Histology
N
% of all tumors
Median age
Rate (95 % CI)
Glioblastoma
49,088
15.8
64
3.19 (3.16–3.22)
Anaplastic astrocytoma
5,374
1.7
54
0.36 (0.35–0.37)
Anaplastic oligodendroglioma
1,687
0.5
49
0.11 (0.11–0.12)
Glioma, malignant, NOS
6,574
2.1
40
0.45 (0.44–0.46)
While traditionally high-grade “malignant” gliomas (grades III and IV) are distinguished from low-grade gliomas (grades I and II), this distinction does not correspond with the known biology of these tumors and is therefore probably outdated. Grade I gliomas are usually circumscribed, with a very low propensity for malignant transformation, and, while not the subject of this chapter, occur predominantly in children and have a molecular pathogenesis that is unrelated to diffuse glioma. In contrast, the diffusely infiltrating gliomas (grades II–IV) are prone to tumor recurrence and progression to higher grades, and their clinical behavior is malignant to varying degrees. As an added complexity, while the histologic diagnosis of glioblastoma (grade IV) is relatively straightforward, the histopathologic distinction of grade II from grade III glioma is ill-defined and subject to considerable inter-observer variability. In addition, the molecular genetics of grade II and III gliomas largely overlap, arguing that these are best considered within a spectrum of a single disease entity. Median survival varies with histologic diagnosis, and ranges from 5 to 7 years for diffuse (grade II) astrocytoma, 3–5 years for anaplastic astrocytoma [3] to 15–16 months for glioblastoma [4]. One-, 2-, 5-, and 10-year survival by histologic diagnosis are summarized in Table 6.2. There is some evidence that average survival times are improving modestly. Median survival for patients with glioblastoma treated in a large randomized trial that defined our current standard of care was 15 months [4], while outcomes in patients treated on clinical trials in the 5 years after that study was published was 20 months [5]. Apart from a possible element of patient selection bias, reasons may include improved treatments at the time of disease recurrence or an improvement in the standard of clinical and supportive patient care over time.
Table 6.2
CBTRUS estimates of 1-, 2-, 5-, and 10-year relative survival rates, 1995–2009.
Histology | 1-year | 2-years | 5-years | 10-year |
|---|---|---|---|---|
Glioblastoma (%) | 35.7 | 13.6 | 4.7 | 2.3 |
Anaplastic astrocytoma (%) | 60.1 | 41.5 | 25.9 | 17.6 |
Anaplastic oligodendroglioma (%) | 81.0 | 66.9 | 49.4 | 34.2 |
Glioma, malignant, NOS | 61.9 % | 50.4 % | 43.3 | 38.3 % |
Standard treatment of glioblastoma includes maximal safe resection, involved field radiation, and concomitant and adjuvant temozolomide. Large retrospective studies have shown that patients who receive a more extensive resection, defined as 78–98 % of contrast enhancing tumor, have improved survival compared to patients who receive a subtotal resection or biopsy [6, 7], so extensive resection is warranted when feasible. The benefit from radiotherapy was defined by randomized trials, which showed a significant improvement in outcomes with radiotherapy compared to chemotherapy alone or best conventional care [8, 9]. A series of randomized studies established the standard dosing and fractionation of 60 Gy in 30 daily fractions [10–17].
A large randomized study defined the role of adjuvant chemotherapy with temozolomide, and found that patients who received concomitant and adjuvant temozolomide had a significant improvement in median survival compared to patients treated with radiation alone (12.1 vs. 14.6 months) [4] The proportion of patients surviving 5-years after diagnosis was five times higher in the temozolomide group (9.8 % vs. 1.9 %) [18]. This “Stupp protocol” has become the standard of care for initial management of glioblastoma. More recently, non-randomized data suggest that bevacizumab may improve outcomes after disease recurrence, with median progression-free survival of 4–6 months and median overall survival of 8–9 months [19, 20], which compared favorably to historical controls [21, 22].
Prospective, randomized trial data defining the utility of these modalities in anaplastic gliomas (WHO grade III) is lacking. These tumors are more heterogeneous in terms of their behavior, genetics, and response to therapy compared to glioblastomas, so the most appropriate up-front treatment has not been established, and may vary depending on the histologic and genetic subtype. Early clinical trials that defined the benefit from radiotherapy included patients with grade III tumors, and based on those data it is generally accepted that radiotherapy improves outcomes in anaplastic glioma, although the number of patients with grade III tumors on those trials was too small to allow a statistically robust subgroup analysis [8, 9]. Most physicians treat patients with anaplastic astrocytoma with radiation and temozolomide per the Stupp protocol [23], and there are retrospective data that suggests a benefit of chemoradiotherapy over radiotherapy alone in patients whose tumors do not harbor a 1p/19q co-deletion [24]. Recent data indicate that in anaplastic oligodendrogliomas, addition of chemotherapy to radiotherapy also benefits patients with non-co-deleted, isocitrate dehydrogenase 1 (IDH1) mutant tumors [25]. The benefit of adjuvant temozolomide, however, has not been confirmed in prospective trials [23, 25–27]. The prospective data that do exist used a more toxic regimen of procarbazine, lomustine, and vincristine, so whether patients with anaplastic tumors benefit from adjuvant temozolomide is still an open question, and large randomized trials in patients with co-deleted [28] and non-co-deleted tumors [29] are ongoing. In order to circumvent the risk of radiation-induced neuro-cognitive deficits, there is a growing interest in treating selected patients with chemosensitive tumors (e.g., those with 1p/19q co-deletion) with chemotherapy alone based on retrospective [24] and prospective [30] data suggesting outcomes similar to radiation alone.
