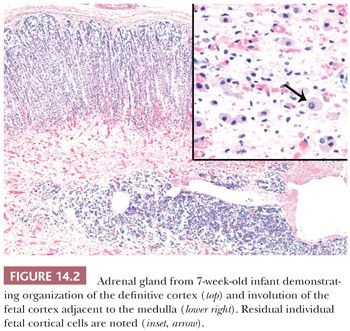
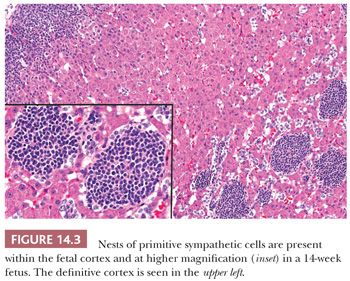
The intra-adrenal and extra-adrenal paraganglia and the sympathetic nervous system are intimately associated during embryonic development and arise from the neural crest. The cortical anlage is invaded on its medial aspect by primitive sympathetic cells (PSCs) and nerve fibers that originate from the contiguous prevertebral and paravertebral sympathetic tissue in the 14-mm embryo (approximately 5 to 6 weeks). Some PSCs, however, may penetrate the anlage without associated nerve fibers (8,9). The PSCs are first apparent as nodular aggregates in the cortex, where they may form rosettes or pseudorosettes. Chromaffin cells are identifiable among the PSCs between the 27- and 33-mm stages (~7 to 8 weeks) and gradually increase in number. The nodules of PSCs peak in number and size between 17 and 20 weeks and then decline (Fig. 14.3). Groups of these cells may, however, persist until birth and may also be apparent in early infancy (see section on neuroblastoma). As the medullary chromaffin cells reach their maximum volume, there is a progressive involution of extra-adrenal chromaffin cells.
There is a complex interaction of multiple genes in the development of the adrenal gland. Steroidogenic factor 1 (SF-1) is a transcription factor that appears to play a central role in the development of the adrenal cortex and steroid-producing gonads (10). The action of SF-1 is thought to be regulated by the transcription factor Wilms tumor 1 (WT1) and the transcriptional coactivator Cbp/p300-interacting transactivator, with Glu/Asp-rich carboxy-terminal domain, 2 (CITED2). SF-1 is thought to activate multiple cortical genes including the nuclear receptor gene dosage-sensitive sex reversal, adrenal hypoplasia critical region, on chromosome X, gene 1 (DAX-1) (11) in the development of the cortex. Chromaffin cell development is influenced by a host of genes, but the transcription factors mammalian achaete scute homolog-1 (MASH1) and paired-like homeobox 2B (PHOX2B) along with the transforming growth factor bone morphometric protein 4 (BMP-4) appear to be key players in the development of the medulla (12). The organization of the cortex and ultimately the adrenal gland is thought to be affected by the interaction of the sonic hedgehog (Shh) gene expression in capsular and mesenchymal cells (13).
ANATOMY AND PHYSIOLOGY
In the normal adult, each gland weighs between 4 and 5 g, although greater weights have been recorded in hospitalized patients dying after prolonged illnesses presumably as a result of prolonged stimulation of the glands during stress (2,14). The latter finding correlates with stress-related lipid depletion mediated by increased secretion of adrenocorticotropic hormone (ACTH). Each adrenal gland measures approximately 5 × 3 × 1 cm. The right gland has a roughly pyramidal shape, whereas the left gland has a crescentic shape. The glands have a tripartite structure, which consists of head (medial), body (middle), and tail (lateral) portions (15,16). The central vein emerges from the gland at the junction of the head and body of the gland. Within the gland itself, the muscle bundles of the central vein are eccentric (Fig. 14.4A) and are oriented toward the medulla.
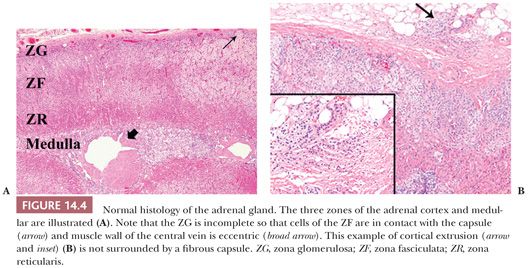
In the fresh state, the outer cortex is bright yellow, whereas the inner cortical zone is brown to tan. The cortex measures approximately 1 mm in thickness in adults and constitutes approximately 90% of the total glandular weight. It consists of the glomerulosa, fasciculata, and reticularis zones (2,17). The glomerulosa comprises up to 15% of the cortical volume and is composed of relatively small lipid-poor cells that synthesize mineralocorticoids. Because this layer is often incomplete, the fasciculata may abut the capsule of the gland directly (Fig. 14.4A). The fasciculata is composed of columns of lipid-rich cells, which synthesize both glucocorticoids and sex steroids. This zone occupies 70% to 80% of the cortical volume. Stimulation of the adrenal by ACTH leads to depletion of lipid stores from the fasciculata (2), so that the cells of this layer become compact and eosinophilic (18). In the process of recovery, the lipid content is gradually replenished (lipid reversion). The remainder of the cortex is composed of the reticularis, which is capable of synthesis of both glucocorticoids and sex steroids. These cells are characterized by eosinophilic cytoplasm, scanty lipid vacuoles, and prominent deposits of lipochrome pigment, which are responsible for the brown color of the reticularis.
Cortical extrusions are found frequently in the adrenal glands of adults. They are characterized by the presence of nodular groups of cortical cells that extend into the periadrenal fat. Typically, they are attached to the adjacent cortex by a small pedicle and are surrounded by a fibrous capsule; however, they may be completely separated from the gland in some instances or may lack a capsule (Fig. 14.4B).
Focal aggregates of lymphocytes are an incidental finding in the adrenal cortices of normal adults and increase in frequency with the age of the patient (19). Most of the lymphocytes are of T lineage (20).
The cells of the glomerulosa contain an abundance of smooth endoplasmic reticulum and small amounts of granular endoplasmic reticulum. The mitochondria in this layer tend to be elongated with lamelliform cristae (2,21). In contrast, the mitochondria of the fasciculata and reticularis zones are round to ovoid with a predominance of tubulovesicular cristae. The intermediate filaments of cortical cells include vimentin and variable amounts of low–molecular-weight cytokeratins (22).
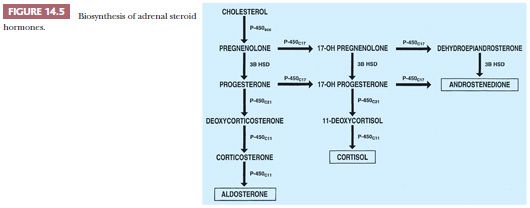
The precursor of all steroid hormones is cholesterol, which is derived from circulating low-density lipoproteins. After internalization into the cortical cells, the lipoproteins are hydrolyzed with the production of cholesterol esters, which yield cholesterol and free fatty acids (23,24). There are four cytochrome P450 enzymes that are involved in the biosynthesis of adrenal steroids (P450scc, P450c17, P450c21, and P450c11). The enzyme 3β-hydroxysteroid dehydrogenase does not belong to the P450 cytochrome family (Fig. 14.5). The synthesis and secretion of glucocorticoids, 18-hydroxysteroids, and androgens are regulated by a complex set of control mechanisms. Hypothalamic corticotropin-releasing hormone (CRH), a 41–amino acid polypeptide, reaches the anterior pituitary gland via the hypophyseal portal system, where it stimulates the release of ACTH (24).
In the adrenal cortex, ACTH stimulates cortical cells by activation of intracytoplasmic cyclases that form cyclic adenosine monophosphate (cAMP) and guanosine monophosphate (GMP) from adenosine triphosphate (ATP) and guanosine triphosphate (GTP), respectively. Both cortisol and ACTH inhibit the release of CRH, and cortisol also inhibits secretion of ACTH. The secretion of ACTH is normally episodic, with the number and duration of episodes increasing to a peak in the early morning and a nadir in the evening. This characteristic pattern is responsible for the circadian rhythm in the secretion of cortisol in healthy individuals (24). The secretion of aldosterone is regulated primarily by the renin-angiotensin system, which operates by volume-regulated changes (24). In addition, aldosterone synthesis and secretion are regulated by potassium ions and, to some extent, by ACTH (25,26).
In the adult, the adrenal medulla occupies 8% to 10% of the gland volume and has an average weight of 0.44 g (14). The major portion of the medulla lies within the head of the gland, whereas the body of the gland contains medullary (chromaffin) cells within its crest and usually within one alar region (15,27). The average corticomedullary ratio is 5:1 in the head of the gland and 14.7:1 in the body. The tail of the adrenal does not normally contain medullary tissue.
