39
Adrenocorticosteroids & Adrenocortical Antagonists
CASE STUDY
A 19-year-old man complains of anorexia, fatigue, dizziness, and weight loss of 8 months’ duration. The examining physician discovers postural hypotension and moderate vitiligo (depigmented areas of skin) and obtains routine blood tests. She finds hyponatremia, hyperkalemia, and acidosis and suspects Addison’s disease. She performs a standard ACTH 1–24 stimulation test, which reveals an insufficient plasma cortisol response, compatible with primary adrenal insufficiency. The diagnosis of autoimmune Addison’s disease is made, and the patient must start replacement of the hormones he cannot produce himself. How should this patient be treated? What precautions should he take?
The natural adrenocortical hormones are steroid molecules produced and released by the adrenal cortex. Both natural and synthetic corticosteroids are used for the diagnosis and treatment of disorders of adrenal function. They are also used—more often and in much larger doses—for treatment of a variety of inflammatory and immunologic disorders.
Secretion of adrenocortical steroids is controlled by the pituitary release of corticotropin (ACTH). Secretion of the salt-retaining hormone aldosterone is primarily under the influence of angiotensin. Corticotropin has some actions that do not depend on its effect on adrenocortical secretion. However, its pharmacologic value as an anti-inflammatory agent and its use in testing adrenal function depend on its secretory action. Its pharmacology is discussed in Chapter 37 and is reviewed only briefly here.
Inhibitors of the synthesis or antagonists of the action of the adrenocortical steroids are important in the treatment of several conditions. These agents are described at the end of this chapter.
 ADRENOCORTICOSTEROIDS
ADRENOCORTICOSTEROIDS
The adrenal cortex releases a large number of steroids into the circulation. Some have minimal biologic activity and function primarily as precursors, and there are some for which no function has been established. The hormonal steroids may be classified as those having important effects on intermediary metabolism and immune function (glucocorticoids), those having principally salt-retaining activity (mineralocorticoids), and those having androgenic or estrogenic activity (see Chapter 40). In humans, the major glucocorticoid is cortisol and the most important mineralocorticoid is aldosterone. Quantitatively, dehydroepiandrosterone (DHEA) in its sulfated form (DHEAS) is the major adrenal androgen. However, DHEA and two other adrenal androgens, androstenedione and androstenediol, are weak androgens and androstenediol is a potent estrogen. Androstenedione can be converted to testosterone and estradiol in extra-adrenal tissues (Figure 39–1). Adrenal androgens constitute the major endogenous precursors of estrogen in women after menopause and in younger patients in whom ovarian function is deficient or absent.
THE NATURALLY OCCURRING GLUCOCORTICOIDS; CORTISOL (HYDROCORTISONE)
Pharmacokinetics
Cortisol (also called hydrocortisone, compound F) exerts a wide range of physiologic effects, including regulation of intermediary metabolism, cardiovascular function, growth, and immunity. Its synthesis and secretion are tightly regulated by the central nervous system, which is very sensitive to negative feedback by the circulating cortisol and exogenous (synthetic) glucocorticoids. Cortisol is synthesized from cholesterol (as shown in Figure 39–1). The mechanisms controlling its secretion are discussed in Chapter 37.
In the normal adult, in the absence of stress, 10–20 mg of cortisol is secreted daily. The rate of secretion follows a circadian rhythm (Figure 39–2) governed by pulses of ACTH that peak in the early morning hours and after meals. In plasma, cortisol is bound to circulating proteins. Corticosteroid-binding globulin (CBG), an α2 globulin synthesized by the liver, binds about 90% of the circulating hormone under normal circumstances. The remainder is free (about 5–10%) or loosely bound to albumin (about 5%) and is available to exert its effect on target cells. When plasma cortisol levels exceed 20–30 mcg/dL, CBG is saturated, and the concentration of free cortisol rises rapidly. CBG is increased in pregnancy and with estrogen administration and in hyperthyroidism. It is decreased by hypothyroidism, genetic defects in synthesis, and protein deficiency states. Albumin has a large capacity but low affinity for cortisol, and for practical purposes albumin-bound cortisol should be considered free. Synthetic corticosteroids such as dexamethasone are largely bound to albumin rather than CBG.