After more than three decades of clinical trials, it is clear that there is significant heterogeneity in the biology and behavior of these tumors and their response to treatment. A better understanding of the histopathologic, genetic, and epigenetic changes that underlie tumor biology will allow for more tailored treatment of these heterogeneous tumors, and this will be the subject of the rest of this review.
Histopathology
Diffuse gliomas are infiltrative glial tumors characterized by increased cellularity, nuclear atypia, and mitotic activity. They are subclassified according to their cellular morphology as either astrocytic, oligodendroglial, or mixed gliomas.
Astrocytomas are composed of cells with elongated or irregular hyperchromatic nuclei and scant cytoplasm. Cell processes form a loose fibrillary matrix and glial fibrillary acidic protein (GFAP) staining highlights both the cytoplasm and cell processes. Proliferative index, as measured by Ki-67 or MIB-1, is generally between 5 and 10 % but is highly variable. Oligodendrogliomas also exhibit GFAP immunoreactivity, but morphologically the cells have rounded hyperchromatic nuclei, perinuclear halos, and few cellular processes. They have a characteristic branching capillary pattern and focal microcalcifications are common (Fig. 6.1). Oligoastrocytomas display features intermediate between astrocytoma and oligodendroglioma. While a biphasic distribution where distinct areas display astrocytic or oligodendroglial differentiation has been described in the literature, this is extremely rare and when found is of uncertain clinical significance. Most commonly mixed oligoastrocytomas represent an indeterminate diffuse variant where the two phenotypes are intermingled [2]. Recent data suggest that from a biologic perspective, mixed oligoastrocytoma is not a distinct entity and as a category is likely composed of a mix of tumors with “oligodendroglioma” biology (e.g., 1p/19q co-deletion) together with tumors with “astrocytoma” biology (e.g., TP53 mutation). These considerations highlight an important concept likely to be introduced into future classification systems of glioma, where key molecular markers are to be used as an important adjunct to conventional histopathologic analysis.
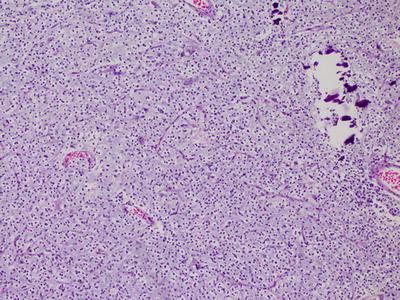
Fig. 6.1
Oligodendroglioma, with characteristic rounded hyperchromatic nuclei, perinuclear halos, and branching capillary pattern.
In addition to the features described above, glioblastomas are defined by the presence of microvascular proliferation and/or necrosis (Fig. 6.2). The cells are poorly differentiated and pleomorphic, and regional heterogeneity is common. Several variants have been described, including small cell glioblastoma, glioblastoma with an oligodendroglioma component, giant cell glioblastoma, and gliosarcoma [2].
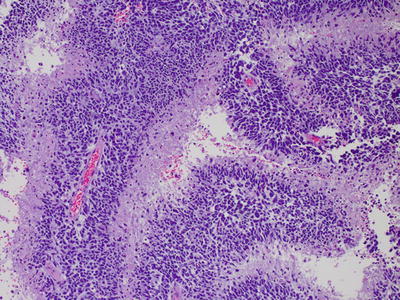
Fig. 6.2
Glioblastoma, with pseudopalisading necrosis.
Small cell glioblastoma is characterized by a monomorphic population of densely packed small, round cells with a high nuclear:cytoplasmic ratio and modest atypia. Proliferative activity is high, and GFAP immunoreactivity can be minimal. Their outcome is similar to standard GBMs [31]. Glioblastomas with an oligodendroglial component contain foci that resemble oligodendroglioma. The presence of areas with both astrocytic differentiation and necrosis differentiates them from anaplastic oligoastrocytomas. Giant cell glioblastomas have numerous multinucleated giant cells along with smaller fusiform cells. There is some data to suggest that the prognosis of glioblastomas with an oligodendroglial component and giant cell glioblastomas may be better compared to standard glioblastomas [32–34]. Gliosarcomas have a mixture of cells with gliomatous and sarcomatous differentiation, either in distinct geographic areas or intermixed. The gliomatous areas show typical features of glioblastoma. Sarcomatous areas often resemble fibrosarcoma, with bundles of spindle cells, but can also show mesenchymal differentiation with cartilaginous, osteoid, myomatous, or lipomatous features. These areas are GFAP negative [2] (Fig. 6.3). The prognosis of gliosarcoma is similar to standard glioblastoma [35].
