved by sclerosis appear to be normal on histologic evaluation, but eventually, with disease progression, the entire glomerular tuft will undergo sclerosis with or without hyalinosis (global involvement) (Fig. 41.26). The segmental process is thought by many investigators to begin in the juxtamedullary glomeruli and usually, but not always, at the vascular pole or hilum of the involved glomerulus, especially near the afferent arteriole. The other glomeruli are normal on histologic examination or may show only prominent glomerular visceral epithelial cells or ectasia of the glomerular capillaries. Glomerular enlargement is a common finding in FSGS. Mesangial prominence and hypercellularity away from the regions of focal and segmental sclerosis may be noted in FSGS, just as in MCNS (see previous discussion). Tubular atrophy, interstitial fibrosis and inflammation, and arteriolar hyaline are usually seen; they become more and more prominent with disease progression.
In 2004, a group of prominent renal pathologists proposed a pathologic classification of FSGS (91), which is now called the Columbia classification:
1. FSGS, not otherwise specified. Segmental glomerular sclerosis without features of the other listed forms; most common variant.
2. Perihilar variant. The segmental sclerosis is localized next to the vascular pole; common in some forms of secondary FSGS, such as in obesity-related FSGS.
3. Cellular variant. Segmental sclerosis with intracapillary hypercellularity with or without foam cells and karyorrhexis; least common variant, somewhat controversial.
4. Tip variant. Segmental sclerosis localized to the origin of the proximal tubule. Controversial to classify as FSGS; many patients respond to steroids and behave as MCNS clinically (see previous section on MCNS).
5. Collapsing variant. Glomerular capillary collapse with overlying hypertrophic glomerular epithelial cells (see the following text).
This morphologic classification appears to have relevant clinical correlation; the tip variant not surprisingly has the best outcome and response to treatment, whereas the collapsing variant has usually the worst response and outcome (92). We will discuss the collapsing variant separately under the section “Collapsing Glomerulopathy.”
Immunofluorescence Characteristics
In the regions of segmental glomerular sclerosis, IgM and C3 are frequently present. This may be related to nonimmune absorption of the large molecule IgM (600,000 to 900,000 Da in molecular weight) in the sclerotic areas, with complement possibly being secondarily activated locally. Diffuse mesangial IgM can be seen, comparable to that in cases of MCNS (see previous discussion). Weak, patchy fluorescence for fibrinogen is occasionally visible in glomerular capillaries (areas of sclerosis).
Electron Microscopic Features
All glomerular capillary walls (even in glomeruli that look normal on LM) display visceral epithelial cell foot process effacement, whereas the sclerotic regions show evidence of increased mesangial matrix, capillary collapse, and prominence of the overlying visceral epithelial cells. Vacuolation and focal detachment of the visceral epithelial cells from the GBM are frequently noted. Discrete, immune-type, electron-dense deposits are not present; however, a few small mesangial, variably electron-dense deposits may be visible. It is important not to misinterpret glomerular hyalinosis as immune deposits. Hyaline is a bulky, amorphous, electron-dense material frequently admixed with lipid droplets (Fig. 41.27). It usually forms right below the GBM in sclerotic areas with collapsed/sclerotic capillary loops and frequently contains lipid droplets. A good clue to their differentiation is to look at glomerular segments with normal-appearing glomerular capillary loops and mesangium away from sclerotic areas. In cases of segmental hyalinosis, no discrete “immune-type” deposits will be seen in these areas, whereas in cases of immune complex GN, deposits will generally be found throughout the glomerulus.
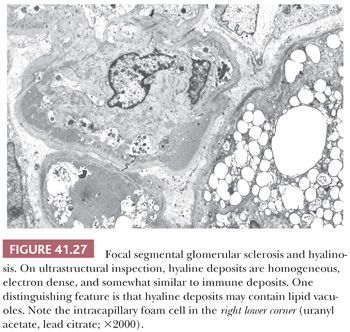
Differential Diagnosis
The differential diagnosis of FSGS and MCNS is discussed in the section on MCNS. Focal segmental sclerosis in glomeruli is a pattern or marker of renal response and not a specific disease entity. Some of the diseases and conditions in which segmental glomerulosclerosis may occur are IgA nephropathy, membranous GN, C3GN, “healed” focal GN, intravenous drug abuse–associated nephropathy, human immunodeficiency virus–associated nephropathy, collapsing glomerulopathy, long-standing hypertension (see benign nephrosclerosis), reduced renal mass (forms of renal hypoplasia/dysplasia and renal ablation/nephrectomy), morbid obesity, Alport syndrome, congenital cyanotic heart disease, sickle cell disease, and chronic pyelonephritis/reflux nephropathy. More and more familial cases are reported with abnormalities/mutations in a variety of genes (93,94). This list is far from complete, and in fact, a segmentally sclerotic glomerulus can be an incidental finding or a late, final common pathway of progressive and severe glomerular injury. Most important in the differential diagnosis is to carefully correlate the clinical history, clinical symptoms, and laboratory findings with the results of LM, IF, and EM. The diagnosis of idiopathic FSGS should be made in the appropriate clinical context only if secondary causes can be excluded.
Outcome
Idiopathic FSGS is a slowly progressive disease. Many patients fail to respond to therapy from the beginning, particularly those with more advanced lesions (85). The more glomeruli affected by segmental or global sclerosis and hyalinosis and the more widespread the interstitial fibrosis with tubular atrophy, the worse the prognosis is. Some investigators suggest that mesangial hypercellularity is an indicator of unfavorable outcome, but others cannot confirm this theory. Clinical indicators of poor outcome include increased serum creatinine at diagnosis, high blood pressure, and severe nephrotic-range proteinuria resistant to therapy. African Americans apparently have a more rapid progression (88). The recurrence rate of FSGS in renal transplants is approximately 30% to 40%. Young age, development of renal failure within 3 years of diagnosis, and, possibly, mesangial prominence predispose to recurrent disease (95). Once there is a recurrence, the recurrence rate in subsequent grafts is more than 80%.
Etiologic Factors and Pathogenesis
The pathogenesis of FSGS may involve immunologic, genetic, toxic, or systemic factors, as well as mesangial dysfunction, coagulation, or hemodynamic factors (85). Whether the primary lesion is mesangial, visceral epithelial, capillary wall, or hemodynamic is still debated. It has been suggested by Brenner et al. (96) that progressive loss of nephron mass may, through loss of normal reflex vasoconstriction of the afferent arteriole, allow (a) hyperperfusion of the remaining glomeruli, with increased capillary pressure across the glomerular capillary wall and subsequent hyperfiltration (hyperperfusion/hyperfiltration injury); and (b) functional mesangial changes and, eventually, development of FSGS irrespective of the pattern of initial glomerular damage. Thus, it may be a common final pathway of progressive glomerular injury. This may be true in many conditions, but recently, it became evident that a proportion of patients with idiopathic FSGS have a circulating glomerular permeability factor (97). Heavy proteinuria immediately or very shortly after transplantation frequently develops in these patients if ESRD develops and they undergo renal transplantation. The circulating permeability factor(s) can be detected with laboratory methods. This has practical importance because the permeability factor(s) can be removed by plasmapheresis. Unfortunately, the exact nature of the permeability factor(s) is still unclear. Numerous candidate factors were proposed, including circulating urokinase receptor (98). However, the clinical relevance of these proposed circulating factors still has to be validated. Genetic associations are gaining more and more relevance as well. At the present time, at least 24 genes have been associated with FSGS (94). Some of these include slit diaphragm proteins such as NPHS1, NPHS2, CD2AP; cytoskeleton proteins such as ACTN4, MYH9, INF2; mitochondrial proteins such as IN2F, COQ2; DNA repair/transcription genes such as WT1, LMX1B; and others (94). In recent years, two candidate genes were implicated to explain the higher incidence of FSGS in African Americans. The MYH9 gene encoding the myosin heavy chain 9, a nonmuscle myosin, was implicated first (99,100). However, later it turned out that an adjacent gene encoding apolipoprotein L1 (APOL1) has a much stronger association with FSGS in African Americans. The APOL1 risk alleles are the G1 and G2 alleles. Interestingly, these are protecting against infection to certain strains of Trypanosoma brucei, a natural gene selection in the African population (101,102). In spite of all of these new discoveries, still most cases of FSGS are sporadic and not familial.