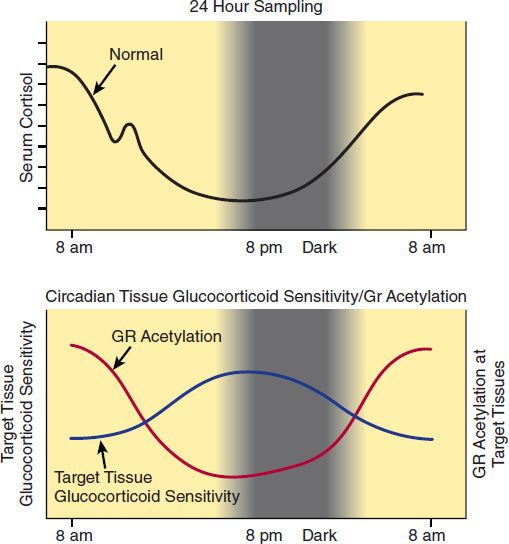
FIGURE 39–2 Circadian variation in plasma cortisol throughout the 24-hour day (upper panel). The sensitivity of tissues to glucocorticoids is also circadian but inverse to that of cortisol, with low sensitivity in the late morning and high sensitivity in the evening and early night (lower panel). The sensitivity of tissues to glucocorticoids is inversely related to that of glucocorticoid receptor (GR) acetylation by the transcription factor CLOCK; the acetylated receptor has decreased transcriptional activity. (Adapted, with permission, from Nader N, Chrousos GP, Kino T: Interactions of the circadian CLOCK system and the HPA axis. Trends Endocrinol Metab 2010;21:277. Copyright Elsevier.)
The half-life of cortisol in the circulation is normally about 60–90 minutes; it may be increased when hydrocortisone (the pharmaceutical preparation of cortisol) is administered in large amounts or when stress, hypothyroidism, or liver disease is present. Only 1% of cortisol is excreted unchanged in the urine as free cortisol; about 20% of cortisol is converted to cortisone by 11-hydroxysteroid dehydrogenase in the kidney and other tissues with mineralocorticoid receptors (see below) before reaching the liver. Most cortisol is metabolized in the liver. About one third of the cortisol produced daily is excreted in the urine as dihydroxy ketone metabolites and is measured as 17-hydroxysteroids (see Figure 39–3 for carbon numbering). Many cortisol metabolites are conjugated with glucuronic acid or sulfate at the C3 and C21 hydroxyls, respectively, in the liver; they are then excreted in the urine.
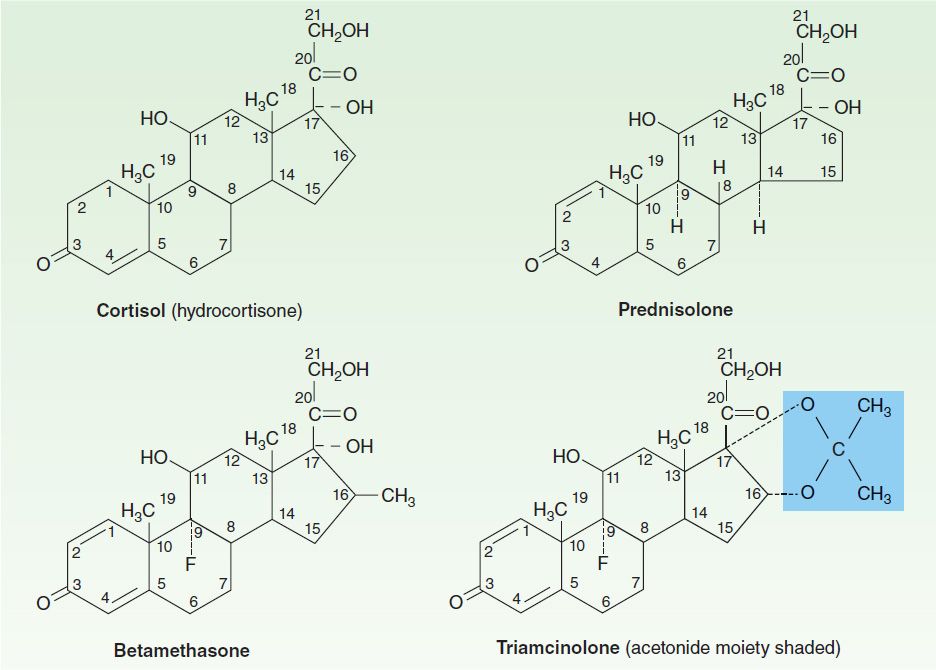
FIGURE 39–3 Chemical structures of several glucocorticoids. The acetonide-substituted derivatives (eg, triamcinolone acetonide) have increased surface activity and are useful in dermatology. Dexamethasone is identical to betamethasone except for the configuration of the methyl group at C16: in betamethasone it is beta (projecting up from the plane of the rings); in dexamethasone it is alpha.