Medullary (chromaffin) cells are typically arranged in small nests and cords that are separated by a rich capillary network. A few medullary cells, particularly those found in the juxtacortical regions, may have enlarged, hyperchromatic nuclei, which increase in number with age (28). In addition, the cytoplasm of medullary cells frequently contains periodic acid-Schiff (PAS)–positive hyaline globules, which also tend to be most prominent in juxtacortical regions (29). The most characteristic feature of medullary cells at the ultrastructural level is the presence of membrane-bound secretory granules in which catecholamines and other products are stored. In specimens initially fixed in glutaraldehyde, norepinephrine granules have very dense cores, which are separated from their limiting membranes by an irregular electron-lucent space or halo. Epinephrine granules have moderately dense cores, which are closely applied to their limiting membranes (30,31). A few medullary cells may also contain intranuclear pseudoinclusions, which represent invaginations of the cytoplasm into the nucleus.
In the past, the catecholamine content of chromaffin cells was demonstrated by a variety of histochemical techniques (32–35), but in current surgical pathology practice, immunohistochemical stains for chromogranins, chromomembrins, synaptophysin, and regulatory peptides are most commonly used (36–39). In addition, antibodies to catecholamine-synthesizing enzymes can be used as markers for medullary cells.
A few ganglion cells are present within the medulla either as single cells or small cell clusters and some may be associated with nerve bundles (6,27). S-100 protein–positive sustentacular cells are present at the peripheries of the medullary cords and nests and are also evident around the ganglion cells. In addition, a cell type that has been termed the small, intensely fluorescent cell or small, granule-containing cell is also seen in the medulla of most mammals. These cells may function as interneurons (40). There may also be small collections of lymphocytes and plasma cells within the medulla, but their significance is unknown.
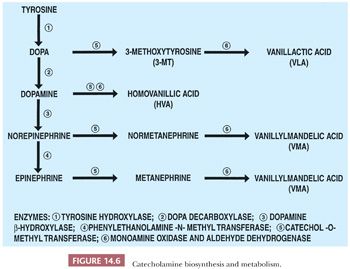
The precursor of the catecholamines is tyrosine, which is converted sequentially to dihydroxyphenylalanine, dopamine, norepinephrine, and epinephrine by a series of well-characterized enzymatic steps (Fig. 14.6) (41). The major catecholamine product of the medulla is epinephrine, which affects the activities of a wide variety of cells and tissues following its interactions with specific receptors.
In addition to the cortical and medullary cells, hybrid cells showing features of both cell types have been described in the rat. Such hybrid cells, at the ultrastructural level, contain chromaffin granules, mitochondria with tubulovesicular cristae, and relatively abundant smooth endoplasmic reticulum (42,43). However, hybrid cells do not apparently occur in normal human adrenal glands, but as will be seen later, adrenocortical neoplasms may demonstrate features of hybrid phenotype.
CONGENITAL ANOMALIES, DEVELOPMENTAL DISORDERS, AND METABOLIC ABNORMALITIES
ACCESSORY ADRENALS, ADRENAL FUSION, AND ADRENAL APLASIA
Congenital anomalies of the adrenal gland rarely come to the attention of the surgical pathologist. The most common is heterotopia (44). Although the term “heterotopia” is commonly used for this condition, a more accurate descriptor is accessory adrenal tissue because, in most cases, orthotopic adrenal gland is also present. Most accessory adrenals consist exclusively of cortical tissue, but a few examples, particularly those in the region of the celiac ganglion, may also contain medulla (45). Accessory adrenal cortex is found most frequently in the retroperitoneal space along the course of the urogenital ridges. In addition, accessory adrenal tissue may also be discovered incidentally just beneath the renal capsule in the upper pole, at the hilar regions of the ovaries and testes, and along the course of the spermatic cord. The studies of MacLennan (46) have shown that adrenal cortical tissue is present in approximately 1% of inguinal hernia sacs from children undergoing inguinal herniorrhaphy. Rare sites of accessory adrenal tissue include pancreas, spleen, liver, mesentery, lung, and brain. Accessory adrenals may undergo hyperplasia in response to increased levels of ACTH and may serve as the site of origin of cortical neoplasms (28,47,48). True heterotopic adrenal glands may be fused with the liver or kidney and are typically surrounded by a common connective tissue capsule (49,50).
Adrenal union (fusion) and adhesion (accreta) are rare anomalies that are distinguished by the presence (adrenal union) or absence (adhesion) of a connective tissue capsule. Fusion is occasionally associated with midline congenital defects, including spinal dysraphism, indeterminate visceral situs, and the Cornelia de Lange syndrome (27). Fusion of the adrenal glands can occur in patients with bilateral renal agenesis.
Aplasia of the adrenal glands has been reported in association with anencephaly; however, in most instances, the adrenal glands are markedly hypoplastic rather than completely absent (51). In approximately 10% of patients with unilateral renal agenesis, the ipsilateral adrenal gland is also absent. Several types of adrenal hypoplasia have been reported (52), and affected infants typically have signs and symptoms of adrenal insufficiency. In so-called primary hypoplasia, the adult cortex is markedly hypoplastic, but the fetal zone is retained and often demonstrates cytomegalic features. This disorder shows an X-linked pattern of inheritance and has been associated with mutations or deletions of the DAX-1 gene (Xp21) (45,53,54). The miniature adult type of hypoplasia may appear sporadically or as an inherited abnormality with an autosomal recessive pattern of inheritance. The adrenal glands have a normal architecture despite their small size.
ADRENAL CYTOMEGALY AND BECKWITH-WIEDEMANN SYNDROME
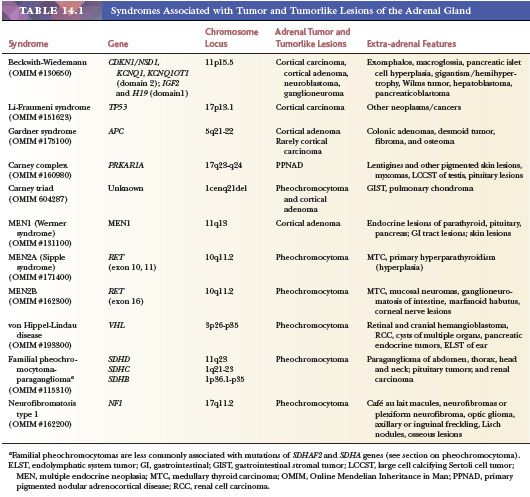
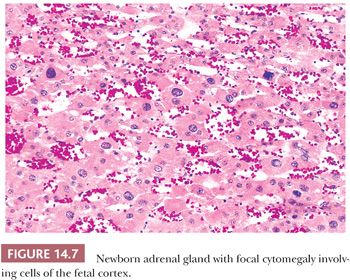
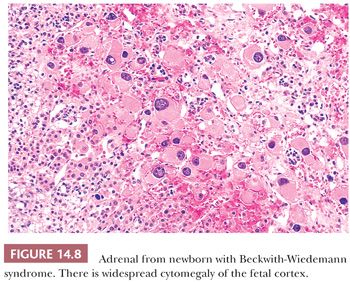
Adrenal cytomegaly is characterized by the presence of focal or diffuse collections of markedly enlarged cortical cells containing hyperchromatic and pleomorphic nuclei, occasionally with nuclear pseudoinclusions within the fetal cortex (Fig. 14.7). Cytomegalic cells may measure up to 150 μm in diameter. This is a common finding that has been observed in the normal fetus and in up to 3% of stillborn infants (55,56). Cytomegalic cells may be particularly prominent in infants with the Beckwith-Wiedemann syndrome (BWS) in which the adrenal glands are typically hyperplastic (Fig. 14.8) (45). Characteristic features of BWS, which include macroglossia, prenatal and postnatal overgrowth (gigantism/hemihyperplasia), abdominal wall defects (exomphalos), and pancreatic islet cell hyperplasia leading to hypoglycemia, permit its early recognition (Table 14.1). Hemorrhagic adrenal cortical macrocysts may also be found (57,58). Patients with BWS are predisposed to the development of malignant tumors including Wilms tumor, adrenocortical carcinoma, neuroblastoma, hepatoblastoma, and pancreaticoblastoma (27,58). Benign adrenal cortical adenomas and ganglioneuromas have also been reported in affected patients (58). An autosomal dominant inheritance is well established in BWS, but approximately 85% of cases are actually sporadic. The molecular basis of this syndrome is complex and involves downregulation of imprinted genes within the chromosome 11p15 regions (IGF2 and H19 at domain 1; CDKN1C, KCNQ1, and KCNQ1OT1 at domain 2) (58). Other syndromes that predispose to adrenal tumors (Table 14.1) are discussed later under the respective sections.