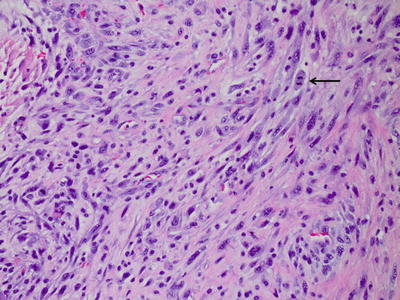
Fig. 6.3
Gliosarcoma, with bundles of elongated spindle cells.
Cytogenetics and Molecular Genetics
1p/19q Co-deletion
Loss of heterozygosity (LOH) of 1p and 19q is a common event in oligodendroglial tumors, occurring in 80–90 % of low-grade oligodendrogliomas and 50–70 % of anaplastic oligodendrogliomas [36]. However, since the distinction of oligodendroglioma from astrocytic tumors is subject to inter-observer variability, the rate of 1p/19q co-deletion relative to histologic diagnosis is only approximate. Several groups have reported that it is mutually exclusive from genetic changes common to astrocytic tumors, including TP53 mutations [37, 38], EGFR amplification [38, 39], and LOH 10q [39, 40]. It is associated with oligodendroglial morphology [41] and is felt to be a reliable diagnostic biomarker of the oligodendroglial phenotype [37, 42]. Tumors with 1p/19q co-deletion often exhibit several other genetic and epigenetic changes including IDH mutations [43, 44], methylguanine methyltransferase (MGMT) promoter methylation [44], CpG island methylator phenotype (G-CIMP) [45], and a proneural gene expression profile [46, 47], which will be discussed in greater detail below.
Recent work has identified candidate tumor suppressor genes on 1p and 19q. Mutations in the homolog of the Drosophila capicua gene (CIC) on 19q13.2 are present in 50–80 % of 1p/19q co-deleted oligodendrogliomas [48–50]. CIC is a downstream transcriptional repressor of receptor tyrosine kinase (RTK) pathways, including EGFR, Ras, Raf, and MAP kinases [48]. The exact mechanism of tumor pathogenesis remains unclear [42]. Mutations in the far-upstream element (FUSE) binding protein 1 (FUBP1) on 1p31.1 are seen in about 20 % of oligodendrogliomas [48, 50]. FUBP1 binds to the FUSE of Myc, a well-known oncogene [51] and negatively regulates Myc expression [52]. Although these are theoretically attractive genes for being involved in the pathogenesis of oligodendrogliomas, no definitive mechanism has yet been identified.
Chromosome 10 Deletion
LOH of chromosome 10 is the most frequent genetic alteration in GBM, and occurs in 60–80 % of cases [53, 54]. Loss of the entire chromosome is common, but partial deletions in three common regions have been described, suggesting that several tumor suppressor genes may exist in chromosome 10 [2, 55]. LOH 10q occurs in both primary and secondary glioblastomas at similar frequencies [53] while LOH 10p is generally seen in primary glioblastomas [56]. One established tumor suppressor in this region is the PTEN (phosphatase and tensin homology) gene on 10q23.3 [57]. PTEN is a phosphatase which inhibits PIP3 signaling, and thereby downregulates the activity of AKT and mTOR and inhibits cell proliferation [58]. It is mutated in 15–40 % of glioblastomas [59, 60] most often in primary glioblastomas [53, 61].
Chromosome 7 Amplification
The most frequent amplification event in glioblastoma is amplification of 7p12 in the region of the EGFR gene [62]. Like PTEN mutation and LOH 10q, EGFR amplification is common in primary glioblastoma, where it occurs in about 40 % of cases [53, 63]. EGFR amplification is significantly less frequent in secondary glioblastoma [63]. About 50–60 % of tumors with EGFR amplification also express a truncated variant of the receptor, EGFR variant III (EGFRvIII), which is constitutively active and ligand-independent [64, 65]. EGFR amplification and EGFRvIII mutation have been associated with an increased proliferation rate, increased invasiveness, resistance to cytotoxic therapy, and worse patient outcomes [66–68]. When other clinical and genetic variables and known prognostic factors are considered in a multivariant analysis, however, it is less clear that EGFR amplification and particularly the EGFRvIII mutation turn out to be independent prognostic factors. The EGFR signaling pathway will be discussed in further detail below.