In some species (eg, the rat), corticosterone is the major glucocorticoid. It is less firmly bound to protein and therefore metabolized more rapidly. The pathways of its degradation are similar to those of cortisol.
Pharmacodynamics
A. Mechanism of Action
Most of the known effects of the glucocorticoids are mediated by widely distributed glucocorticoid receptors. These proteins are members of the superfamily of nuclear receptors, which includes steroid, sterol (vitamin D), thyroid, retinoic acid, and many other receptors with unknown or nonexistent ligands (orphan receptors). All these receptors interact with the promoters of—and regulate the transcription of—target genes (Figure 39–4). In the absence of the hormonal ligand, glucocorticoid receptors are primarily cytoplasmic, in oligomeric complexes with chaperone heat-shock proteins (hsp). The most important of these are two molecules of hsp90, although other proteins (eg, hsp40, hsp70, FKBP5) are certainly involved. Free hormone from the plasma and interstitial fluid enters the cell and binds to the receptor, inducing conformational changes that allow it to dissociate from the heat shock proteins. The dimeric ligand-bound receptor complex then is actively transported into the nucleus, where it interacts with DNA and nuclear proteins. As a homodimer, it binds to glucocorticoid receptor elements (GREs) in the promoters of responsive genes. The GRE is composed of two palindromic sequences that bind to the hormone receptor dimer.
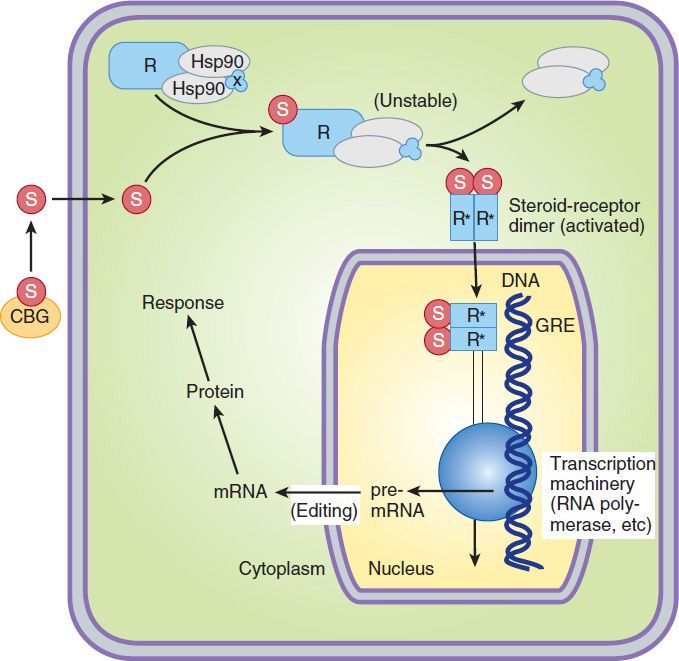
FIGURE 39–4 A model of the interaction of a steroid, S (eg, cortisol), and its receptor, R, and the subsequent events in a target cell. The steroid is present in the blood in bound form on the corticosteroid-binding globulin (CBG) but enters the cell as the free molecule. The intracellular receptor is bound to stabilizing proteins, including two molecules of heat-shock protein 90 (hsp90) and several others including FKBP5, denoted as “X” in the figure. This receptor complex is incapable of activating transcription. When the complex binds a molecule of cortisol, an unstable complex is created and the hsp90 and associated molecules are released. The steroid-receptor complex is now able to dimerize, enter the nucleus, bind to a glucocorticoid response element (GRE) on the regulatory region of the gene, and regulate transcription by RNA polymerase II and associated transcription factors. A variety of regulatory factors (not shown) may participate in facilitating (coactivators) or inhibiting (corepressors) the steroid response. The resulting mRNA is edited and exported to the cytoplasm for the production of protein that brings about the final hormone response. An alternative to the steroid-receptor complex interaction with a GRE is an interaction with and altering the function of other transcription factors, such as NF-κB in the nucleus of cells.