THE ISSUE OF FOCAL GLOBAL GLOMERULOSCLEROSIS IN IDIOPATHIC NEPHROTIC SYNDROME
Focal global glomerulosclerosis associated with INS is considered to be a separate entity in some studies. These patients appear to be responsive to steroids and have outcomes similar to those of MCNS; some of them, however, progress to ESRD (103). We have strong doubts that seeing one, or a few, globally sclerotic glomeruli admixed with normal glomeruli in nephrotic patients warrants a separate diagnosis. It is possible to see a few obsolescent glomeruli (up to 10% in adults younger than 40 years of age) in any condition, even in normal kidneys and MCNS (Fig. 41.16) (104). Thus, it is important to decide in such instances whether the glomerulus is truly sclerotic secondary to a glomerular disease process or just obsolescent secondary to aging or nephrosclerosis (for comparison, see Figs. 41.16 and 41.26). A useful hint, when the glomerulus in question contains hyalinosis, is that hyalinosis is not usually present in obsolescent glomeruli. If true sclerotic glomeruli are seen, particularly in association with hyalinosis, a primary glomerular disease, such as FSGS (idiopathic or secondary), should be considered. One has to remember that in conditions with the pattern of FSGS, one might miss the diagnostic segmental lesions but see globally sclerotic glomeruli. If FSGS only involves 10% of the glomeruli and the biopsy specimen only contains 10 glomeruli, the sampling will easily miss the lesions.
COLLAPSING GLOMERULOPATHY INCLUDING HUMAN IMMUNODEFICIENCY VIRUS–ASSOCIATED NEPHROPATHY AND IDIOPATHIC COLLAPSING GLOMERULOPATHY
Human immunodeficiency virus (HIV)–associated nephropathy (HIVAN) and collapsing glomerulopathy are discussed as a variant of FSGS in most textbooks and reviews, including the Columbia classification of FSGS (91). There are some similarities between the glomerular morphologic findings in collapsing glomerulopathy and idiopathic FSGS; still, we believe that the morphologic features and, in particular, the clinical behavior of HIVAN are different enough to discuss it separately. Also, HIVAN and collapsing glomerulopathy in HIV-negative patients are so similar that they most likely represent very closely related diseases; therefore, we will discuss them together. The HIV-negative form of collapsing glomerulopathy can be associated with a variety of diseases and conditions, such as infection (parvovirus B19, cytomegalovirus); autoimmune diseases, such as SLE; thrombotic microangiopathies; other forms of obliterative vascular changes; medications (such as bisphosphonates and α-interferon); permeability factors; and genetic conditions (105–110). Still, many times, no underlying condition can be identified, and the disease is best classified as idiopathic collapsing glomerulopathy.
Epidemiology
HIV-positive African American male patients are most often affected with HIVAN, but African American women also may develop the disease (111–113). After the introduction of highly active antiretroviral therapy, the incidence of HIVAN is declining, and the HIV-negative form of collapsing glomerulopathy is now more common than HIVAN. HIVAN is unusual in whites. The HIV-negative form of collapsing glomerulopathy also primarily affects African Americans (114–116) with the exception of pamidronate (a bisphosphonate) treatment–associated collapsing glomerulopathy, which can occur equally in all races (117).
Clinical Symptoms
The disease is characterized by severe, usually nephrotic-range proteinuria with a relatively short clinical history. Renal impairment is evident at the time of diagnosis in most patients, and progression to renal failure is usually rapid. HIVAN is usually a late manifestation of HIV infection. Collapsing glomerulopathy in HIV-negative patients has very similar clinical features.
Light Microscopic Features
The characteristic glomerular lesion is a predominantly collapsing type of focal glomerulosclerosis that is segmental or, more frequently, global (Fig. 41.28A) (111–116,118). The collapsing capillary loops are best appreciated by PAS or methenamine silver stains. The glomerular epithelial cells, including podocytes, are particularly prominent; they are swollen, typically fill the entire Bowman space, and may contain large protein droplets in the cytoplasm. There is some disagreement whether these hypertrophic glomerular epithelial cells are parietal or visceral (podocytes) epithelial cells; probably both are represented (119). These large epithelial cells filling the Bowman space resemble crescents; therefore, the lesion is frequently designated as “pseudocrescent” (105). There is no increase in mesangial matrix. Unlike idiopathic FSGS, the tubulointerstitial changes are also very characteristic. There is usually quite striking interstitial edema or fibrosis with tubular loss and atrophy at the time of diagnosis. A distinctive (but not specific) finding is microcystic dilatation of a few tubules, with a scalloping outline (Fig. 41.28B). Large numbers of protein resorption droplets are frequently seen in hypertrophic proximal tubular epithelial cells. A prominent interstitial inflammatory cell infiltrate, which consists mainly of lymphocytes and macrophages, occasionally with many plasma cells, is almost invariably present. In HIV-negative forms of collapsing glomerulopathy, the tubulointerstitial diseases may be less prominent.
Immunofluorescence Characteristics
As in FSGS, the findings on IF are negative or nonspecific unless the collapsing glomerulopathy is associated with SLE.
Electron Microscopic Features
In HIVAN, a distinctive but nonspecific change is the presence of endothelial tubuloreticular inclusions (Fig. 41.29). These inclusions are most likely not part of the renal disease process but rather a response to HIV infection. They reflect high α-interferon levels and are also called interferon footprints. They are also frequently seen in lupus nephritis and occasionally, in lesser amounts, in other conditions. In collapsing glomerulopathy of HIV-negative patients, these tubuloreticular inclusions are typically absent. Discrete, electron-dense, immune-type deposits are not part of HIVAN. In collapsing glomerulopathy associated with SLE, electron-dense, immune-type deposits are frequently seen.
Differential Diagnosis
HIVAN must be distinguished from other forms of FSGS, including secondary forms of FSGS (120). Changes characteristically seen in HIV-positive and HIV-negative collapsing glomerulopathy, and less frequently in other forms of FSGS, are collapsing glomerulosclerosis with prominent podocytes, prominent chronic and acute tubulointerstitial disease with microcystic dilatation of tubules, and endothelial tubuloreticular inclusions on EM. The only morphologic difference between HIVAN and the HIV-negative form of collapsing glomerulopathy is the presence of tubuloreticular inclusions in HIVAN (tubuloreticular inclusions are also seen in SLE-associated collapsing glomerulopathy). One has to be careful not to misinterpret the hypertrophic glomerular epithelial cells filling the Bowman space, the so-called pseudocrescents, as true crescents. Unlike in crescentic GN, in HIVAN/collapsing glomerulopathy, the glomerular capillary loops are collapsed, not disrupted; the Bowman capsule is intact; there is usually no fibrin in the Bowman space; and the hypertrophic podocytes frequently contain large protein droplets. Of course, any kind of renal disease, such as immune complex GN, can develop in HIV-positive patients, just as in the general population; they should not be diagnosed as HIVAN.