STORAGE DISEASES
Adrenoleukodystrophy (Addison-Schilder disease; OMIM #300100) is a rare, X-linked recessive disorder characterized by progressive demyelination of the central and peripheral nervous system and by adrenal cortical insufficiency (59). The disorder is caused by mutations of the ABCD1 gene on chromosome Xq28, which encodes an ATP-binding transporter adrenoleukodystrophy protein (ALDP) that is localized in the peroxisomal membrane. Several mutations have been identified and result in the defective oxidation of very long fatty acids (60). The diagnosis can be established by the presence of hexacosanoate and other long-chain fatty acids in cultured skin fibroblasts (61). The adrenal glands in this disorder are grossly atrophic with weights ranging from 1 to 2 g or less, with ballooning and striation of the cells of the inner zona fasciculata and zona reticularis. Groups of ballooned cells form nodules that may undergo degenerative changes with the formation of large cortical vacuoles. At the ultrastructural level, there is proliferation of smooth endoplasmic reticulum and the presence of lamellar inclusions with a trilaminar structure (59). An adult variant with an onset in the second or third decades is known as adrenomyeloneuropathy, a condition that can be associated with unexplained adrenal insufficiency in the absence of neurologic manifestations at first clinical presentation (62). A third form of the disease has been reported in women who are carriers of the abnormal gene.
Wolman disease (primary familial xanthomatosis; OMIM #278000) is a rare autosomal recessive lipid storage disorder caused by deficiency of lysosomal acid lipase (63). The disease is characterized by the accumulation of triglycerides and cholesterol esters in a variety of tissues including the liver, spleen, and adrenal glands. Most affected individuals die by the age of 6 months. Typically, the adrenal glands are markedly enlarged even though they often retain their normal configurations. The glands often demonstrate multiple foci of calcification in association with necrosis and fibrosis (63). The adrenal gland may also be involved in storage diseases (e.g., Niemann-Pick disease, Gaucher disease, etc.), which may result in adrenal enlargement and hypofunction.
CONGENITAL ADRENAL HYPERPLASIA
The syndromes of congenital adrenal hyperplasia (congenital adrenogenital syndromes) result from a series of autosomal recessive enzymatic defects in the biosynthesis of adrenal steroids (64). This group of disorders is caused by deficient activity of one of the following: P450c21 (21-hydroxylase), P450c11 (11β-hydroxylase), 3β-hydroxysteroid dehydrogenase, P450c17 (17-hydroxylase/17,20-lyase), or P450scc. On clinical examination, affected patients may have abnormalities of sexual development, salt wasting, hypertension, or acute adrenal insufficiency, depending on the specific defect. In females, common abnormalities associated with these defects include masculinization in utero, postpubertal masculinization, and primary amenorrhea. Males may show signs of precocious puberty or pseudohermaphroditism. The biochemical, genetic, and molecular features of these disorders are discussed in detail elsewhere (64–67).
The most common deficiency affects the enzyme 21-hydroxylase (P450c21; OMIM #201910) and is responsible for approximately 95% of cases of congenital adrenal hyperplasia (64). The worldwide incidence of classic 21-hydroxylase deficiency is 1 in 14,500 births, with a heterozygote frequency of approximately 1 in 60. Affected individuals have evidence of cortisol deficiency, aldosterone deficiency with salt wasting (65% of cases), and excess adrenal androgen production with evidence of virilization (30% of cases). Excess adrenal androgen production is a consequence of the accumulation of 17-hydroxypregnenolone, which is subsequently metabolized to androgenic steroids. Nonclassic 21-hydroxylase deficiency (attenuated or acquired) has a frequency of 1 in 100 in certain parts of the United States and is one of the most common autosomal recessive disorders. Affected individuals have mild degrees of cortisol deficiency, normal aldosterone production, and excess production of adrenal androgens. This form of 21-hydroxylase deficiency is most often diagnosed in childhood or early adulthood. A small proportion of individuals with 21-hydroxylase deficiency may have no apparent symptoms (cryptic form).
Deficiency of 11β-hydroxylase (P450c11; OMIM #202010) accounts for approximately 5% of all cases of congenital adrenal hyperplasia and is associated with increased production of androgens and deoxycorticosterone (65–67). As a result, affected individuals typically exhibit signs of hyperandrogenism and hypertension. Deficiency of 17α-hydroxylase is responsible for approximately 1% of cases. External genitalia are female in both sexes. Increased levels of deoxycorticosterone are responsible for the hypertension seen in these patients.
Adrenal hyperplasia in the congenital adrenogenital syndromes results from inadequate production of glucocorticoids, leading to stimulation of the cortex by increased pituitary ACTH production. The adrenal glands become markedly enlarged with a characteristic cerebriform appearance and a tan-brown color (27). On microscopy, cortical cells are lipid depleted. However, in lipoid congenital adrenal hyperplasia (OMIM #201710), the adrenal glands are pale yellow and are characterized on microscopic examination by vacuolated cells, occasionally with formation of cholesterol clefts and an accompanying giant cell reaction. Initially, these individuals were thought to have a deficiency of 20,22-desmolase (P450scc) because of the inability to convert cholesterol to pregnenolone. However, the defect has been shown to be due to mutations encoding the steroidogenic acute regulatory protein (68,69). These disorders are usually treated by replacement of the deficient steroid hormone and surgical correction of ambiguous genitalia or hypospadias.
Adrenal cortical adenomas and carcinomas may develop, although rarely, in the setting of congenital adrenal hyperplasia (70,71). Testicular tumors can also arise in affected patients. These testicular or adrenal lesions are not autonomous neoplasms because they are dependent on the presence of elevated levels of ACTH (72). They are commonly bilateral and are most typically located in the hilar regions of the testes. The cell of origin is unknown, but the component cells of these lesions contain abundant eosinophilic cytoplasm, lacking crystalloids of Reinke. In the series reported by Rutgers et al. (72), the diagnosis of congenital adrenal hyperplasia was made only after the appearance of testicular tumors in 18% of the cases. Examples of ovarian steroid cell tumors have also been described in the setting of congenital adrenal hyperplasia (73,74), as well as giant myelolipomas (27,75) (see later section on myelolipoma).
HYPOFUNCTIONAL STATES
Hypofunction may occur as the result of a primary disorder of the adrenal cortex or as a secondary change caused by a disorder of the pituitary–hypothalamic axis.
PRIMARY HYPOFUNCTION
Autoimmune Adrenalitis
Inflammation of the adrenals may result from autoimmune mechanisms, infection, or a variety of other causes (76). Approximately 75% of cases have an autoimmune origin. In idiopathic (autoimmune) Addison disease, the glands are markedly atrophic, and the residual cortical tissue is infiltrated by chronic inflammatory cells, including lymphocytes and plasma cells. As noted in the previous section, focal aggregates of lymphocytes are a normal finding with increasing frequency in the adrenal gland of older patients (20) and should not be misconstrued as evidence of adrenalitis. All layers of the cortex are involved in autoimmune adrenalitis, but the medulla is unaffected. Typically, the capsules of the glands are fibrotic. Both humoral and cell-mediated immune mechanisms have been implicated in the development of autoimmune adrenal hypofunction. Autoantibodies to cortical cells are present in 50% of all patients and in more than 70% of women with newly diagnosed disease. The major targets for autoantibody reactivity are the adrenal cytochrome P450 enzymes (77). Affected patients can also have antibodies to gonadal, gastric parietal, and thyroid follicular cells.
Adrenocortical hypofunction may be associated with hypofunction of other endocrine glands (78). The type I polyglandular autoimmune syndrome is associated with mucocutaneous candidiasis, hypoparathyroidism, adrenal insufficiency, autoimmune thyroiditis, and diabetes mellitus. Alopecia may also be present. This form of the disease has also been termed autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED). Mutations in the APECED or autoimmune regulatory (AIRE) gene (21q22.3) have been implicated in the development of this syndrome (79–83). The type II polyglandular autoimmune syndrome is characterized by adrenal insufficiency, autoimmune thyroiditis, and insulin-dependent diabetes mellitus. The type I syndrome is inherited as an autosomal recessive trait, whereas the pattern of inheritance of the type II syndrome is usually dominant and is associated with HLA-DR3 and CTLA-4 (81). Type III and IV syndromes have also been described. The type III syndrome involves autoimmune thyroid disease and other autoimmune diseases including type I diabetes mellitus, but there is usually lack of adrenal gland involvement (82). Type IV syndrome involves two or more organs and involves the adrenal but without involvement of the thyroid (81).
Tuberculosis and Fungal Diseases
Infectious disorders, including tuberculosis and fungal diseases (e.g., histoplasmosis, North and South American blastomycosis, coccidiomycosis, cryptococcosis), can affect both the cortical and medullary regions of the adrenal glands. Although tuberculosis is now a rare cause of adrenal insufficiency in the United States and Western Europe, it is a common cause in parts of the world where tuberculosis is endemic. In contrast to the shrunken appearance of the adrenal glands in idiopathic Addison disease, the glands in mycobacterial infection are typically enlarged and replaced by caseous material. Infection with Mycobacterium avium-intracellulare, on the other hand, is typically associated with the presence of confluent masses of histiocytes containing the acid-fast organisms.