In addition to binding to GREs, the ligand-bound receptor also forms complexes with and influences the function of other transcription factors, such as AP1 and nuclear factor kappa-B (NF-κB), which act on non-GRE-containing promoters, to contribute to the regulation of transcription of their responsive genes. These transcription factors have broad actions on the regulation of growth factors, proinflammatory cytokines, etc, and to a great extent mediate the anti-growth, anti-inflammatory, and immunosuppressive effects of glucocorticoids.
Two genes for the corticoid receptor have been identified: one encoding the classic glucocorticoid receptor (GR) and the other encoding the mineralocorticoid receptor (MR). Alternative splicing of human glucocorticoid receptor pre-mRNA generates two highly homologous isoforms, termed hGRα and hGRβ. Human GRα is the classic ligand-activated glucocorticoid receptor which, in the hormone-bound state, modulates the expression of glucocorticoid-responsive genes. In contrast, hGRβ does not bind glucocorticoids and is transcriptionally inactive. However, hGRβ is able to inhibit the effects of hormone-activated hGRα on glucocorticoid-responsive genes, playing the role of a physiologically relevant endogenous inhibitor of glucocorticoid action. It was recently shown that the two hGR alternative transcripts have eight distinct translation initiation sites; ie, in a human cell there may be up to 16 GRα and GRβ isoforms, which may form up to 256 homodimers and heterodimers with different transcriptional and possibly nontranscriptional activities. This variability suggests that this important class of steroid receptors has complex stochastic activities. In addition, rare mutations in hGR may result in partial glucocorticoid resistance. Affected individuals have increased ACTH secretion because of reduced pituitary feedback and additional endocrine abnormalities (see below).
The prototype GR isoform is composed of about 800 amino acids and can be divided into three functional domains (see Figure 2–6). The glucocorticoid-binding domain is located at the carboxyl terminal of the molecule. The DNA-binding domain is located in the middle of the protein and contains nine cysteine residues. This region folds into a “two-finger” structure stabilized by zinc ions connected to cysteines to form two tetrahedrons. This part of the molecule binds to the GREs that regulate glucocorticoid action on glucocorticoid-regulated genes. The zinc fingers represent the basic structure by which the DNA-binding domain recognizes specific nucleic acid sequences. The amino-terminal domain is involved in the transactivation activity of the receptor and increases its specificity.
The interaction of glucocorticoid receptors with GREs or other transcription factors is facilitated or inhibited by several families of proteins called steroid receptor coregulators, divided into coactivators and corepressors. The coregulators do this by serving as bridges between the receptors and other nuclear proteins and by expressing enzymatic activities such as histone acetylase or deacetylase, which alter the conformation of nucleosomes and the transcribability of genes.
Between 10% and 20% of expressed genes in a cell are regulated by glucocorticoids. The number and affinity of receptors for the hormone, the complement of transcription factors and coregulators, and post-transcription events determine the relative specificity of these hormones’ actions in various cells. The effects of glucocorticoids are mainly due to proteins synthesized from mRNA transcribed from their target genes.
Some of the effects of glucocorticoids can be attributed to their binding to mineralocorticoid receptors. Indeed, MRs bind aldosterone and cortisol with similar affinity. A mineralocorticoid effect of the higher levels of cortisol is avoided in some tissues (eg, kidney, colon, salivary glands) by expression of 11β-hydroxysteroid dehydrogenase type 2, the enzyme responsible for biotransformation to its 11-keto derivative (cortisone), which has minimal action on aldosterone receptors.