The definition of the cellular variant of FSGS is somewhat individual depending on investigators. Some use this terminology in all HIV-negative patients who show any degree of glomerular capillary collapse with prominent podocytes (cellular lesion), including the cases of collapsing glomerulopathy we alluded to previously (121). Others, including us, prefer to use the term “cellular lesion” or cellular variant of FSGS if the lesion is mostly segmental and is not associated with the prominent tubulointerstitial injury seen in HIVAN/collapsing glomerulopathy. However, the issue is controversial because there is a continuum of collapsing changes from the discrete segmental lesion in primary FSGS through the global collapsing lesion in HIVAN.
Outcome
ESRD develops in most patients with HIVAN within weeks or months of clinical onset. Highly active antiretroviral therapy may ameliorate not only the systemic HIV disease but also HIVAN (122). The outcome of HIV-negative collapsing glomerulopathy is also dismal but is somewhat better than that of HIVAN (109,110). If the underlying etiology is known and can be eliminated, the outcome obviously will improve, such as in pamidronate-induced collapsing glomerulopathy (117).
Etiology and Pathogenesis
The pathogenesis of HIVAN is still not precisely clear. Some experimental and human studies suggest that HIVAN is the direct result of renal parenchymal infection by HIV (123,124). The preponderance in African American men indicates a genetic predisposition. In fact, recently, it has been discovered that APOL1 risk alleles associate with HIVAN (125).
The pathogenesis of HIV-negative collapsing glomerulopathy is more obscure. As pointed out earlier, several conditions and diseases can be associated with HIV-negative collapsing glomerulopathy, including parvovirus infection, pamidronate, SLE, genetic disorders, and others (105,106,110,117,126). Interestingly, it appears that collapsing glomerulopathy associated with SLE is also related to the APOL1 risk alleles. It is quite plausible that other forms of collapsing glomerulopathy have the same gene associations. In both HIV-negative and -positive forms, the podocyte appears to be the primary target in the glomerulus, resulting in a dysregulated, dedifferentiated podocyte phenotype (105,109,127). However, some data indicate that parietal epithelial cells also proliferate and undergo hypertrophy in collapsing glomerulopathy (119).
MEMBRANOUS GLOMERULONEPHRITIS
Membranous GN is another morphologic pattern that can be either primary or associated with a variety of disorders (see the section on pathogenesis) (128). In recent years, it became evident that patients with primary membranous GN have circulating IgG4 antibodies directed against the phospholipase A2 receptor (PLA2R) (129,130). After this discovery, many investigators propose that membranous GN without apparent underlying etiology should be called primary rather than idiopathic. However, we have to note that not all patients with membranous GN without apparent underlying etiologies have circulating anti-PLA2R antibodies (130).
Epidemiology
Membranous GN is one of the two most common causes of INS in nondiabetic adults, particularly in the elderly, in most parts of the world. It is relatively rare in children.
Clinical Symptoms
The condition usually begins with the insidious development of marked proteinuria (usually with the development of the nephrotic syndrome). Microscopic hematuria and hypertension may be present. Circulating anti-PLA2R antibodies can be detected in 70% to 80% of patients with primary/idiopathic MGN (129–131).
Light Microscopic Features
In early-stage cases, the glomeruli may appear normal, and the diagnosis can be rendered only on the basis of IF and EM. The histologic picture is usually characteristic, particularly in more advanced stages. There is diffuse global thickening of the glomerular capillary walls (Fig. 41.30A,B) as the result of electron-dense deposits on the epithelial side of the GBM, with or without the formation of methenamine silver–positive projections of GBM-like material (so-called spikes) between these deposits (132,133). Note that the spikes (basement membrane surrounding the deposits), and not the deposits, are silver positive. If the glomerular capillary wall is cut en face or if the disease is advanced and the nonargyrophilic deposits are engulfed by basement membrane material, a moth-eaten or Swiss cheese–like appearance of the thickened GBM is noted in methenamine silver–stained sections on tangential sections of the GBMs (Fig. 41.30B). Masson trichrome stain sometimes reveals the fuchsinophilic subepithelial deposits if they are large enough. The glomeruli usually are not hypercellular. In some cases, however, mesangial prominence with or without hypercellularity may be noted, as will superimposed focal segmental sclerosis (the latter is especially obvious in advancing stages).
Immunofluorescence Characteristics
Diffuse, granular, glomerular capillary wall deposits of IgG (100% of cases), C3 (75% of cases), and sometimes other immunoreactants are virtually diagnostic of this disease (Fig. 41.31). Mesangial deposits may be present. The glomerular deposits in idiopathic membranous GN are usually IgG4 predominant (7). PLA2R (Fig. 41.32) can be detected along the glomerular capillary loops in the subepithelial deposits using indirect IF (134,135). In our and others’ experience, this is an easy staining method that can be performed on paraffin sections and is comparable in terms of sensitivity to the serum enzyme-linked immunosorbent assay test (135).
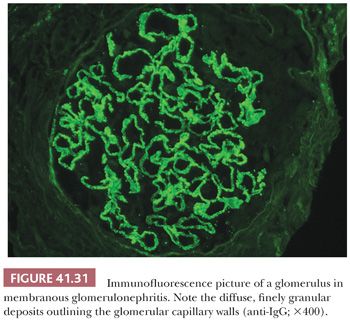
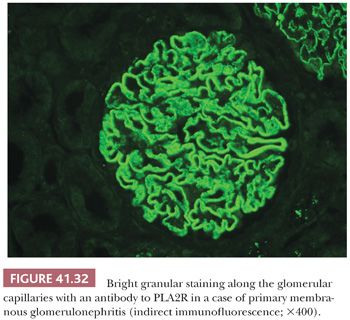
Electron Microscopic Features
EM is the best method for diagnosing membranous GN. There are numerous discrete, glomerular, subepithelial, electron-dense, “immune-type” deposits close to each other throughout the capillary walls (Fig. 41.33). As the disease progresses, basement membrane–like material will protrude between the deposits (spike formation). Mesangial electron-dense deposits, although not characteristic, may develop even in the absence of lupus nephritis.
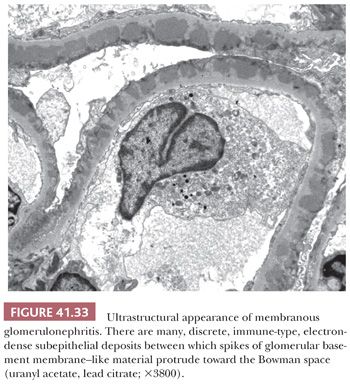
Staging of Membranous Glomerulonephritis
A widely used staging system was introduced by Ehrenreich and Churg (132), reflecting the morphologic stage of the disease based particularly on ultrastructural examination. In stage I, there are subepithelial deposits without spikes. Stage II has basement membrane spikes between the subepithelial deposits. By stage III, most deposits are engulfed by the thickened basement membrane; some deposits are still electron dense, whereas others have an electron-lucent appearance (“washed out” deposits). In stage IV, there are vague, washed-out deposits within the markedly thickened GBM. Stages III and IV may give the GBM a so-called tram-track appearance if impregnated with silver. There are many overlaps between these stages; the stages do not necessarily follow each other temporarily, and it is debatable whether this staging of membranous GN has a prognostic value.
Differential Diagnosis
Membranous GN is a morphologic pattern noted in association with many disorders. These secondary forms account for 20% to 50% of cases in many series (128). Until recently, primary and secondary forms could not be differentiated based on morphologic findings alone; however, now, using antibodies to the IgG subclasses and to PLA2R, most cases of primary MGN can be identified; in primary MGN cases, the glomerular deposits are predominantly IgG4 positive and they stain for PLA2R (7,134). Also, most patients with primary MGN have circulating anti-PLA2R IgG4 antibodies in the serum. However, differentiating between the secondary forms is still difficult. There are several secondary associations (128,133):
1. SLE (class V lupus nephritis). In lupus-associated membranous GN, there are always mesangial deposits. Ultrastructural examination usually reveals endothelial tubuloreticular inclusions. IF characteristically shows a “full house” pattern (in addition to IgG and C3, other immunoreactants, such as IgA, C1q, and IgM, are also present).