IDH1 and IDH2
Mutations in isocitrate dehydrogenase-1 and -2 (IDH1 and -2) were first identified as important driver mutations in a subset of glioblastomas by Parsons et al. in 2008 [69]. IDH catalyzes the oxidative carboxylation of isocitrate to α-ketoglutarate (α-KG) within the citric acid cycle. The IDH family of genes code for enzymes that catalyze the NADP/NAD-dependent oxidative decarboxylation of isocitrate to alpha-ketoglutarate, with subsequent NADPH/NADH release [5]. These enzymes are found both in the cytoplasm (IDH1), and in the mitochondria (IDH2 and IDH3). IDH mutations are present in 50–75 % of anaplastic tumors and 75–85 % secondary glioblastomas, but are uncommon in primary glioblastomas, occurring in about 5 % [43, 44, 69–71]. IDH mutations frequently occur in association with other mutations common to secondary glioblastomas, such as TP53 mutations, 1p/19q co-deletions, and methylated MGMT promoter, and are inversely associated with alterations in PTEN, EGFR, and LOH 10, which are common to primary glioblastomas [70, 72, 73].
IDH1 mutations are point mutations at position 395 (G395A) of the IDH1 gene (codon 132 of the IDH1 binding site), most commonly with replacement of arginine with histidine (IDH1-R132H) [69], which accounts for >90 % of IDH1 mutations [43, 69, 71]. IDH2 mutations are less common, accounting for only 4–5 % of IDH mutations, and appear largely in the setting of 1p/19q co-deleted tumors [43, 71] IDH2 are also point mutations at a homologous codon (172) within the binding site [42]. Collective data suggest that IDH mutation is a very early lesion in the pathogenesis of lower grade (grade II–III) diffuse glioma. Co-deletion of 1p/19q is almost invariably observed in the setting of an IDH mutation. Interestingly, IDH-mutated but non-1p/19q-co-deleted tumors are nearly always TP53-mutated, suggesting that either co-deletion or TP53 mutation occur after IDH mutation and either of these aberrations is required for most cases of glioma pathogenesis. There is a mutant-specific commercially available antibody to the IDH1-R132H protein which is a reliable diagnostic biomarker [74]. From a practical and clinical perspective, this immunohistochemical test has several diagnostic uses, which include mutation detection and distinction of diffuse glioma from entities not associated with IDH mutation (for example circumscribed glioma and ependymoma). In addition, in the setting of a differential diagnosis of diffuse glioma versus reactive conditions (astrogliosis, treatment effects), R132H-specific immunohistochemistry can be very useful when positive. It is important to remember that absence of staining/mutation is not always helpful, since not all diffuse gliomas are IDH-mutated.
The pathogenesis of IDH mutations is still under active investigation. The mutation causes reduced enzymatic activity in the conversion of isocitrate to α-KG [75], but since the remaining allele produces functional enzyme it is likely that it is a gain of function which is pathogenic [76]. The mutated enzyme has been shown to catalyze the reduction of α-KG to 2-hydroxyglutarate (2-HG), and 2-HG levels are elevated in IDH1 mutated tumors, suggesting that 2-HG is an oncometabolite [77]. This is supported by the fact that patients with inborn errors of metabolism leading to accumulation of 2-HG in the brain have an elevated risk of brain tumors [78]. 2-HG is a competitive inhibitor of α-KG-dependent dioxygenases including histone demethylases and the TET family of hydroxylases [79], which are involved in DNA methylation [80]. Recent data shows that IDH mutation causes DNA hypermethylation over serial passages and is sufficient to establish the G-CIMP hypermethylated phenotype [81]. Mutations in IDH genes are not specific to gliomas, and have been described in other neoplasms, including acute myelogenous leukemia (AML) and cartilaginous neoplasms, among others. IDH mutations in these tumors, unlike gliomas, have not been shown to have a more favorable outcome.
The absence or presence of IDH mutation in diffuse gliomas largely accounts for the prior designation of primary and secondary pathways to glioblastoma, respectively. Lower grade diffuse gliomas (grade II–III) are largely IDH mutant, while grade IV gliomas (glioblastomas) are largely IDH wild type. However, a subset of grade II–III gliomas are IDH wild type and likely represent a precursor lesion to glioblastoma. In addition, while IDH-mutant glioblastomas are rare in the setting of a new glioma diagnosis, they likely represent malignant progression from a previously undiagnosed lower grade IDH-mutant glioma. Although indistinguishable histologically, many distinctions exist between IDH-mutant and IDH wild-type diffuse gliomas in terms of methylation pattern, DNA copy number aberrations, gene expression profiles, and somatic mutational profiles. Future progress in the classification and management of diffuse gliomas will likely benefit by treating IDH-mutant and IDH wild-type tumors as separate clinico-pathological entities.