Viruses
Cytomegalovirus (CMV) has been identified in the adrenal glands of a large proportion of patients dying of acquired immunodeficiency syndrome (84). Adrenal cortical necrosis associated with CMV infection can be severe enough to result in acute adrenal insufficiency in some instances. Both herpes simplex and varicella zoster may also involve the adrenal glands and may lead to adrenal cortical insufficiency when they are associated with extensive cortical necrosis.
Amyloidosis
Rarely, amyloid deposition can result in cortical hypofunction (85). Typically, adrenal involvement is associated with extensive systemic amyloid disease of the AA type. The adrenal glands may have a normal shape and size or may be enlarged. In severe cases, the glands are pale tan to yellow. Microscopically, amyloid deposits affect the fasciculata and reticularis zones and are typically present between the cortical cells and capillary endothelium. The cortical cells ultimately become atrophic as a result of the progressive deposition of intercellular amyloid. In patients with AL disease, the amyloid deposits are predominantly vascular in distribution.
Adrenal Hemorrhage
Adrenal hemorrhage may develop in a segmental fashion or may involve the entire adrenal (86). This syndrome may be seen in association with sepsis and shock caused by meningococcal infection or infection with other bacteria, including Haemophilus influenzae, Streptococcus pneumoniae, and Pseudomonas aeruginosa (Waterhouse-Friderichsen syndrome). Typically, the glands are enlarged and hemorrhagic, with necrosis of both cortical and medullary tissue. Adrenal hemorrhage in the Waterhouse-Friderichsen syndrome is regarded as the consequence rather than the cause of shock. Anticoagulant therapy may also be associated with adrenal hemorrhage. Corticomedullary necrosis of milder degrees has been reported in association with hypotension and shock (87). In patients with segmental lesions, examination of the capsular vessels and sinusoids will reveal evidence of thrombus formation. Affected cortical areas show a pattern of ischemic necrosis that ultimately heals by the process of fibrosis.
Miscellaneous Causes of Hypofunction
Rarely, adrenal hypofunction may result from bilateral involvement by tumor, most commonly metastatic carcinoma (see section on secondary tumors). A rare case of hypofunction has been reported secondary to involvement by Erdheim-Chester disease, a non-Langerhans histiocytosis that typically involves bone but in which extraskeletal manifestations are present in up to 50% of cases. In this condition, there is diffuse enlargement of the adrenal gland secondary to infiltration by foamy histiocytes (88).
SECONDARY HYPOFUNCTION
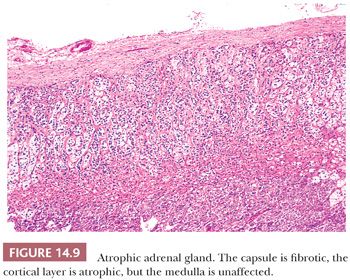
Adrenocortical atrophy may be found in association with lesions primarily affecting the adenohypophysis or hypothalamus, leading to diminished secretion of ACTH (2). The administration of exogenous corticosteroids will produce similar changes as a result of suppression of ACTH. The adrenal glands in secondary hypofunctional states are considerably smaller than normal, although the overall configurations of the glands are retained. Typically, the cortex is bright yellow as a result of lipid accumulation in the cortical cells, the capsule is fibrotic, and the medulla is unaffected (Fig. 14.9). The zona glomerulosa is usually of normal thickness in these cases.
HYPERFUNCTIONAL STATES
ADRENOCORTICAL HYPERPLASIA
Hyperplasia of the adrenal cortex, which represents an increased cortical mass resulting from stimulation of the cortex by ACTH derived from the pituitary gland or from a variety of extrapituitary sources, can be associated with a wide variety of clinical syndromes. Cortical hyperplasia can also selectively involve the zona glomerulosa in patients with so-called idiopathic hyperaldosteronism, as discussed in a subsequent section.
Pituitary/Hypothalamic-Based Hyperplasia (Cushing Disease)
Hyperplasia may be the result of stimulation of the adrenal glands by ACTH-producing pituitary adenomas or of hypothalamic stimulation of the pituitary ACTH cells by CRH (89,90). Basophilic pituitary adenomas were originally observed in association with hypercortisolism by Harvey Cushing in 1932, and this association has been termed Cushing disease or ACTH-dependent Cushing syndrome. Immunohistochemical studies have shown that ACTH-producing pituitary adenomas are considerably more common than had been recognized on light microscopy alone, and many of them have been classified as microadenomas.
Adrenocortical hyperplasia in patients with ACTH-dependent Cushing syndrome can be either diffuse or nodular, and combinations of these two forms of hyperplasia are common. In diffuse hyperplasia, glandular weights may be increased only slightly (2). In more advanced cases, the combined average weight varies between 12 and 24 g. The glands have rounded contours, rather than the sharp outlines typical of normal glands. The inner portion of the cortex is widened and often appears pale brown or tan. The outer layers of the cortex are typically yellow. On microscopy, the inner brown zone corresponds to lipid-depleted cells of the fasciculata, whereas the cells of the outer cortex are more characteristically vacuolated (91). The glomerulosa in adults with Cushing disease is often difficult to identify, but in children, the glomerulosa may also appear slightly hyperplastic (92).
In some cases, the cortex may appear nodular with individual nodules measuring less than 0.5 or 1.0 cm in diameter, depending on varying criteria used by different authors (27). This type of change has been referred to as diffuse and micronodular hyperplasia. If the nodules exceed 1 cm in diameter, the hyperplasia is defined as the diffuse and macronodular type.
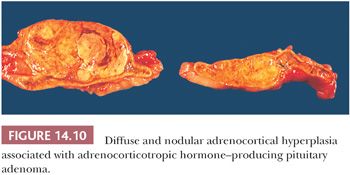
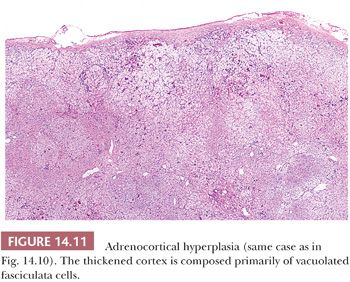
In diffuse and nodular (micro- or macro-) hyperplasia, multiple cortical nodules are superimposed on a diffusely hyperplastic cortex (Figs. 14.10 and 14.11). Formation of nodules is often asymmetric, and although one adrenal gland may show diffuse and nodular cortical hyperplasia, the contralateral adrenal gland may appear diffusely hyperplastic. The nodules are often composed of admixtures of clear- and compact-type cells. In contrast to the atrophic cortex adjacent to a functioning adenoma, the cortex between or adjacent to the nodules in nodular hyperplasia is diffusely hyperplastic.
Adrenocortical Hyperplasia Associated with Paraneoplastic (Ectopic) Production of Adrenocorticotropic Hormone or Corticotropin-Releasing Hormone
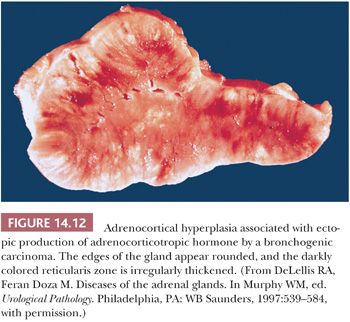
Hyperplasia can be found in association with a variety of neoplasms producing ACTH or CRH (90,93). In most series, bronchial carcinoids and small cell carcinomas account for the majority of cases. Other tumors associated with the paraneoplastic ACTH syndrome include pancreatic endocrine neoplasms, medullary thyroid carcinoma, thymic carcinoids, and pheochromocytomas. In patients with the paraneoplastic ACTH syndrome associated with bronchogenic small cell carcinoma or other tumors, the adrenals are usually larger (average combined weight of 20 to 30 g) than those seen in association with diffuse hyperplasia stemming from pituitary ACTH overproduction. The cortex is diffusely hyperplastic and appears tan-brown throughout its width (Fig. 14.12) (94). On microscopy, there is evidence of diffuse hyperplasia of the fasciculata cells, which are characterized by a compact or lipid-depleted appearance (95). Foci of nuclear enlargement and atypia of reticularis cells may be noted, and these features may be particularly striking adjacent to metastatic foci in the glands (28). Both bronchial carcinoids and small cell bronchogenic carcinomas may also produce CRH. Exceptionally, secretion of both ACTH and CRH from the same tumor has been documented (96).