2. Neoplasms. Carcinomas of the lung, gastrointestinal tract, and breast are found, particularly in patients older than 60 years of age (128).
3. Chronic infections (hepatitis B, sometimes hepatitis C, syphilis).
4. Toxins and therapeutic agents (gold, mercury, d-penicillamine, rarely NSAID).
5. Several diseases, such as rheumatoid arthritis, thyroiditis, inflammatory bowel disease, and others.
6. De novo membranous GN in renal allografts could be difficult to differentiate from recurrent membranous GN. However, in recurrent membranous GN, the deposits are usually IgG4 dominant and PLA2R positive, whereas in de novo MGN, the deposits are usually IgG1 dominant and PLA2R negative (136,137).
Other glomerular patterns are easy to differentiate by IF and EM. Early-stage membranous GN (stage I) may be indistinguishable from MCNS by LM only. In advanced stages, LM features may resemble those of diffuse diabetic glomerulosclerosis or other advanced sclerosing glomerular diseases with glomerular capillary wall thickening and sometimes FSGS.
Outcome
Membranous GN is typically a very slowly progressive disease. The overall 10-year renal survival is between 77% and 90% (138). Segmental glomerular sclerosis, prominent interstitial fibrosis with tubular atrophy, very heavy and persistent proteinuria, and high blood pressure portend a more rapid progression. It appears that levels of circulating anti-PLA2R correlate with clinical disease and, therefore, measuring the serum levels can be a marker of response to treatment (131). Secondary forms may regress clinically if the underlying cause can be eliminated. Renal vein thrombosis is a fairly typical complication of membranous GN, as well as of other glomerular diseases (less commonly), leading to long-standing nephrotic syndrome. The recurrence rate of membranous GN in renal transplants appears to be relatively low, but this rate is difficult to assess because membranous GN is the most common de novo glomerular disease in renal allografts.
Etiologic Factors and Pathogenesis
There is good evidence, at least in experimental models, that many, if not most, examples of membranous GN represent in situ immune complex formation (i.e., circulating antibody binding to antigens normally present, or previously localized, along the glomerular capillary walls) rather than the deposition of circulating immune complexes along the glomerular capillary wall (139). As pointed out earlier, most patients with a primary MGN have circulating antibodies to PLA2R (129,130). However, other candidate antigens such as aldolase reductase, superoxide dismutase 2, α-enolase, neutral endopeptidase, and bovine serum albumin were all implicated, at least in some cases (130). Whether these antibodies have truly a pathogenetic role has to be confirmed. The causes of secondary forms were listed earlier.
DIABETIC NEPHROPATHY
Epidemiology
Diabetic nephropathy is the most common cause of ESRD in the United States. More than 40% of the dialysis population developed ESRD of diabetic nephropathy (140). It is estimated that by the year 2035, more than 590 million people in the world will be diabetic (141).
Clinical Symptoms
Microalbuminuria is the first warning sign of renal disease in patients with diabetes mellitus. With time, proteinuria becomes more and more severe and less selective. Eventually, most patients with diabetic nephropathy will experience nephrotic-range proteinuria and renal failure. Hypertension is common. Microscopic hematuria may occur, but it is not typical. Advancing renal disease usually shows good correlation with fundus changes, particularly in type 1 diabetes mellitus. This correlation is not very good in type 2 diabetes mellitus. In fact, most kidney biopsy samples with evidence of diabetic glomerulosclerosis are from patients with renal disease in the absence of eyeground changes (diabetic retinopathy) or from patients with an atypical clinical course, which may be an indication for renal biopsy.
Light Microscopic Features
There are no substantial histologic differences in the renal changes caused by type 1 and type 2 diabetes (142,143). In early-stage disease, only glomerular enlargement with or without mild mesangial expansion may be seen. The characteristic lesions of diabetic nephropathy are seen in the glomeruli and arterioles (Fig. 41.34).
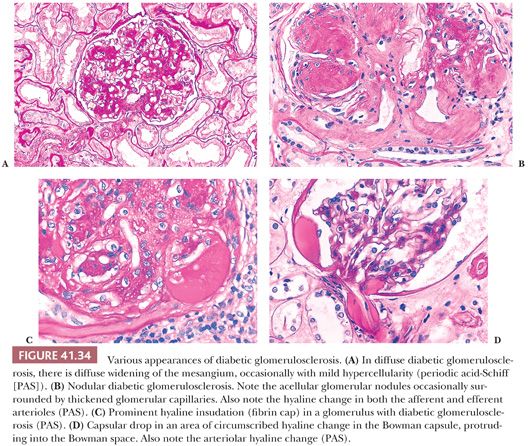
Diffuse diabetic glomerulosclerosis is characterized by an increase in the amount of mesangial matrix in all mesangial regions of all glomeruli, associated with diffuse thickening of the GBM. GBM thickening may not be detected by LM because the GBM must be at least two or three times the normal thickness to be recognized as thickened by LM (Fig. 41.34A).
Nodular diabetic glomerulosclerosis was first described by Kimmelstiel and Wilson in 1936, hence the name Kimmelstiel-Wilson nodule. There are sclerotic nodules in one or more intercapillary or mesangial regions of the glomerular tuft (Fig. 41.34B). These usually acellular nodules are often peripheral and laminated (with methenamine silver stain) and consist, on morphologic inspection, of material with the same staining properties as mesangial matrix and GBM. The acellular nodule may be surrounded by dilated glomerular capillary loops (microaneurysms). Nodular glomerulosclerosis is almost always superimposed on preexisting diffuse diabetic glomerulosclerosis. The number of nodules tends to increase with the clinical duration of diabetes.
The insudative glomerular lesions consist of the so-called fibrin cap (Fig. 41.34C) and the capsular drop (Fig. 41.34D). Originally named exudation, the term insudation more correctly describes the phenomenon. Both insudative lesions are the result of accumulation of plasma proteins (hyalinosis) between the glomerular endothelium and the GBM in the fibrin cap and between the parietal epithelial cell and Bowman capsule in the capsular drop (another lesion that Kimmelstiel thought was pathognomonic of diabetes mellitus). These insudative lesions are common in more severe patterns of advancing diabetic glomerulosclerosis most likely reflecting worsening glomerular hyperperfusion/hyperfiltration injury secondary to progressive nephron loss. Neither insudative lesion is pathognomonic of diabetic nephropathy, but both are more common and more severe in this disorder than in any other disease.
Diabetic glomerulosclerosis may be complicated by superimposed glomerular disease. Postinfectious GN or active infection–associated GN (such as Staphylococcus infection–associated GN) may be superimposed on diabetic glomerulosclerosis (18,144). We have seen several such cases in diabetic patients with infected leg ulcers. Membranous GN is another glomerular disease that is commonly superimposed on diabetic nephropathy. The association of membranous GN with diabetes mellitus may be coincidental, however, because membranous GN is the most common form of GN in the elderly, the population most often affected by type 2 diabetes mellitus.
The renal arterioles show insudative changes similar to those found in the glomeruli (arteriolar hyalinosis). Frequently, both afferent and efferent arterioles (the efferent hyalinization is quite characteristic of diabetes) show signs of prominent hyalinosis (Fig. 41.34B). Fibrous intimal thickening is a routine finding in arteries. These changes may be seen in the absence of hypertension and develop concomitantly with the glomerular changes; of course, diabetes mellitus and hypertension usually occur together.