ATRX
Alpha Thalassemia/Mental Retardation Syndrome X-Linked (ATRX) is a component of the SWI/SNF complex of chromatin remodeling proteins and is involved in gene regulation. Jiao et al. have recently described mutations in ATRX in gliomas, occurring in more than one third of astrocytomas (71 %, grades II and III), 57 % of secondary glioblastomas, and 68 % of mixed oligoastrocytomas. The frequency in primary glioblastoma and oligodendroglioma was much lower (4 % and 14 %, respectively). ATRX mutation was almost always seen in the presence of a TP53 mutation (94 %) and IDH mutations (99 %) in adult gliomas [82].
Epigenetics and Gene Expression Profiling
MGMT
O6-MGMT is a DNA repair protein on 10q26 that removes alkyl groups from the O6 position of guanine, thereby counteracting the activity of alkylating agents such as temozolomide and nitrosoureas [83]. It can be epigenetically silenced via methylation of 5′-CpG islands within the transcription factor binding sites [84]. MGMT is methylated in about 40 % of primary glioblastomas, 70 % of secondary glioblastomas, and 50 % of anaplastic astrocytomas [85, 86]. MGMT methylation is strongly associated with 1p/19q co-deletion [87, 88] and IDH mutations [44, 86]. MGMT methylation is predictive of response to temozolomide [89] and nitrosoureas [90], consistent with their mechanism of action. There is emerging evidence, however, that MGMT methylation is also an independent prognostic marker and associated with improved outcome in patients treated with radiotherapy alone [30, 72, 91], suggesting that this epigenetic change is associated with biological effects beyond DNA alkylator damage repair [92]. MGMT methylation is also associated with the G-CIMP hypermethylated phenotype [45] and it is possible that these genetic and global epigenetic changes underlie the prognostic effect of MGMT methylation. That said, whether MGMT methylation has the same prognostic significance in patients with IDH-mutant tumors as has been shown in patient cohorts with largely IDH wild-type glioblastomas, is not entirely clear.
Glioma-CpG-Island Methylator Phenotype (G-CIMP)
Noushmehr et al. have identified a subgroup of glioblastomas characterized by a distinct pattern of hypermethylation of CpG islands in a subset of glioma-specific genes, including genes involved in cell adhesion, regulation of transcription, metabolic processes, and nucleic acid synthesis [93]. This glioma-CpG-island methylator phenotype (G-CIMP) leads to transcriptional silencing of specific genes via methylation of promoter regions. It is associated with younger age, the “proneural” gene expression profile, a high frequency of IDH1 and TP53 mutations, and a low frequency of PTEN, NF1, and EGFR mutations. As outlined above, there is evidence to suggest that IDH mutations are sufficient to cause this glioma-specific methylation phenotype.
Gene Expression Subtypes
The Cancer Genome Atlas (TCGA) has identified four gene expression subtypes within glioblastoma including proneural, neural, classical, and mesenchymal subtypes [94]. The frequency of copy number alterations and mutations by subtype in this cohort are described in Tables 6.3 and 6.4.
Table 6.3
Copy number alterations in glioblastoma subtypes in TCGA samples.
Proneural (%) | Neural (%) | Classical (%) | Mesenchymal (%) | Gene(s) in ROI | |
|---|---|---|---|---|---|
Low and high level amplified events | |||||
7p11.2 | 54 | 96 | 100 | 95 | EGFR |
7q21.2 | 46 | 96 | 92 | 89 | CDK6 |
7q31.2 | 54 | 92 | 86 | 91 | MET |
7q34 | 52 | 92 | 86 | 91 | |
High level amplification events | |||||
7p11.2 | 17 | 67 | 95 | 29 | EGFR |
4q12 | 35 | 13 | 5 | 9 | PDGFRA |
Homozygous and hemizygous deletion events | |||||
17q11.2 | 6 | 17 | 5 | 38 | NF1 |
10q23 | 69 | 96 | 100 | 87 | PTEN |
9p21.3 | 56 | 71 | 95 | 67 | CDKN2A/CDKN2B |
13q14 | 52 | 46 | 16 | 53 | RB1 |
Homozygous deletion events | |||||
9p21.3 | 41 | 54 | 92 | 53 | CDKN2A/CDKN2B |
Table 6.4
Distribution of mutated genes in glioblastoma subtypes.