Adrenocortical Hyperplasia Associated with Hyperaldosteronism
Primary hyperaldosteronism is characterized by the excessive secretion of aldosterone from the adrenal glands and is associated with suppression of plasma renin activity with resultant hypokalemia and hypertension. At least six subtypes of primary hyperaldosteronism have been recognized, including aldosterone-producing adenoma, idiopathic hyperaldosteronism, primary adrenal hyperplasia, aldosterone-producing adrenal cortical carcinoma, aldosterone-producing ovarian tumor, and familial hyperaldosteronism (FH) (97). FH is subdivided into three groups: FH-I (glucocorticoid-remediable hyperaldosteronism), FH-II (aldosterone-producing adenoma and idiopathic hyperaldosteronism), and FH-III (hypertension and massive adrenocortical hyperplasia) (98).
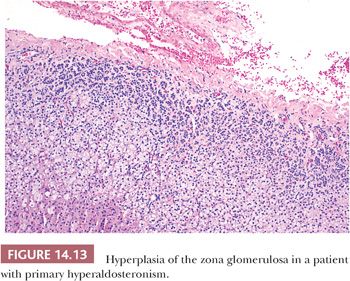
Primary hyperaldosteronism is most often associated with adrenocortical adenomas; however, in approximately 40% of cases, the only apparent adrenal abnormality is hyperplasia of the zona glomerulosa with or without the formation of micronodules (16,27,99,100). Generally, biochemical abnormalities in patients with hyperplasia are less severe than in those with adenomas. On histologic examination, hyperplasia of the glomerulosa is characterized by thickening of this cell layer, with tonguelike projections of the glomerulosa extending toward the fasciculata (Fig. 14.13). Micronodules, when present, are usually composed of clear fasciculata-type cells (16,28) and are thought to be a consequence of the associated hypertension. In approximately 10% of cases, it may not be possible to distinguish a micronodule from a true adenoma associated with aldosterone production. In FH-III, the resected adrenal is massively enlarged (combined weights up to 82 g) with hyperplasia of a single cortical compartment with features of fasciculata or transitional cells but atrophy of the glomerulosa (98).
MACRONODULAR HYPERPLASIA (MASSIVE MACRONODULAR ADRENOCORTICAL DISEASE)
In macronodular hyperplasia with marked adrenal enlargement, the adrenal glands may together weigh up to 180 g, and individual nodules may measure up to 4.0 cm in diameter (45,101,102). Because of the very large size of the glands and confluence of adjacent nodules, the resected adrenal glands may be mistaken for neoplasms. Nodules can be composed of clear cells, compact cells, or admixtures of these cell types. Macronodular hyperplasia is corticotropin (ACTH) independent, and this entity can (rarely) involve a single gland (103). The cortex between the nodules is often atrophic, as might be expected in an ACTH-independent process. This entity, which has also been referred to as massive macronodular adrenocortical disease (MMAD), has a bimodal age distribution. A small proportion of patients may present during the first year of life, and this form of the disease may be associated with the McCune-Albright syndrome. Most patients present clinically in the fifth decade with Cushing syndrome, but a subset of patients may have subclinical disease and are discovered to have enlarged adrenals on imaging studies during the workup for other disorders. Males and females are equally affected. Rare examples of familial MMAD have also been reported. This disorder is associated with aberrant (ectopic) expression and regulation of various G-protein–coupled receptors (104). Assie et al. (105) recently reported germline and somatic mutations in both alleles of the armadillo repeat-containing 5 (ARMC5) gene in tumors from 18 of 33 patients with macronodular hyperplasia with Cushing syndrome. Louiset et al. (106) demonstrated intra-adrenal corticotropin production and release by a subset of adrenocortical cells in tissue from patients with bilateral disease and Cushing syndrome. Based on these reported findings, Lacroix (107) has concluded that corticotropin-independent macronodular adrenal hyperplasia seems an inappropriate concept and suggests that screening of affected patients along with family members for ARMC5 mutation would be appropriate.
PRIMARY PIGMENTED NODULAR ADRENOCORTICAL DISEASE—MICROADENOMATOUS HYPERPLASIA OF THE ADRENAL GLAND
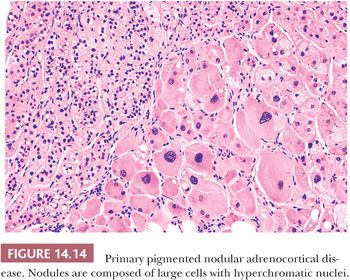
Primary pigmented nodular adrenocortical disease (PPNAD) is a rare disorder characterized by the presence of multiple pigmented nodules of cortical cells with intervening atrophic cortical tissue usually seen in association with features of ACTH-independent Cushing syndrome (108–111). The glands may be either smaller than normal or enlarged. Individual nodules, which can vary in color from gray to black, typically measure from 1 to 3 mm in diameter, although larger nodules measuring up to 3 cm in diameter may also be evident (27). The nodules are composed of large, granular eosinophilic cells that often contain enlarged hyperchromatic nuclei with prominent nucleoli (Fig. 14.14). Because of the atypical nuclear features, this entity has also been referred to as micronodular dysplasia. The cells are generally filled with lipochrome pigment, which is responsible for their dark color on gross examination.
The origin of PPNAD is unknown, but numerous theories have been advanced to describe its origin (112). PPNAD arises sporadically or in a familial form that can be associated with Carney complex type 1 (CNC1; OMIM #160980 ), which includes cardiac myxomas, spotty pigmentation, neurofibromas, testicular Leydig or Sertoli cell tumors, mammary myxoid fibroadenomas, and cerebral hemangiomas (110,113). CNC1 may occur sporadically or may be inherited as an autosomal dominant trait. The gene encoding the protein kinase A (PKA) type Iα regulatory subunit, PRKAR1A, has been mapped to 17q22-24, and loss of heterozygosity (LOH) studies from patients with CNC1 have revealed mutations in this gene in approximately 50% of affected individuals. No mutations have been found on 2p16. Studies of sporadic and isolated cases of CNC have also revealed inactivating mutations of PRKAR1A. The wild-type alleles could be inactivated by somatic mutations consistent with the hypothesis that the gene belongs to the tumor suppressor class (114,115).
Horvath et al. (116) reported mutations in the gene encoding phosphodiesterase 11A4 (PDE11A4) in cases of PPNAD and other forms of micronodular adrenocortical hyperplasia. LOH and other analyses showed susceptible genes at the 2q31-2q25 locus (116). Phosphodiesterases (PDEs) regulate cyclic nucleotide levels. The same group subsequently reported that missense mutations of PDE11A were present frequently in patients from the general population with adrenal cortical hyperplasia and adenoma, speculating that PDE11A genetic defects may be associated with adrenal pathology in a wider clinical spectrum (117).
ADRENAL NEOPLASMS
In addition to CNC and BWS, several other familial syndromes predispose to the development of adrenal tumors (Table 14.1). These include Li-Fraumeni syndrome; Gardner syndrome; Lynch syndrome; multiple endocrine neoplasia (MEN) types 1, 2A, and 2B; familial pheochromocytoma-paraganglioma; neurofibromatosis type 1; and von Hippel-Lindau disease (118–121). Unlike BWS, which predisposes to both cortical and medullary tumors, the other syndromes give rise to either cortical or medullary tumors. Although BWS presents early in life, the manifestations of the other syndromes usually occur later in life, but features of MEN2B, which include oral, ocular, and gastrointestinal ganglioneuromatosis associated with a marfanoid habitus, may present at birth or early infancy. Although Carney triad originally included extra-adrenal paraganglioma, gastrointestinal stromal tumor and pulmonary chondroma, examples of adrenocortical adenomas and pheochromocytoma (Table 14.1) are also seen in a subset of these patients (122, 122a).
ADRENOCORTICAL NEOPLASMS
ADRENOCORTICAL ADENOMAS
Adrenocortical adenomas are a heterogeneous group of benign neoplasms that can differentiate toward any of the cortical layers (27,123). The prevalence of cortical adenomas greater than 1 cm from autopsy series ranges from 1.5% to 7% (118). Functional tumors are most commonly associated with overproduction of aldosterone (Conn syndrome), followed by glucocorticoids (Cushing syndrome). Virilizing and rarely feminizing tumors (adrenogenital syndromes) are less common (118). Mixed syndromes can also develop. However, tumors without apparent functional activity are the most common cortical adenomas and are usually identified by imaging studies in the workup of patients for nonadrenal disorders (118).