Tubular changes include protein resorption droplets in the proximal tubules and tubular atrophy. The basement membranes of atrophic tubules in diabetic nephropathy can be conspicuously prominently thickened. Interstitial fibrosis and chronic interstitial inflammation may be so pronounced as to suggest an underlying chronic pyelonephritis or acute interstitial nephritis of other etiologies. These long-term tubulointerstitial changes can take place in the absence of infection and are probably related to the accompanying severe vascular disease, ischemia, and the progressive renal disease. However, severe acute and chronic pyelonephritis is not an unusual complication of diabetic nephropathy. Papillary necrosis, with or without pyelonephritis, is most often seen in diabetic patients (see discussion of analgesic nephropathy).
Recently, a new consensus classification of diabetic nephropathy was developed commissioned by the Renal Pathology Society (145). According to this classification, there are four classes of diabetic nephropathy:
Class I: There is GBM thickening seen by EM but no other obvious light microscopic criteria for diabetic glomerulosclerosis.
Class II: Mesangial expansion (formerly known as diffuse diabetic glomerulosclerosis). In class II, diabetic nephropathy subclasses are differentiated based on the severity of the mesangial expansion; class IIA represents cases with mild mesangial expansion in at least 20% of the glomeruli, and class IIB represents cases with severe mesangial expansion in at least 25% of the glomeruli in an absence of Kimmelstiel-Wilson nodules.
Class III: Includes cases where at least one glomerulus contains a convincing Kimmelstiel-Wilson nodule.
Class IV: Advanced diabetic glomerulosclerosis with over 50% globally sclerotic glomeruli.
This classification has good interobserver reproducibility, but its clinical value remains to be proven and validated with further studies.
Immunofluorescence Characteristics
A mild to moderate, nonspecific, linear, glomerular capillary loop staining for IgG is typical. The binding of the IgG antibody to the GBM is thought to be nonspecific and is probably a result of passive imbibition of serum proteins into the thickened, glycosylated GBM. Similar linear staining is almost always seen with antibodies to albumin. No anti-GBM antibody activity can be detected in these patients. Mild, linear IgG and albumin staining may be seen along tubular and Bowman capsular basement membranes as well, particularly along the basement membranes of atrophic tubules. Nonspecific deposition of the macromolecule IgM and C3 is evident in areas of sclerosis and hyalinosis.
Electron Microscopic Features
Thickening of the GBM and mesangial volume increase are the earliest lesions detectable (Fig. 41.35) (146); they become more prominent with disease progression. In nodular glomerulosclerosis, the nodules may show a fibrillary substructure (diabetic fibrillosis), which is probably a degenerative feature of the increased glomerular extracellular matrix and should not be misinterpreted as amyloidosis or fibrillary GN (Fig. 41.36) (147). Hyaline deposits have a similar appearance, as described for FSGS. Discrete, “immune-type,” electron-dense deposits are not seen; if present, they are suggestive of a superimposed immune complex GN. Foot process effacement is variable and does not show a very good correlation with the degree of proteinuria. Usually, patients with very heavy nephrotic syndrome have widespread foot process effacement, but foot process effacement may be relatively mild in many patients who have gradually developing nephrotic-range proteinuria.
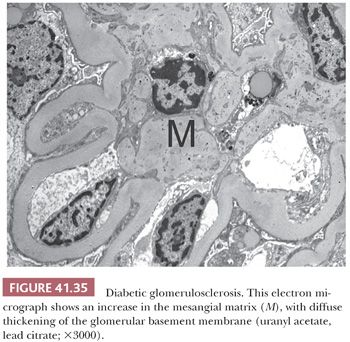
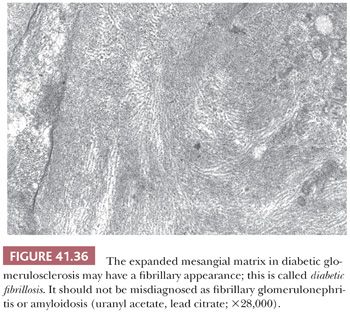
Differential Diagnosis
Sclerotic mesangial nodules are highly characteristic of diabetes mellitus, although similar lesions have been observed in some other conditions, such as monoclonal Ig (usually κ light chain) deposition disease, advanced MPGN, and congenital cyanotic heart disease. Amyloidosis can produce mesangial nodules, but these nodules are not sclerotic (they are methenamine silver negative) and consist of Congo red positive amyloid (Table 41.6). Idiopathic nodular GN is another possibility, if all of the other listed conditions can be excluded (32).
The sclerotic nodules in light chain deposition disease, advanced MPGN, and so-called idiopathic nodular GN differ from those in nodular diabetic glomerulosclerosis. They are usually greater in number and are more uniform in size within each glomerulus and throughout all glomeruli than in diabetic glomerulosclerosis. Moreover, glomerular and arteriolar hyalinosis is usually less severe than in diabetic nephropathy, and there is less thickening of the GBM in these nondiabetic conditions.
The differential diagnosis of diffuse diabetic glomerulosclerosis is more diverse. Primarily, immune complex GN should be excluded, including the mesangial glomerulonephritides (IgA, class II lupus nephritis, etc.), membranous GN, and MPGN. Long-standing hypertension may cause histologic changes similar to diabetic nephropathy, such as prominent arteriolar hyaline change, glomerular sclerosis and hyalinosis, and chronic tubulointerstitial changes. Monoclonal Ig deposition disease may also be associated with diffuse mesangial expansion.
Outcome
The speed of progression to ESRD from the clinical onset of microalbuminuria is very individual and may take anywhere from 3 to 20 years. Increasing proteinuria and worsening high blood pressure are signs of progressive disease. Recently, substantial efforts have been made to identify biomarkers of diabetic nephropathy that could be useful clinically to predict disease progression (148). Numerous biomarkers have been proposed, but none of them were developed into clinically useful tests so far. Recurrence of diabetic glomerulosclerosis, in the form of thickened GBMs, occurs in most long-surviving renal allografts (unless a type 1 diabetic patient received simultaneous kidney and pancreas allografts); however, the reappearance of typical Kimmelstiel-Wilson nodules is rarely seen within 5 years after transplantation.
Etiologic Factors and Pathogenesis
Although the exact cause of diabetic nephropathy is unclear, a basic feature of diabetes mellitus is an abnormality of the microcirculation. This microangiopathy is characterized on morphologic examination by thickening of the glomerular capillary basement membranes and in functional terms by increased permeability. Even the basement membranes of epithelial and nonepithelial cells may be abnormal (such as those of renal tubules, breast ducts, or Schwann cells). Increased production or decreased degradation of glomerular extracellular matrix, mesangiolysis with subsequent sclerosis and microaneurysm formation, glycosylation of glomerular extracellular matrix, genetic susceptibility, growth factors, and hemodynamic abnormalities, including glomerular hyperperfusion or hyperfiltration injury, may all have important effects (149).
GLOMERULAR DISEASES ASSOCIATED WITH ISOLATED HEMATURIA
One of the most perplexing and controversial problems involved in the use of renal biopsy is the finding of asymptomatic isolated hematuria. The most crucial initial determination to be made in evaluating patients with hematuria is identifying the likely site of bleeding. Many times, isolated hematuria is not related to a renal parenchymal disease and a urologic workup is warranted. Historically, the most helpful indicators of a glomerular source of bleeding have been cola-colored urine, proteinuria, and red cell or heme granular casts. Dysmorphic red blood cell morphology has also been suggested as a clue to the renal/glomerular origin of the blood. If phase-contrast microscopy and, less optimally, bright-field microscopy demonstrate misshapen, dysmorphic erythrocytes in the urine, “glomerular bleeding” is likely (150). On percutaneous renal biopsy, the finding of erythrocytes in either Bowman space or tubular lumina suggests that at least some of the hematuria is of renal (and probably glomerular) origin. One has to keep in mind that red blood cells may be pushed into tubules and the Bowman capsule during the biopsy process.