The GR also interacts with other regulators of cell function. One such molecule is CLOCK/BMAL-1, a transcription factor dimer expressed in all tissues and generating the circadian rhythm of cortisol secretion (Figure 39–2) at the suprachiasmatic nucleus of the hypothalamus. CLOCK is an acetyltransferase that acetylates the hinge region of the GR, neutralizing its transcriptional activity and thus rendering target tissues resistant to glucocorticoids. As shown in Figure 39–2, lower panel, the glucocorticoid target tissue sensitivity rhythm generated is in reverse phase to that of circulating cortisol concentrations, explaining the increased sensitivity of the organism to evening administration of glucocorticoids. The GR also interacts with NF-κB, a regulator of production of cytokines and other molecules involved in inflammation.
Prompt effects such as initial feedback suppression of pituitary ACTH occur in minutes and are too rapid to be explained on the basis of gene transcription and protein synthesis. It is not known how these effects are mediated. Among the proposed mechanisms are direct effects on cell membrane receptors for the hormone or nongenomic effects of the classic hormone-bound glucocorticoid receptor. The putative membrane receptors might be entirely different from the known intracellular receptors. For example, recent studies implicate G protein-coupled membrane receptors in the response of glutamatergic neurons to glucocorticoids in rats. Furthermore, all steroid receptors (except the MRs) have been shown to have palmitoylation motifs that allow enzymatic addition of palmitate and increased localization of the receptors in the vicinity of plasma membranes. Such receptors are available for direct interactions with and effects on various membrane-associated or cytoplasmic proteins without the need for entry into the nucleus and induction of transcriptional actions.
B. Physiologic Effects
The glucocorticoids have widespread effects because they influence the function of most cells in the body. The major metabolic consequences of glucocorticoid secretion or administration are due to direct actions of these hormones in the cell. However, some important effects are the result of homeostatic responses by insulin and glucagon. Although many of the effects of glucocorticoids are dose-related and become magnified when large amounts are administered for therapeutic purposes, there are also other effects—called permissive effects—without which many normal functions become deficient. For example, the response of vascular and bronchial smooth muscle to catecholamines is diminished in the absence of cortisol and restored by physiologic amounts of this glucocorticoid. Similarly, the lipolytic responses of fat cells to catecholamines, ACTH, and growth hormone are attenuated in the absence of glucocorticoids.
C. Metabolic Effects
The glucocorticoids have important dose-related effects on carbohydrate, protein, and fat metabolism. The same effects are responsible for some of the serious adverse effects associated with their use in therapeutic doses. Glucocorticoids stimulate and are required for gluconeogenesis and glycogen synthesis in the fasting state. They stimulate phosphoenolpyruvate carboxykinase, glucose-6-phosphatase, and glycogen synthase and the release of amino acids in the course of muscle catabolism.
Glucocorticoids increase serum glucose levels and thus stimulate insulin release and inhibit the uptake of glucose by muscle cells, while they stimulate hormone-sensitive lipase and thus lipolysis. The increased insulin secretion stimulates lipogenesis and to a lesser degree inhibits lipolysis, leading to a net increase in fat deposition combined with increased release of fatty acids and glycerol into the circulation.
The net results of these actions are most apparent in the fasting state, when the supply of glucose from gluconeogenesis, the release of amino acids from muscle catabolism, the inhibition of peripheral glucose uptake, and the stimulation of lipolysis all contribute to maintenance of an adequate glucose supply to the brain.
D. Catabolic and Antianabolic Effects
Although glucocorticoids stimulate RNA and protein synthesis in the liver, they have catabolic and antianabolic effects in lymphoid and connective tissue, muscle, peripheral fat, and skin. Supraphysiologic amounts of glucocorticoids lead to decreased muscle mass and weakness and thinning of the skin. Catabolic and antianabolic effects on bone are the cause of osteoporosis in Cushing’s syndrome and impose a major limitation in the long-term therapeutic use of glucocorticoids. In children, glucocorticoids reduce growth. This effect may be partially prevented by administration of growth hormone in high doses, but this use of growth hormone is not recommended.