Cushing Syndrome
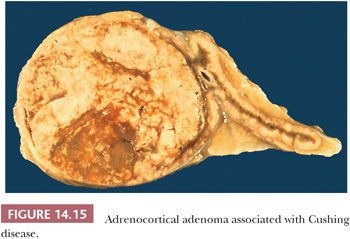
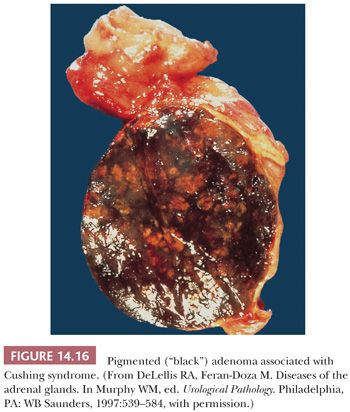
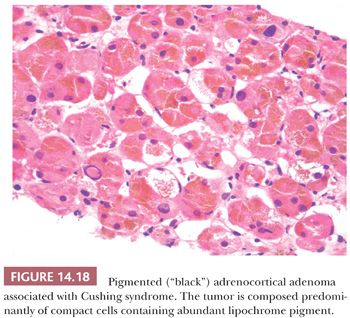
Tumors associated with Cushing syndrome are typically unilateral and present as sharply circumscribed masses that usually weigh less than 60 g and measure 3 to 4 cm in average diameter. Tumors weighing more than 100 g should be examined with particular care to rule out malignancy (16,28,45). On cross section, adenomas vary from yellow to brown (Fig. 14.15), and occasional examples of heavily pigmented (black) adenomas have been reported (Fig. 14.16). Necrosis is rare in the absence of previous arteriographic or venographic study, but cystic change is relatively common, particularly in larger tumors.
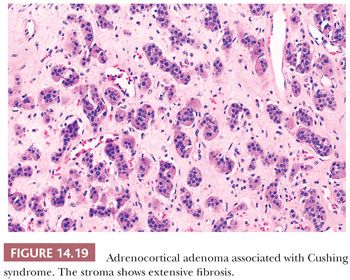
Microscopically, adenomas have pushing borders with a pseudocapsule derived from compression of the adjacent cortex or expansion of the adrenal capsule (18). They are most often composed of small nests, cords, or alveolar arrangements of vacuolated (clear) cells that most closely resemble those of the normal fasciculata. Generally, adenoma cells are somewhat larger than normal cortical cells. Variable numbers of compact-type cells are also evident (Fig. 14.17A,B). The reticulin framework is preserved in adenomas (Fig. 14.17C). Black adenomas are composed exclusively of lipochrome-rich compact cells (Fig. 14.18) (2). Foci of spindle cell growth may be evident in some cases, and occasional adenomas may exhibit considerable fibrosis (Fig. 14.19). The nuclear-to-cytoplasmic ratio is generally low, although a few single cells and small cell groups may have enlarged hyperchromatic nuclei. Typically, the nuclei are vesicular with small, distinct nucleoli. Mitotic activity is rare in adenomas. In fine-needle aspiration biopsy specimens, the cells are round to polyhedral with round nuclei and foamy cytoplasm. Numerous naked nuclei in a background of granular to foamy material may be prominent.
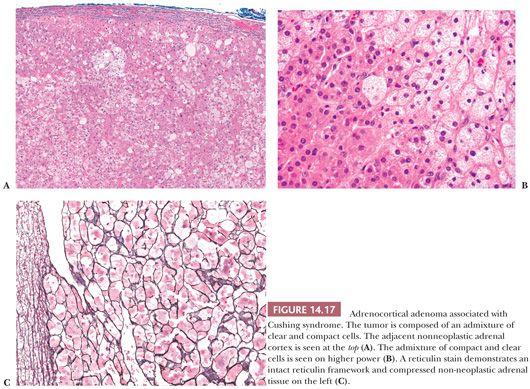
On ultrastructural examination, adenoma cells most closely resemble the cells of the normal fasciculata or reticularis (2). The cytoplasm typically contains abundant smooth endoplasmic reticulum and variable numbers of lipid droplets. The mitochondria are round to ovoid with a predominance of tubulovesicular or exclusively vesicular cristae (21). Lamelliform cristae may also be present. The immunohistochemical profile of adenomas is discussed along with that of adrenocortical carcinomas.
Foci of myelolipomatous change or calcification may be seen, particularly in larger adenomas (28). The cortex adjacent to functional adenomas and in the contralateral adrenal is typically atrophic, with cortical cells that have a clear or vacuolated cytoplasm. The atrophy, however, does not involve the glomerulosa (100).
X chromosome inactivation analyses have shown that some adenomas are clonal, whereas others are polyclonal. Monoclonal adenomas are larger than polyclonal lesions and have a higher prevalence of nuclear pleomorphism (124). Recently, a subset of cortisol producing adenomas have been shown to harbor activating somatic mutations of protein kinase A catalytic subunit (PRKACA) gene by independent investigators (124a–c).
Conn Syndrome
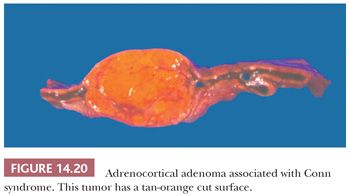
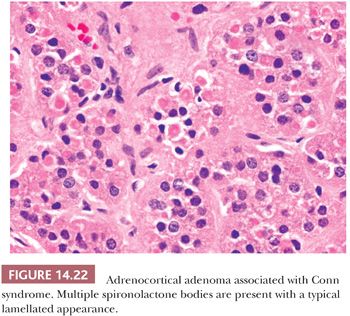
Initial studies suggested that adrenocortical adenomas were responsible for Conn syndrome in up to 90% of cases; however, more recent studies indicate that adenomas are present in a considerably smaller proportion of cases. Most adenomas associated with hyperaldosteronism measure less than 2.0 cm in diameter and are round to ovoid in configuration (125). Most commonly, they are unilateral, although bilateral tumors have been reported. They are characteristically bright yellow similar to tumors associated with Cushing syndrome (Fig. 14.15). However, they may have a tan orange gross appearance (Fig. 14.20). These tumors are generally, but not always, demarcated from the adjacent cortex by a pseudocapsule. The tumor cells are usually arranged in small nests and cords. They can resemble cells of the glomerulosa, fasciculata, or reticularis or combine the features of both glomerulosa and fasciculata cells (hybrid cells) (Fig. 14.21) (28,125). Rarely, black adenomas and myxoid adenomas have been associated with Conn syndrome (126–128). In the cases of myxoid adenomas, the tumors tend to demonstrate a pseudoglandular pattern (127,128). In patients treated with spironolactone, some cells within the adenoma may contain lamellated eosinophilic inclusions (spironolactone bodies) measuring up to 10 μm in diameter. They are often demarcated from the adjacent cytoplasm by a clear halo (Fig. 14.22). Ultrastructurally, spironolactone bodies most closely resemble myelin figures.
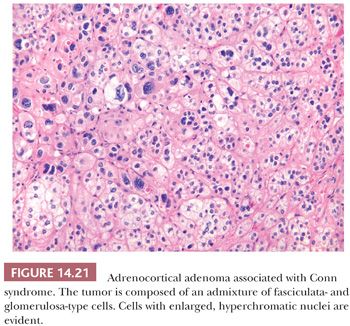
Although most of the tumor cells have relatively small vesicular nuclei with small but distinct nucleoli, some tumors exhibit considerable variation in nuclear size and shape (Fig. 14.21). At the ultrastructural level, the mitochondria manifest tubular or vesicular cristae, although some may have lamelliform cristae typical of the zona glomerulosa. The fasciculata adjacent to aldosterone-secreting adenomas is of normal thickness. However, hyperplasia of the zona glomerulosa may be present in association with these tumors (125).
Adrenogenital Syndromes
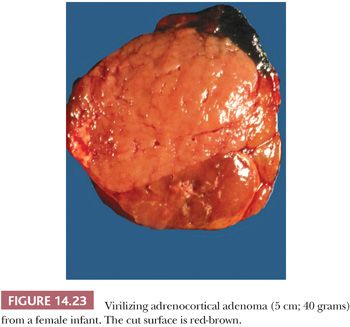
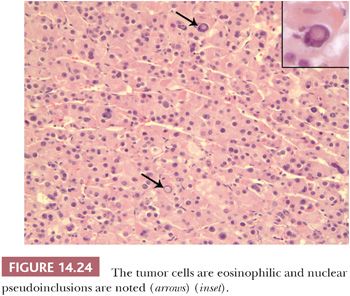
Benign adrenocortical tumors may be associated with syndromes of virilization or feminization, but the presence of a pure adrenogenital syndrome, particularly feminization, should raise the possibility of malignancy. Some authors, in fact, consider all feminizing cortical neoplasms to be potentially malignant. Virilizing adenomas are generally larger than those found in the context of pure Cushing syndrome, and a few adenomas associated with adrenogenital syndromes have weighed up to 500 g (16,27,28). Similar to tumors associated with glucocorticoid overproduction, virilizing adenomas are sharply circumscribed or encapsulated; however, they tend to be red-brown rather than yellow on cross section (Fig 14.23) (28). Smaller tumors have an alveolar pattern of growth, whereas larger tumors tend to have more solid or diffuse growth patterns. Although most tumor cells have a low nuclear-to-cytoplasmic ratio, single cells and small cell groups may exhibit nuclear enlargement and hyperchromasia. The cytoplasm is usually eosinophilic and granular (Fig. 14.24). Rare virilizing tumors contain Reinke crystalloids and have been termed Leydig cell adenomas similar to their testicular counterparts (129,130). On ultrastructural examination, the mitochondria are of the tubulolamellar type. Sex steroid–producing adenomas are not associated with atrophy of the adjacent cortex or the contralateral adrenal gland.