IMMUNOGLOBULIN A NEPHROPATHY (BERGER DISEASE) AND HENOCH-SCHÖNLEIN PURPURA (ANAPHYLACTOID PURPURA)
These two diseases are most likely related, and based on renal biopsy, there is no way to differentiate them. Thus, we will discuss them together. Because glomerular IgA may be seen in several other glomerular diseases, IgA nephropathy (also called Berger disease, after Jean Berger, who described it in 1968) is defined as an immune complex GN with the predominance or codominance of IgA in the glomerular mesangial regions in the absence of clinical evidence of HSP, SLE, or chronic liver disease (151–154). HSP is a systemic disease with vasculitis and multiorgan involvement, but the renal disease is quite similar to, if not the same as, IgA nephropathy. For this reason, many investigators consider them to be the same disease, representing a spectrum in which IgA nephropathy is the renal-limited form (153,155,156).
Epidemiology
IgA nephropathy is probably the most common glomerular disease in the world. This is particularly true in some countries, where it represents more than 20% to 30% of the conditions found on renal biopsy. In Europe, although slightly less frequently encountered, it is still the predominant glomerular disease (151–153). For unknown reasons, this is not the case in the United States, where membranous GN and FSGS are more often diagnosed. IgA nephropathy is less common in African Americans than in whites. The disease typically develops in young adults, but it may be found in any age group. HSP is much less common than IgA nephropathy and is mainly a disease of children (155).
Clinical Symptoms
Hematuria is invariably present, frequently only of microscopic extent, but the disease occasionally manifests with gross hematuria and the nephritic syndrome. Episodes of isolated gross hematuria may occur during the course of disease. Proteinuria can be very mild, but significant proteinuria (>0.5 g per 24-hour urine) is not unusual. In some cases, even nephrotic-range proteinuria is present. Renal function is usually normal at the time of diagnosis, particularly in patients who present with gross hematuria. In HSP, the renal symptoms are identical but are associated with the classic characteristic, defining signs of the systemic disease, including palpable purpura (due to leukocytoclastic vasculitis), abdominal pain, joint pain, and other symptoms.
Light Microscopic Features
IgA nephropathy does not have a truly characteristic light microscopic appearance; it shows a variety of patterns. Perhaps the most typical and classic glomerular lesion is a mesangioproliferative GN with focal or DMH and widening (Fig. 41.37). Other patterns seen on LM include FSGS with or without hyalinosis, focal and segmental proliferative or necrotizing GN, essentially normal-appearing glomeruli, and rarely, crescentic GN and a membranoproliferative pattern (151,157,158).
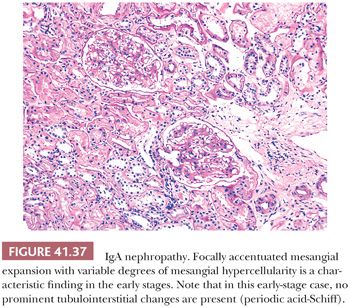
Because of the heterogeneity of light microscopic lesions in IgA nephropathy, there were several attempts to classify the disease. In our opinion, the best classification in the past was created by Dr. Mark Haas (157). According to the Haas classification, IgA nephropathy was subclassified into five subclasses (I—minimal or no mesangial hypercellularity without glomerular sclerosis, II—focal segmental glomerular sclerosis without active cellular proliferation, III—focal proliferative GN, IV—diffuse proliferative GN, V—chronic sclerosing IgA nephropathy with over 40% globally sclerotic glomeruli or over 40% interstitial fibrosis and tubular atrophy). More recently, the working group of the International IgA Nephropathy Network and the Renal Pathology Society came up with the Oxford classification of IgA nephropathy (159,160). The participants came to an agreement to score six types of lesions in biopsies with IgA nephropathy: (a) mesangial cellularity, (b) segmental glomerular sclerosis, (c) endocapillary hypercellularity, (d) cellular/fibrocellular crescents, (e) percentage of interstitial fibrosis and tubular atrophy, and (f) arteriosclerosis (160). The clinical correlations revealed that the mesangial hypercellularity score, the segmental glomerular sclerosis score, the endocapillary hypercellularity score, and the tubular atrophy/interstitial fibrosis score had independent value in predicting renal outcome (159). However, subsequent validation studies do not all agree on the validation of the Oxford classification, and this classification at the present time is best considered a working classification, which most likely will undergo substantial revisions (161).
Immunofluorescence Characteristics
The only way to diagnose IgA nephropathy is to perform IF on a renal biopsy sample. By definition, IF studies reveal diffuse, global, and predominant, or at least codominant, mesangial IgA fluorescence in all cases (e.g., no immunoreactant shows stronger fluorescence than IgA) (Fig. 41.38). IgG and IgM, usually of lesser intensity, are noted in approximately one-half to two-thirds of cases. C3 is found in more than three-fourths of cases. C1q fluorescence is usually absent; if strong C1q staining in association with glomerular IgA deposition is noted, the possibility of underlying SLE should always be considered. FRA is also often seen, as is properdin. In addition to mesangial fluorescence, granular glomerular capillary wall staining for IgA may be found, especially in more aggressive and more advanced cases.
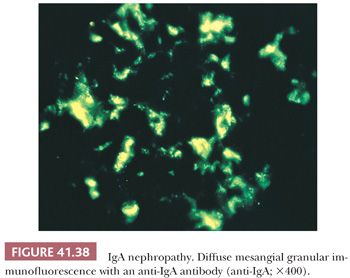
Electron Microscopic Features
Virtually, all biopsy specimens show discrete, electron-dense, “immune-type” deposits in the glomerular mesangial matrix (Fig. 41.39). In some (usually early stage) cases, they are preferentially located immediately below the basement membrane covering the mesangium (these are also called paramesangial deposits). Peripheral glomerular capillary wall deposits can be noted. In an early review by Navas-Palacios et al. (162) of 10 series (335 patients with IgA nephropathy), only 13% of patients had glomerular subepithelial deposits, 14% had intramembranous deposits, and 17% had subendothelial deposits. These deposits are usually small and are not as abundant and large as the peripheral glomerular capillary wall deposits seen in SLE in our experience, especially in children. Other investigators have found more numerous and larger glomerular capillary wall deposits in more severe forms of IgA nephropathy. If large subepithelial deposits (“humps”) are noted, the possibility of an underlying infectious process (particularly Staphylococcus infection) should always be considered.
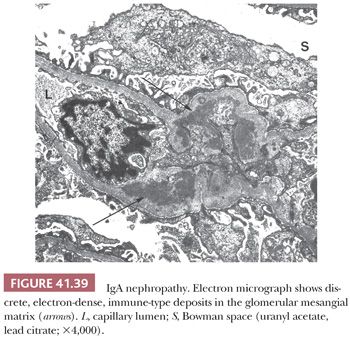
Differential Diagnosis
Because of the variable light microscopic appearances, the differential diagnosis at the light microscopic level may include any glomerular disease. IF is essential to the differential diagnosis. Other diseases in which mesangial IgA deposition can be prominent include lupus nephritis, Staphylococcus infection, and hepatic glomerulosclerosis. In lupus nephritis, IgA may be codominant, but it is rarely the predominant immunoreactant. In lupus nephritis, there typically is a “full house” IF (i.e., most immunoreactants, including C1q, are positive), and EM frequently reveals glomerular endothelial tubuloreticular inclusions. Still, in some cases, only clinical and laboratory data characteristic of SLE provide us with the correct diagnosis.
As detailed earlier in this chapter, in Staphylococcus infection, particularly in infection with MRSA, a proliferative GN with IgA-predominant deposits may occur. The glomeruli show a variable degree of hypercellularity, and there is usually prominent acute tubular necrosis with many red blood cells in the tubules. IF almost always shows strong glomerular C3 deposits and frequent glomerular capillary deposits. By EM, subepithelial deposits (“humps”) always point towards an underlying Staphylococcus infection in an IgA-dominant immune complex GN.