E. Anti-Inflammatory and Immunosuppressive Effects
Glucocorticoids dramatically reduce the manifestations of inflammation. This is due to their profound effects on the concentration, distribution, and function of peripheral leukocytes and to their suppressive effects on the inflammatory cytokines and chemokines and on other mediators of inflammation. Inflammation, regardless of its cause, is characterized by the extravasation and infiltration of leukocytes into the affected tissue. These events are mediated by a complex series of interactions of white cell adhesion molecules with those on endothelial cells and are inhibited by glucocorticoids. After a single dose of a short-acting glucocorticoid, the concentration of neutrophils in the circulation increases while the lymphocytes (T and B cells), monocytes, eosinophils, and basophils decrease. The changes are maximal at 6 hours and are dissipated in 24 hours. The increase in neutrophils is due both to the increased influx into the blood from the bone marrow and to the decreased migration from the blood vessels, leading to a reduction in the number of cells at the site of inflammation. The reduction in circulating lymphocytes, monocytes, eosinophils, and basophils is primarily the result of their movement from the vascular bed to lymphoid tissue.
Glucocorticoids also inhibit the functions of tissue macrophages and other antigen-presenting cells. The ability of these cells to respond to antigens and mitogens is reduced. The effect on macrophages is particularly marked and limits their ability to phagocytose and kill microorganisms and to produce tumor necrosis factor-α, interleukin-1, metalloproteinases, and plasminogen activator. Both macrophages and lymphocytes produce less interleukin-12 and interferon-γ, important inducers of TH1 cell activity, and cellular immunity.
In addition to their effects on leukocyte function, glucocorticoids influence the inflammatory response by inhibiting phospholipase A2 thereby reducing the synthesis of arachidonic acid, the precursor of prostaglandins and leukotrienes, and of platelet-activating factor. Finally, glucocorticoids reduce expression of cyclooxygenase-2, the inducible form of this enzyme, in inflammatory cells, thus reducing the amount of enzyme available to produce prostaglandins (see chapters 18 and 36).
Glucocorticoids cause vasoconstriction when applied directly to the skin, possibly by suppressing mast cell degranulation. They also decrease capillary permeability by reducing the amount of histamine released by basophils and mast cells.
The anti-inflammatory and immunosuppressive effects of glucocorticoids are largely due to the actions described above. In humans, complement activation is unaltered, but its effects are inhibited. Antibody production can be reduced by large doses of steroids, although it is unaffected by moderate doses (eg, 20 mg/d of prednisone).
The anti-inflammatory and immunosuppressive effects of these agents are widely useful therapeutically but are also responsible for some of their most serious adverse effects (see text that follows).
F. Other Effects
Glucocorticoids have important effects on the nervous system. Adrenal insufficiency causes marked slowing of the alpha rhythm of the electroencephalogram and is associated with depression. Increased amounts of glucocorticoids often produce behavioral disturbances in humans: initially insomnia and euphoria and subsequently depression. Large doses of glucocorticoids may increase intracranial pressure (pseudotumor cerebri).
Glucocorticoids given chronically suppress the pituitary release of ACTH, growth hormone, thyroid-stimulating hormone, and luteinizing hormone.
Large doses of glucocorticoids have been associated with the development of peptic ulcer, possibly by suppressing the local immune response against Helicobacter pylori. They also promote fat redistribution in the body, with increase of visceral, facial, nuchal, and supraclavicular fat, and they appear to antagonize the effect of vitamin D on calcium absorption. The glucocorticoids also have important effects on the hematopoietic system. In addition to their effects on leukocytes, they increase the number of platelets and red blood cells.
Cortisol deficiency results in impaired renal function (particularly glomerular filtration), augmented vasopressin secretion, and diminished ability to excrete a water load.
Glucocorticoids have important effects on the development of the fetal lungs. Indeed, the structural and functional changes in the lungs near term, including the production of pulmonary surface-active material required for air breathing (surfactant), are stimulated by glucocorticoids.
SYNTHETIC CORTICOSTEROIDS
Glucocorticoids have become important agents for use in the treatment of many inflammatory, immunologic, hematologic, and other disorders. This has stimulated the development of many synthetic steroids with anti-inflammatory and immunosuppressive activity.
Pharmacokinetics
Pharmaceutical steroids are usually synthesized from cholic acid obtained from cattle or steroid sapogenins found in plants. Further modifications of these steroids have led to the marketing of a large group of synthetic steroids with special characteristics that are pharmacologically and therapeutically important (Table 39–1; Figure 39–3).
TABLE 39–1 Some commonly used natural and synthetic corticosteroids for general use.