Gene | Proneural (%) | Neural (%) | Classical (%) | Mesenchymal (%) |
|---|---|---|---|---|
TP53 | 54 | 21 | 0 | 32 |
PTEN | 16 | 21 | 23 | 32 |
NF1 | 5 | 16 | 5 | 37 |
EGFR | 16 | 26 | 32 | 5 |
IDH1 | 30 | 5 | 0 | 0 |
PIK3R1 | 19 | 11 | 5 | 0 |
RB1 | 3 | 5 | 0 | 13 |
ERBB2 | 5 | 16 | 5 | 3 |
EGFRvIII | 3 | 0 | 23 | 3 |
PIK3CA | 8 | 5 | 5 | 3 |
PDGFRA | 11 | 0 | 0 | 0 |
The proneural subtype has an oligodendroglial signature, and is characterized by alterations in PDGFRA and IDH1 as well as TP53 mutations. Copy number changes that are common in classic glioblastoma, such as chromosome 7 amplification and chromosome 10 loss, are significantly less frequent in the proneural subtype. Oligodendrocytic development genes, including PDGFRA, NKX2-2, and Olig-2, are highly expressed. Patients with proneural tumors are younger and have a better prognosis, and secondary glioblastomas are more common in this subtype. It is important to realize that the improved prognosis observed by the proneural subclass is likely due to its inclusion of IDH-mutant tumors, which represent a subset of this transcriptomal class.
The neural subtype is associated with both oligodendrocytic and astrocytic gene signatures, and is also characterized by expression of neuronal markers, including NEFL, GABRA1, SYT1, and SLC12A5. Its expression patterns are most similar to normal brain.
The Classical subtype has an astrocytic signature, and is characterized by chromosome 7 amplification and chromosome 10 loss, EGFR amplification and EGFRvIII mutation, and increased expression of Notch and Sonic hedgehog (SHH) signaling pathway constituents. TP53 mutations are not seen in this subtype.
The mesenchymal subtype has a cultured astroglial signature, and microglial markers such as CD68, PTPRC, and TNF are highly expressed. NF1 mutations and PTEN loss are common in this subtype. Phenotypically, these tumors have more abundant necrosis, perhaps related to high expression of genes in the tumor necrosis factor (TNF) and NF-κB pathways. Both mesenchymal (CHI3L1, MET) and astrocytic markers (CD44, MERTK) are expressed.
While the TCGA classification is the most widely cited, it is important to realize that the TCGA classification system for gene expression analysis has not been validated using an unsupervised analysis from an independent data set. However, examination of additional analyses in the literature would suggest that, with some variation in nomenclature, the mesenchymal and proneural subtypes appear to be reproducibly identified in gene expression datasets [95, 96]. In addition, while the mesenchymal subtype is generally restricted to IDH wild-type glioblastoma, the proneural subtype is observed in diffuse gliomas of all three WHO grades. In addition, IDH-mutated gliomas are almost invariably proneural, but a subset of IDH wild-type tumors can also cluster in the proneural class.
Prognostic Stratification
Traditionally, prognostic stratification for malignant gliomas has been based on clinical and histologic features of the tumors. Regressive partitioning analysis has identified age, histologic grade, performance status, and extent of resection as important prognostic factors [97]. Outcomes stratified by these variables are markedly different, and vary from 5 months in the worst prognostic group to almost 60 months in the best prognostic group [97]. The significance of these prognostic markers has been born out in multiple studies [3, 6, 7, 21, 98–100], and they continue to be the foundation for estimating prognosis.
There is evidence that several of the genetic and molecular features of these tumors discussed above have prognostic significance, and they are increasingly being incorporated into prognostic schema. It has long been recognized, for instance, that 1p/19q loss is associated with a better response to chemotherapy [101, 102] and radiotherapy [25], as well as with longer survival [25, 27, 101]. As discussed above, there is also evidence that MGMT methylation is both a predictive [89, 90] and prognostic [30, 72, 91] biomarker. Both tests are now commercially available, and are routinely used as part of the diagnostic evaluation of these tumors.
More recently IDH1 and 2 mutations have emerged as another important prognostic factor. Several groups have established that gliomas with IDH mutations have a relatively favorable prognosis [43, 69], and that IDH mutations are more prognostic than grade [73, 103] As outlined above, current evidence suggests that IDH status defines two biologically and clinically distinct types of diffuse glioma, and IDH-mutant tumors are less aggressive than IDH wild-type gliomas when matched for histological grade. In contrast, EGFR overexpression is associated with worse prognosis and therapeutic resistance [104, 105]. Although TP53 mutations have emerged as a useful diagnostic biomarker given their higher incidence in secondary glioblastoma compared to primary glioblastoma [63], they do not appear to have prognostic significance in glioblastoma [106].
Molecular Signaling Pathways
The PI3K kinase pathway is altered in the majority of glioblastomas through a variety of mechanisms. Activating mutations or amplifications of multiple RTKs which activate this pathway, including EGFR, Her2, PDGFRα, or MET have been described [107]. PI3K is activated by RTKs and leads to increased cell proliferation and survival via activation of multiple downstream effectors including Akt and mTOR [58, 108]. Activating mutations of the pathway components Ras, PI3K, and Akt are often identified [107]. Inactivating mutations or deletions of NF1, a negative regulator of Ras, or PTEN, a negative regulator of PI3K, are also common [53, 107, 109]. Constituents in this pathway and the frequency of alterations in glioblastoma are summarized in Fig. 6.4a.