Hepatic glomerulosclerosis may have morphologic characteristics similar to those of IgA nephropathy. A history of chronic liver disease, usually with cirrhosis, is helpful distinguishing this condition. In hepatic glomerulosclerosis, usually the IgA deposits are weak to moderate and are not accompanied by prominent C3 deposition or IgG deposition. Also, the patients rarely have the clinical symptomatology of a GN. Much less commonly, mesangial IgA deposits may occur in inflammatory bowel disease, psoriasis, and lymphoproliferative disorders. As mentioned earlier, the only way HSP can be differentiated from IgA nephropathy is in terms of the presumed systemic disease in the former. One has to remember that HSP is a rare disease in adults, and if an adult patient, particularly if a diabetic patient presents with symptoms of apparent HSP, an underlying Staphylococcus infection should be considered.
Outcome
Disease progression is usually quite slow, but IgA nephropathy can progress to ESRD; in fact, it is a major cause of ESRD. Glomerulosclerosis, widespread crescents, and interstitial fibrosis/tubular atrophy are the main morphologic signs associated with poor outcome (151–153,158,162,163). Severe obliterative microvascular changes of thrombotic microangiopathy also portend a poor outcome (164). However, such patients usually have more advanced chronic renal injury and difficult-to-control hypertension. High blood pressure, severe proteinuria, and elevated serum creatinine levels at the time of diagnosis are clinical indicators of more rapid disease progression (152–154). Secondary forms of IgA (with the exception of Staphylococcus infection–associated GN) are usually clinically milder than primary forms.
It appears that the prognosis of HSP in children is better than that of IgA nephropathy. Many children with HSP experience complete remission and can be considered cured. Follow-up biopsies frequently show histologic signs of improvement in renal disease, although mesangial IgA deposits rarely disappear entirely (155). The recurrence rate of mesangial IgA deposits in renal allografts is approximately 50% in patients with IgA nephropathy and HSP, but the recurrent disease rarely causes graft failure (34,35). Although glomerular IgA deposits recur in HSP patients after renal transplantation, the recurrence of systemic disease is exceptional.
Etiologic Factors and Pathogenesis
The pathogenesis of IgA nephropathy is still unclear and may be based on many factors, including immunologic abnormalities, viral/food antigens, inability to clear IgA immune deposits, hereditary predisposition, and perhaps most importantly, abnormal glycosylation of IgA (152–154). There is growing evidence that in IgA nephropathy, the circulating IgA1 molecules are deficient in galactose in their carbohydrate side chains (O-glycans). These galactose-deficient IgA1 molecules elude the IgA1 catabolic pathway in the liver and reach the glomerular capillary tufts and eventually will get stuck in the mesangium. However, it appears that for the galactose-deficient IgA1 molecule to be nephritogenic, it has to form immune complexes (154).
THIN BASEMENT MEMBRANE DISEASE (OR NEPHROPATHY)
Epidemiology
Many patients with persistent, asymptomatic, microscopic hematuria may have widespread thinning of their GBMs. The disease may be familial or occur sporadically; it is found with equal frequency in adults and children in approximately 1% of the population (165,166).
Clinical Symptoms
The characteristic clinical picture is symptomatic persistent or recurrent microscopic hematuria. Proteinuria is absent or mild. Renal function is normal.
Light Microscopic Features
Normal renal tissue is noted on histologic examination in most cases. Red blood cells are usually present in tubular lumina.
Immunofluorescence Characteristics
IF using the routine panel of antibodies shows negative results. Sometimes, mild mesangial IgM and/or C3 deposition can be seen, which is most likely nonspecific. IF, using antibodies to the α3 and α5 chains of type IV collagen, reveals diffuse linear glomerular capillary staining (normal finding; see later section on Alport syndrome) (167,168).
Electron Microscopic Features
Widespread thinning of the GBM is required to make this diagnosis (Fig. 41.40) (165,169). Unfortunately, data in the literature on normal GBM thickness are somewhat variable, and the GBM thickness changes with age and differs between sexes. At birth, the GBM is approximately 150 nm thick, becomes thicker rapidly, and reaches a plateau by 9 to 10 years of age. Thereafter, there is only a very mild, gradual increase in thickness. Data in the literature regard normal GBM thickness in adults to be between 300 and 480 nm (168,169). In studies where men and women were evaluated separately, men had a GBM approximately 50 nm thicker than that in women. Interlaboratory variances are significant; thus, it is desirable for each laboratory to determine its own age- and sex-matched normal value. Some researchers measure the lamina densa only, whereas others measure the thickness of the entire GBM. In our experience, the latter measurement is easier to perform and is more reliable. Dische (169) suggests a cutoff value of normal minus 1.5 standard deviations (169). Based on earlier discussion, it is obvious that the diagnosis is somewhat arbitrary, and criteria may differ from laboratory to laboratory. We agree with Ivanyi et al. (170) that a considerable number of patients with isolated glomerular hematuria have only segmental GBM thickening. We even encounter biopsies from such patients with normal or thickened GBM.
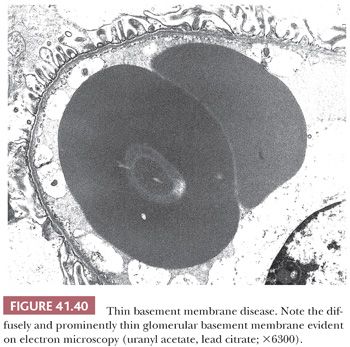
Differential Diagnosis
Segmental thinning of the GBM is a common nonspecific finding in a variety of renal diseases, such as FSGS. IgA nephropathy can easily be distinguished based on IF. Alport syndrome (hereditary nephritis) may be an important differential diagnostic consideration in some cases. There is evidence that Alport syndrome in some children initially manifests as thin basement membrane disease before the development of diagnostic GBM changes of Alport syndrome. Heterozygous patients for the X-linked and autosomal recessive forms of Alport syndrome may also have thin GBM as the only morphologic manifestation. Careful evaluation of the family history and the newly available monoclonal antibodies to the α3 and α5 chains of type IV collagen (see later discussion) are helpful in differential diagnosis (167). Fortunately, genetic testing is now available for Alport syndrome, which can help in the differentiation of ambiguous cases. In some cases of benign persistent hematuria, no GBM abnormalities can be identified. In such cases, further, more detailed urologic workup is recommended.
Outcome
Thin basement membrane disease, not representing Alport syndrome, appears to be a benign condition, and most patients do not show progressive renal disease. However, it appears that a few patients may have a slowly progressive renal disease (166).
Etiologic Factors and Pathogenesis
Thin basement membrane disease is clearly the result of heterogeneous abnormalities of the GBM. Recent data indicate abnormalities in the gene coding the α4 or α3 chains of type IV collagen (167,171,172). However, it is evident that in many patients, no abnormalities of these genes can be identified (173).
ALPORT SYNDROME
Epidemiology
The condition is rare and, in 80% of cases, shows an X-linked inheritance. The remaining patients have autosomal recessive and, exceptionally, autosomal dominant inheritance (172).
Clinical Symptoms
In affected men, microscopic hematuria initially develops, which later, with disease progression, is complicated by proteinuria and renal failure. Women usually have only microscopic, asymptomatic hematuria. A sensorineural hearing deficit is common. Ocular defects (anterior lenticonus), diffuse leiomyomatosis, and hematologic abnormalities (e.g., giant platelets, granulocyte abnormalities) are other, less typical associations (166).