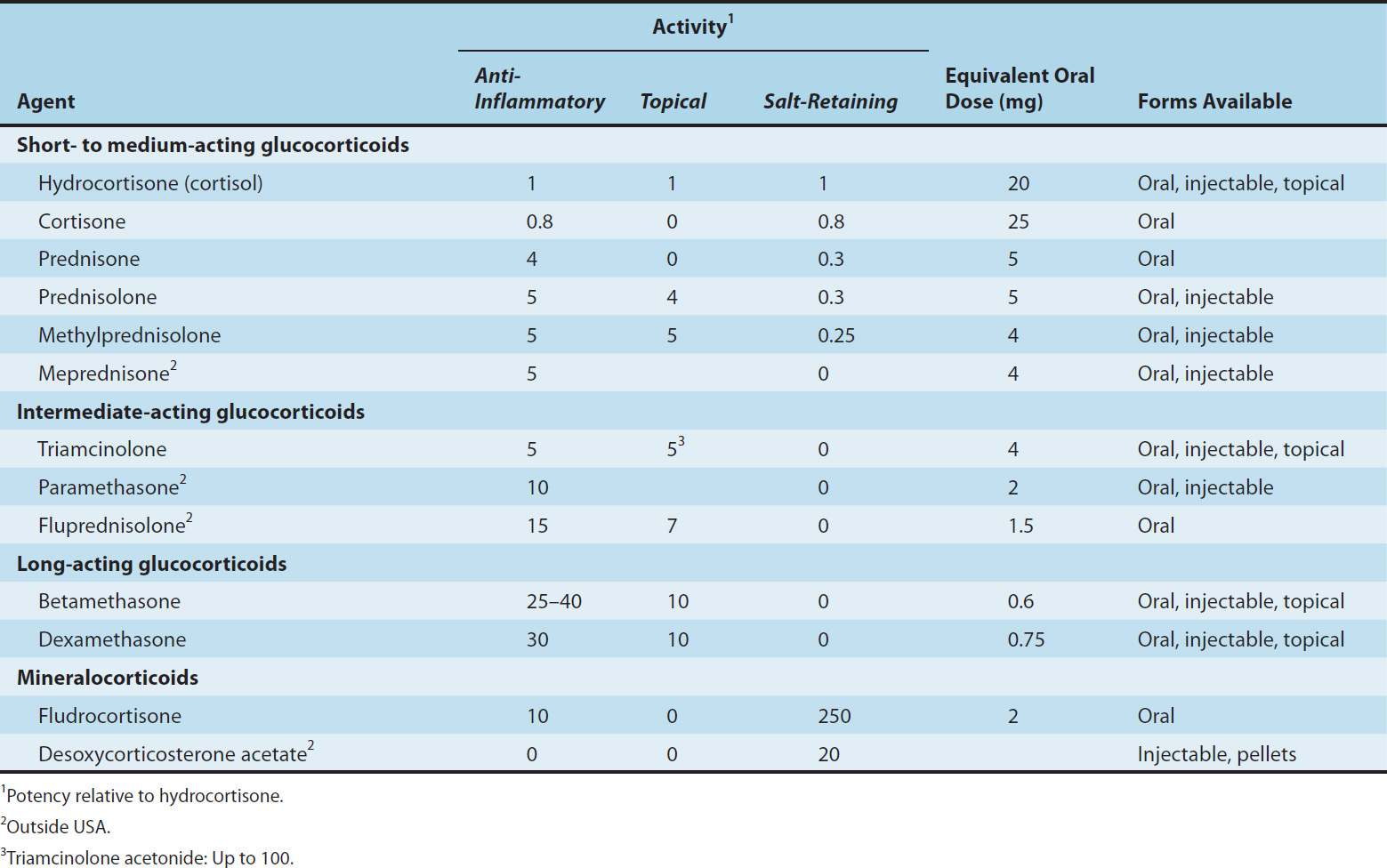
The metabolism of the naturally occurring adrenal steroids has been discussed above. The synthetic corticosteroids (Table 39–1) are in most cases rapidly and completely absorbed when given by mouth. Although they are transported and metabolized in a fashion similar to that of the endogenous steroids, important differences exist.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