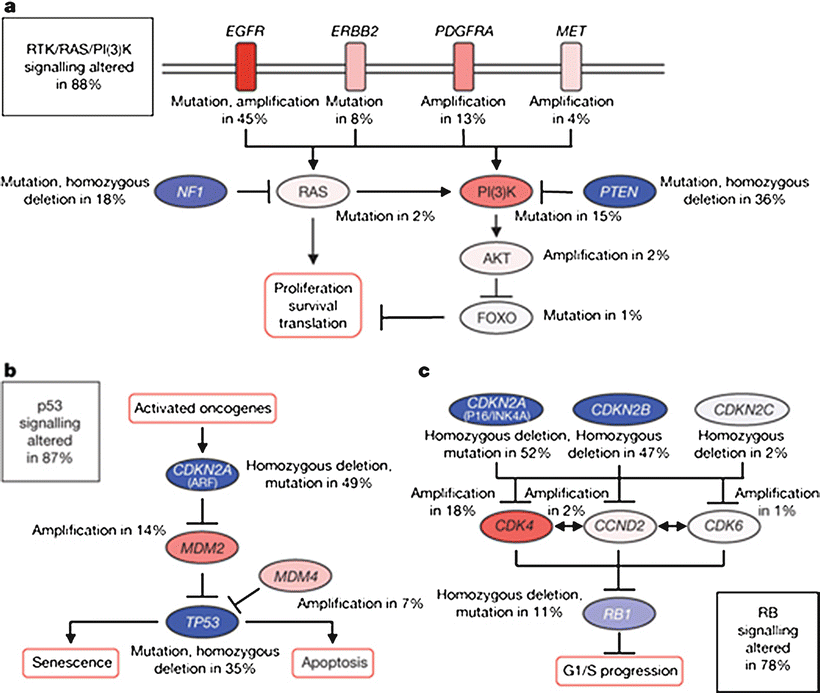
Fig. 6.4
Common signaling pathway alterations in glioblastoma (From Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455(7216):1061-1068, with permission)
TCGA identified alterations in the TP53 tumor suppressor pathway in 87 % of glioblastomas [107]. Disruption of this pathway leads to clonal expansion and genetic instability, as outlined above [110, 111]. Alterations in this pathway can occur via inactivating mutations or deletions in TP53 itself, or amplification of negative regulators of TP53, MDM2, and MDM4. These two events are generally mutually exclusive [107, 112]. Homozygous deletion of the CDKN2A gene, which leads to loss of p14ARF, an inhibitor of MDM2, also leads to functional loss of p53 activity [113]. A summary of this pathway and frequency of alterations in glioblastoma is provided in Fig. 6.4b.
The Rb pathway is altered in 78 % of glioblastomas [107]. Rb is a tumor suppressor that controls cell cycle progression from G1 to S phase [113]. Alterations in this pathway are most often due to deletions or mutations of Rb, amplification of its negative regulator CDK4, or deletions of p16INK4a or CDKN2B, which are in turn inhibitors of CDK4 [113]. Of note, p16INK4a is also translated from the CDKN2A gene, underscoring the connectedness of the TP53 and Rb pathways [114]. This pathway is summarized in Fig. 6.4c.
There has been increasing interest in stem-like cell (GSCs) signaling pathways in recent years. GSCs are characterized by their ability for self-renewal, their ability to differentiate into multiple lineages, and their tumorgenicity [115, 116]. GSCs have been identified as an important cause of treatment resistance in malignant gliomas [117, 118]. Two main stem cell signaling pathways that have inconsistently been implicated in GSC biology are the Notch pathway and the SHH pathway. Jagged and Delta-like ligands bind to Notch, which leads to γ-secretase-mediated cleavage of the intracellular domain of the Notch receptor [119]. Notch-IC then translocates to the nucleus where it functions as a transcriptional activator of several physiologic processes, including angiogenesis, specification of cell fate, and regulation of differentiation [119, 120]. SHH binds to the patched homolog receptor, and thereby releases the membrane protein Smoothened homolog, resulting in the activation of Gli proteins [121]. Gli proteins are transcription factors that modulate several target genes including MYC and CCND1 [121]. Different studies have implicated one, both or neither of these pathways in GSC biology. It is therefore likely that these pathways have variable activities and roles in different GSC lines from different tumors. These two pathways are summarized in Fig. 6.5.