Light Microscopic Features
LM can initially show normal results. In affected men, glomerulosclerosis, which may be segmental, will develop later. Interstitial fibrosis with tubular atrophy is seen in conjunction with glomerulosclerosis. The presence of interstitial foam cells in the absence of nephrotic-range proteinuria is characteristic (but not specific) of Alport syndrome (Fig. 41.41). In fact, all light microscopic changes in Alport syndrome are nonspecific.
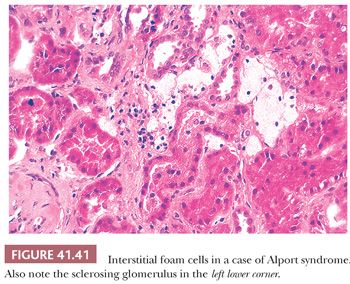
Immunofluorescence Characteristics
Antibodies of the conventional IF panel do not show specific immunostaining. In normal (non–Alport-affected) kidneys, fluorescein (or peroxidase)-labeled monoclonal antibodies to the α3, α4, and α5 chains of type IV collagen stain the GBM, the basement membranes of distal convoluted tubules, and Bowman capsule in a linear fashion. In X-linked Alport syndrome, this basement membrane staining is usually, but not always, absent (168,174) (Fig. 41.42). The staining in heterozygous women may show a mosaic pattern (interrupted or segmentally weaker staining), but this feature should be interpreted with caution (175). In autosomal recessive form of Alport syndrome, similarly to the X-linked variant, there is no staining for the α3 antibody, but the α5 antibody may stain the Bowman capsule and distal TBMs (168). Appropriate controls should include, at a minimum, a renal biopsy specimen with proven Alport syndrome and renal tissue with normal basement membranes. Anti-GBM antibodies purified from patients with anti-GBM disease or from those with Alport syndrome in whom anti-GBM disease develops after transplantation can also be used, but they are less specific. These immunohistochemical methods should be considered auxiliary at the present time, and the diagnosis of Alport syndrome should not be rendered based only on these immunohistochemical findings. IF on skin biopsies with antibodies to the α5 chain of type IV collagen may be useful in identifying affected patients; the epidermal basement membrane lacks staining in patients with Alport syndrome (in autosomal recessive Alport syndrome, α5 chain staining may be retained along the epidermal basement membrane) (168,176).
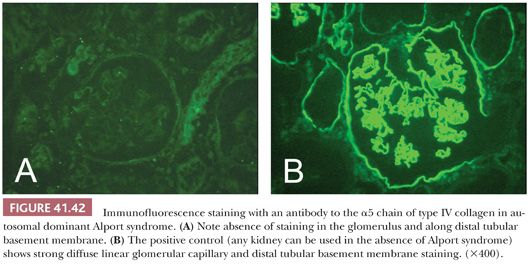
Electron Microscopic Features
Diffuse or widespread splitting, splintering, and lamellation of the lamina densa and the entire GBM, with rarefactions, are the most characteristic findings (Fig. 41.43) (168,174–176). Occasionally, electron-dense concretions are evident, as is thinning of the GBM. The exact percentage of glomerular capillaries that must show these changes is unclear, but some have suggested that 50% is a good diagnostic figure. The irregular basement membrane is usually thicker than normal. Unfortunately, these changes are not always diffuse, and at early stages and particularly in young children, only segmental irregularities with GBM thinning are seen. It is important that recent studies correlating ultrastructural and genetic findings indicate that not all patients with the genetic mutations of Alport syndrome have ultrastructural GBM abnormalities (174).
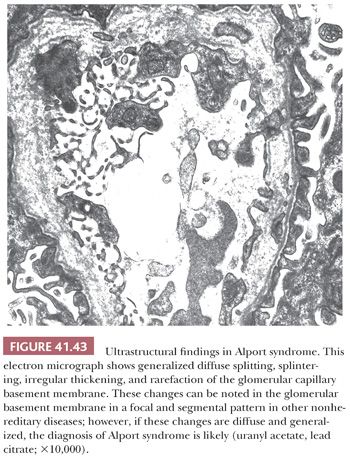
Differential Diagnosis
In a classic, fully developed case, diagnosis is straightforward. However, distinguishing Alport syndrome from thin basement membrane disease in children and in female carriers may be difficult. The most important differential diagnostic approach is to carefully evaluate the family history. The immunohistochemical stains detailed earlier are also useful. Focal and segmental irregularities of the GBM similar to those in Alport syndrome can be seen in a variety of sclerosing glomerular diseases, such as FSGS; in immune complex glomerulonephritides after resorption of capillary loop deposits; and in other renal conditions (177). A large Italian study indicated that the combined use of collagen expression analysis and EM makes the diagnosis of Alport syndrome possible in only 92% of cases (genetic studies were used as “gold standard”) (174). As pointed out earlier, genetic testing for Alport syndrome is now available; therefore, in questionable cases, genetic testing should be recommended.
Outcome
ESRD invariably develops in men in adulthood, usually between the ages of 30 and 50 years, but this can be quite variable. Women, who are heterozygous for the X-linked form, usually have an indolent course, but approximately 10% of women develop ESRD (175). Anti-GBM disease may develop in patients undergoing transplantation for Alport syndrome, as a consequence of antibody formation against the α5 chain of type IV collagen in patients who genetically lack normal α5 chains. Fortunately, this happens only in a few patients (46). Disease progression can be slowed down with appropriate renoprotective measures (166).
Etiology and Pathogenesis
The absence of the normal α5 chain in type IV collagen is the result of a single mutation or several mutations in the COL4A5 gene on the long arm of the X chromosome (172). The α5 chain probably forms a network with the α3 and α4 chains; thus, these are not expressed in the basement membranes either. The abnormal GBM most likely will be more susceptible to injury and unable to normally regenerate, and glomerulosclerosis will result. Rare autosomal recessive and, exceptionally, autosomal dominant forms of Alport syndrome also exist.
OTHER FINDINGS IN PATIENTS WITH ASYMPTOMATIC MICROSCOPIC HEMATURIA
Often in the pathologic study of tissues from patients with isolated or benign essential hematuria, none of the previously cited conditions is found. Some investigators have reported mesangial IgM and/or IgG, as well as C3 deposition (in the absence of IgA), in otherwise fairly unremarkable glomeruli in these patients (178,179). It appears that the glomerulopathies with non-IgA mesangial Ig deposition have a good prognosis. Some patients have loin pain in association with microscopic hematuria; at times, this may also occur with episodes of gross hematuria. Usually, no light microscopic glomerular abnormalities can be seen in biopsy specimens taken from patients with the loin pain hematuria syndrome, but ultrastructural GBM abnormalities are common (180,181). Often, there are patients in whom hematuria appears to be of renal origin, but no renal morphologic abnormality can be found. All of these conditions appear to have a benign clinical course. As noted earlier, it is important in the percutaneous renal biopsy report to note whether red blood cells are present in Bowman space or tubules to indicate that the hematuria is probably of glomerular origin.
WARFARIN (ANTICOAGULATION)-RELATED NEPHROPATHY
We have recently characterized a condition in patients on warfarin therapy who unexpectedly developed acute kidney injury after overanticoagulation (182,183). Clinically, these patients, in addition to acute kidney injury, have severe, usually gross hematuria. Renal biopsy findings reveal prominent acute tubular necrosis with numerous occlusive red blood cell casts in the tubules (Fig. 41.44). An underlying renal/glomerular disease is a predisposing factor, and almost all patients have some degree of underlying chronic kidney disease. Unfortunately, the outcome is frequently dismal, and many patients do not recover adequate renal function. It appears that renal recovery depends on the degree of underlying chronic injury and the general condition of the patient. According to our recent experience, the disease can be associated not only with warfarin but other anticoagulants as well; therefore, the condition is best designated as anticoagulation-associated nephropathy.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