Nonfunctional Adrenocortical Adenomas and Cortical Nodules
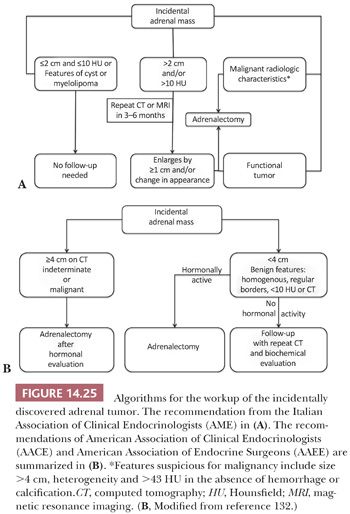
Adrenocortical nodules are common findings in patients without clinical or biochemical evidence of steroid hormone hypersecretion (27). Autopsy studies have revealed cortical nodules in approximately 25% of individuals. The nodules are also commonly detected with abdominal CT and MRI scans, and they have been grouped among the lesions classified as “incidentalomas.” Subsequent workup of these lesions is based on their size and attenuation characteristics on unenhanced CT, which is measured in Hounsfield units (HU). Tumors with high lipid content have low attenuation that is more typical of adenomas. Lesions that are small and 10 HU or less are likely to be benign, whereas those that are greater than 10 HU are considered indeterminate. Chemical shift MRI that also relies on the lipid content to characterize adrenal tumors is of comparable sensitivity and specificity to unenhanced CT (131). PET scan and other imaging modalities may be utilized, but CT and MRI are the imaging methods recommended by the Italian Association of Clinical Endocrinologists (AME), American Association of Clinical Endocrinologists (AACE), and American Association of Endocrine Surgeons (AAEE) (Fig. 14.25) (131,132).
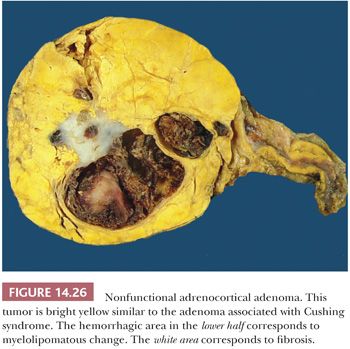
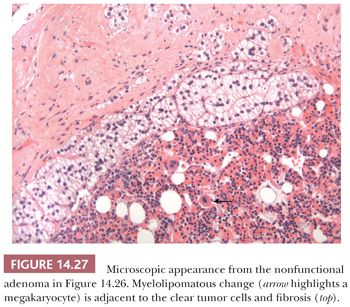
Although cortical nodules are commonly multicentric and bilateral, single nodules measuring up to 2 to 3 cm in diameter may be evident. Smaller nodules are often nonencapsulated, whereas the larger single nodules may be surrounded by a fibrous capsule. The adenomas and nodules are bright yellow (Fig. 14.26) with foci of brownish discoloration and are composed of fasciculata-type cells predominantly. Myxoid areas or myxoid-predominant tumors can be seen, as in cases with Conn syndrome and adrenocortical carcinomas (127,128) (see later section on adrenocortical carcinoma). Similarly, the neoplastic cells can be arranged in a pseudoglandular pattern. Occasionally, foci of myelolipomatous change (Fig. 14.27) or ossification may be evident within the nodules. In contrast to functional adenomas associated with glucocorticoid production, the cortex adjacent to nonfunctional nodules is not atrophic.
Nodules are most frequently encountered in the adrenals of elderly individuals or in patients with essential hypertension or diabetes mellitus. The nodules most likely represent foci of compensatory cortical hyperplasia that have developed in response to focal atrophy of the cortex induced by narrowing of adrenal capsular arterioles (133). Nonfunctional cortical nodules may be particularly prominent in patients with aldosterone-secreting adenomas, presumably as a result of the associated hypertension. Cortical nodularity may also be evident in the zona reticularis. Nodules arising in this zone often appear brown to black because of the presence of lipofuscin pigment. Incidental pigmented nodules may be found in up to one-third of normal adult adrenal glands.
Oncocytic Adenoma (Oncocytoma)
Tumors with oncocytic features (oncocytomas) develop rarely as primary adrenocortical neoplasms. Although some behave as benign neoplasms, others may be malignant, as discussed in the section on adrenocortical carcinoma. Sasano et al. (134) first described three adrenocortical oncocytomas which were unassociated with clinical syndromes of steroid excess and which lacked steroid synthetic hormones, as determined by immunohistochemical analysis. Subsequent larger series of cases (135,136) have also shown that the majority of oncocytomas are nonfunctional, but some tumors may be associated with virilization (136,137) or Cushing syndrome (138). A case of a giant oncocytoma arising in retroperitoneal accessory adrenal tissue has been described (139). Although oncocytic adrenocortical tumors are uncommon adrenocortical lesions, there has been a significant emphasis in the literature recently because the criteria for malignancy in these lesions differ from nononcocytic adrenocortical tumors (see section on adrenocortical carcinoma).
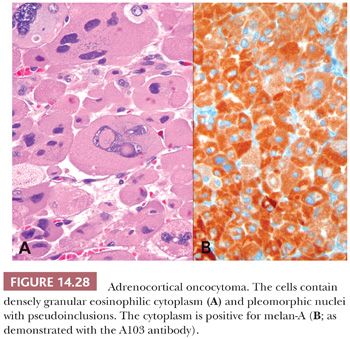
Typically, adrenocortical oncocytomas are dark brown, similar to oncocytomas at other sites. They have abundant granular eosinophilic cytoplasm (Fig. 14.28), which corresponds to the presence of numerous mitochondria with both lamellar and tubulovesicular cristae and small, electron-dense inclusions. Smaller eosinophilic cells (“small oncocytes”) may be present and may be quite frequent in some cases (140). The nuclei are enlarged, irregularly shaped, and vesicular with coarse chromatin and prominent nucleoli. Nuclear pseudoinclusions may be seen. Mitotic activity is minimal to absent, there is no evidence of necrosis, and vascular space invasion is absent. The cells have a tendency to be arranged in diffuse sheets, but variable trabecular, alveolar, and microcystic architecture can be seen (140).
Adrenocortical tumors can contain variable proportions of oncocytic cells. As a result, Bisceglia et al. (141) suggested that oncocytic adrenocortical tumors be classified as pure when the tumor was composed of 90% oncocytic cells and mixed when the tumors were composed of 50% to 90% oncocytic cells. Subsequently, Duregon et al. (142) classified oncocytic adrenocortical tumors as being pure, mixed, having focal oncocytic features, or being conventional adrenocortical tumors when the oncocytic cells constituted more than 90%, 50% to 90%, 10% to 49%, and less than 10% of the tumor cells, respectively. In the latter study, the authors demonstrated the presence of 4977-bp mitochondrial DNA “common deletion” by real-time polymerase chain reaction (PCR) and fluorescence in situ hybridization (FISH) in 9 of 12 oncocytic adrenocortical adenomas. This deletion is seen in oncocytic neoplasms from other organs, hence the designation “common deletion.” However, this finding was not entirely specific because it was demonstrated in some normal adrenocortical cells and some examples of conventional adrenocortical adenomas.
ADRENOCORTICAL CARCINOMAS
Adrenocortical carcinomas are rare tumors with an incidence of one per million population per year (118). They account for 0.05% to 0.2% of all malignancies. There is a bimodal age distribution, with a small peak in the first two decades and a larger peak in the fifth decade (118,143). Adrenocortical carcinomas develop somewhat more commonly in women than men in most large clinical series (118), although some studies have demonstrated a slight male predominance. There is no racial predilection, but there is a suggestion that there is an increased rate among blacks (118,143).
Adrenocortical carcinomas have been reported in approximately 1% of patients with the Li-Fraumeni syndrome, with most affected individuals harboring p53 mutations at chromosome locus 17p13. These tumors, in fact, may be the only manifestation of this disorder in childhood (144). The frequency of cortical malignancies is also increased in patients with BWS (Table 14.1) and congenital adrenal hyperplasia. Recently, Raymond et al. (121) demonstrated that the prevalence of Lynch syndrome in patients with adrenocortical carcinoma was similar to that of colorectal and endometrial carcinoma in that population and therefore concluded that adrenocortical carcinoma is a Lynch syndrome–associated malignancy.