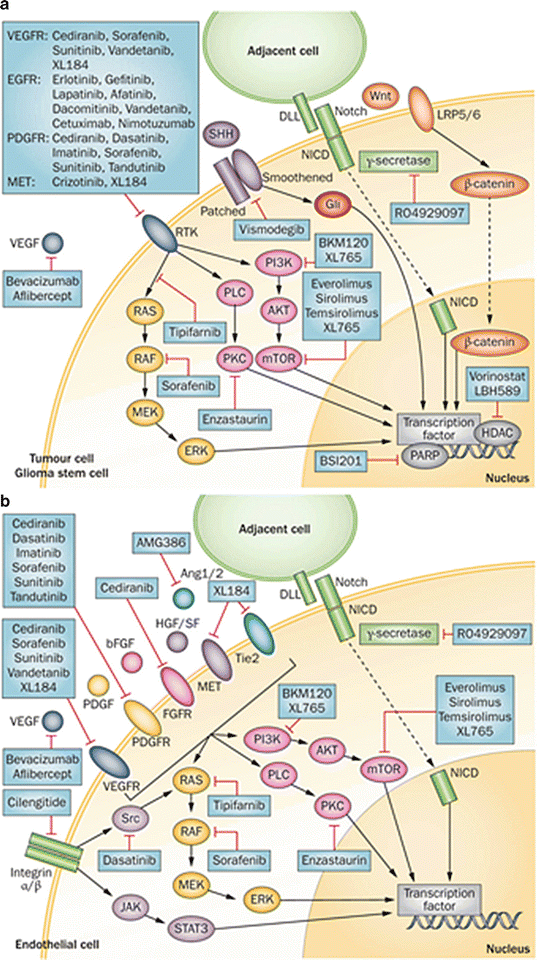
Fig. 6.5
Cell signaling pathways in malignant glioma cells (a) and tumor-associated endothelial cells (b), and agents under investigation to target these pathways (From Tanaka S, Louis DN, Curry WT, Batchelor TT, Dietrich J: Diagnostic and therapeutic avenues for glioblastoma: no longer a dead end? Nat Rev Clin Oncol 2013, 10(1):14-26, with permission)
Finally, it has long been recognized that gliomas are highly angiogenic tumors, so angiogenic signaling pathways represent a major area of glioma research. The vascular endothelial growth factor (VEGF) signaling pathway is pivotal to the development of tumor-associated blood vessels [120]. VEGF is secreted by tumor cells and binds to the VEGF receptor (VEGFR) on tumor-associated endothelium. This stimulates a signaling cascade through the PI3K/Ras/MAPK pathway, leading to endothelial cell proliferation and migration [120, 122]. Other pro-angiogenic factors and their receptors can stimulate these pathways, including platelet-derived growth factor (PDGF), fibroblast growth factor 2 (FGF-2), hypoxia-inducible factor 1-alpha (HIF-1α), hepatocyte growth factor (HGF), and angiopoetin-1 and -2, via their receptors PDGFR, FGFR, Met, and Tie2 and may be important causes of resistance to VEGF targeted therapy [120, 123, 124]. These pathways are summarized in Fig. 6.5.
Molecular Targeted Therapies
The elucidation of these important signaling pathways in malignant glioma has stimulated extensive research in the use of molecular targeted therapies. The most successful of the targeted therapies to date is bevacizumab, a humanized monoclonal antibody against VEGF-A ligand, which was approved by the FDA in 2009 for the treatment of recurrent glioblastoma [125]. Treatment with bevacizumab in phase II trials resulted in radiographic responses in 28–57 % of patients, median progression-free survival of 4–6 months, and median overall survival of 8–10 months [19, 20, 126], which compared favorably to historical controls [21, 22].
Two recent phase III trials examined the utility of bevacizumab added to chemoradiation as initial therapy for newly diagnosed glioblastoma. They showed an improvement in progression-free survival, but not overall survival, compared to controls, many of whom received bevacizumab at recurrence [127–129]. These data suggest that bevacizumab is not indicated as part of initial therapy. It should be noted that overall survival in both arms of these two trials was 16–17 months, which was a modest improvement compared to the 14.6 months seen in earlier phase III studies with radiation and temozolomide alone [4].
The success of bevacizumab has spurred interest in other VEGF targeted therapies, including decoy receptors, pan-VEGFR tyrosine kinase inhibitors (TKIs), multitargeted TKIs, integrin inhibitors, protein kinase C-β inhibitors, and inhibitors of other pro-angiogenic pathways. Although some have shown modest antitumor activity [130, 131], many have proven ineffective alone [132–135], or in combination with other targeted agents [136, 137], and none has proven more efficacious than bevacizumab to date. Ongoing clinical trials are exploring the activity of anti-angiogenic agents in combination with other targeted agents [138, 139], and in combination with bevacizumab [140]. Anti-angiogenic agents tested in clinical trials are summarized in Table 6.5.
Table 6.5
Anti-angiogenic agents under investigation.
Agent | Target | References |
|---|---|---|
Bevacizumab | VEGF-A | |
Aflibercept | VEGF-A, VEGF-B, PlGF | [135] |
Cediranib | VEGFR-1,2,3, PDGFR-α,β, FGFR-1, c-Kit | |
Sorafenib | VEGFR-2,3, PDGFR-α,β, BRAF, c-Kit, Ras | Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|