There is considerable variability in the reported functional status of adrenocortical carcinomas. Approximately 80% of tumors are functional, most commonly secreting glucocorticoids (45%), glucocorticoids and androgenic steroids (45%), and androgenic steroids alone (10%). Rarely, mineralocorticoid production may be present (118). However, occasional single institution studies have reported a much higher proportion (up to 75%) of carcinomas being unassociated with syndromes of hormone overproduction (145). Some patients may present with abdominal pain, and up to 30% may have a palpable abdominal mass. In some instances, patients may show signs of hypoglycemia as a result of the production of insulin-like growth factors by the tumor or hypercalcemia as a result of the production of parathyroid hormone–related peptide.
Gross Morphology
As a rule, cortical carcinomas of conventional type are large tumors weighing more than 100 g in adults; most often, tumor weight is in excess of 750 g (16,146–149). Rarely, however, tumors weighing less than 50 g will metastasize, whereas a small proportion of tumors weighing more than 1000 g may not (27). Benign tumors associated with sex steroid overproduction, however, can weigh considerably more than 100 g. Tumor weight is also a useful predictor of malignancy in children. Tumors weighing more than 500 g in a series of 23 cases reported by Cagle et al. (150) were malignant, whereas only a single tumor weighing less than 500 g pursued a malignant course.
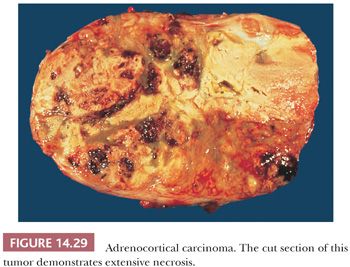
On gross examination, most cortical malignancies have a nodular appearance with individual nodules varying from pink to yellow-tan, depending on their lipid content. Carcinomas associated with feminization or virilization tend to be red-brown, whereas those associated with Cushing syndrome are more often yellow-tan. Foci of necrosis, hemorrhage, and calcification are common, particularly in large tumors (Fig. 14.29). The larger tumors often invade contiguous structures, including the kidney and liver.
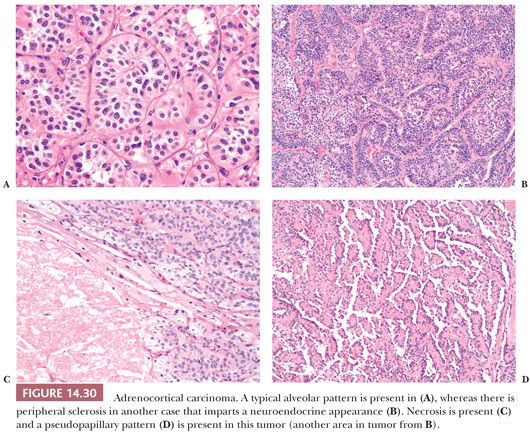
Microscopic Morphology
Adrenocortical carcinomas have alveolar (Fig. 14.30A,B), trabecular, or solid patterns of growth, and admixtures of these patterns are common (27,28,45). Necrosis, particularly in large tumors, may be extensive (Fig. 14.30C). Foci of myxoid change (127,128), pseudoglandular patterns, pseudopapillary patterns (Fig. 14.30D), and spindle cell growth may be prominent in some tumors (27). Papotti et al. (151) have reported that the majority of myxoid adrenocortical tumors, even when conventional criteria are not met, have a malignant biologic behavior (see later). Depending on their lipid content, the cytoplasm may vary from vacuolated to eosinophilic. Some tumors may have eosinophilic globular inclusions resembling those seen in pheochromocytomas. Rare cases may exhibit adenosquamous differentiation (152). There may be considerable variation in the appearance of the nuclei. In some instances, they may appear relatively small and uniform (Fig. 14.31A), whereas in others, they may exhibit pronounced atypia manifested by pleomorphism (Fig. 14.31B), coarse chromatin, and multiple prominent nucleoli. Mitotic activity, including atypical forms (Fig. 14.31C), is often prominent. Nuclear pseudoinclusions, representing invaginations of the cytoplasm into the nucleus, may be particularly striking in some cortical carcinomas.
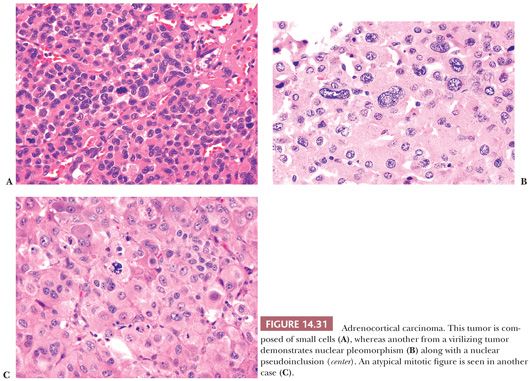
A subset of adrenocortical carcinomas may be composed of oncocytic cells (136,140–142,153–155). Although some of these tumors may be associated with Cushing syndrome or feminizing features (136), others may be nonfunctional. Criteria for malignancy in these tumors differ from those of conventional adrenocortical carcinomas and are described later. More recently, examples of rhabdoid adrenocortical carcinomas composed of cells with eosinophilic cytoplasmic inclusions and eccentric nuclei with prominent nucleoli have been reported (156). The latter tumors are positive with markers for primary adrenocortical tumors but stain weakly with antimitochondrial antibodies unlike oncocytic tumors (156).
Very rarely, adrenocortical carcinomas may contain sarcomatous foci (carcinosarcoma). In the case reported by Fischler et al. (157), the sarcomatous component had features of rhabdomyosarcoma and stained positively for muscle-specific actin and desmin. Although this tumor was associated with virilization, the case reported by Decorato et al. (158) was nonfunctional. Thway et al. (159) reported an example of oncocytic adrenocortical carcinosarcoma in a 45-year-old man with pleomorphic rhabdomyosarcoma that metastasized to the mesentery, hilar lymph nodes, lungs, and brain over the 11-month terminal course from diagnosis. Furthermore, Kao et al. (160) recently reported an example of adrenocortical carcinosarcoma with oncocytic and primitive neuroectodermal-like features.
Cytopathologic Features
In fine-needle aspiration biopsy samples, cortical carcinomas generally contain single cells and poorly cohesive cell clusters in a necrotic background. There may be considerable nuclear atypia and mitotic activity, but some cortical carcinomas appear deceptively bland (161). According to Ren et al. (162), common cytologic features include hypercellularity, necrosis, nuclear pleomorphism, mitotic figures, and prominent nucleoli. Twenty percent of their cases exhibited all five features, whereas necrosis and/or mitoses were found in all cases.
Immunohistochemistry and Electron Microscopy
There are considerable differences in the reported incidences of keratin positivity in cortical carcinomas, depending on the specificities of the antibodies and the types of tissue preparation (Fig. 14.32). With microwave retrieval methods, cytokeratin immunoreactivity is present focally in up to 60% of adrenal cortical neoplasms (163,164). Interestingly, all four of the oncocytic carcinomas reported by Hoang et al. (154) were positive for cytokeratins using AE1/AE3 and CAM 5.2 antibodies. Vimentin is evident in most cortical carcinomas following microwave-induced antigen retrieval. Some cortical carcinomas may, therefore, exhibit a vimentin-positive, cytokeratin-negative phenotype, whereas others may coexpress cytokeratins and vimentin (165).
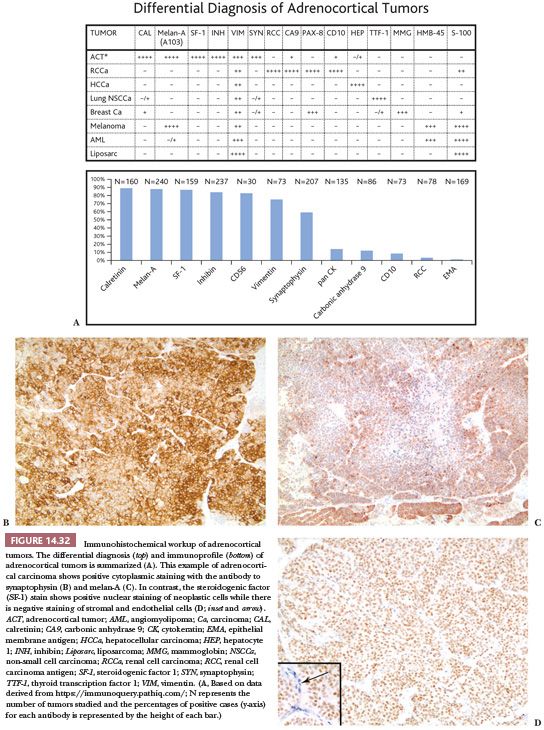
Several other approaches have been used for the identification of adrenocortical neoplasms, including antibodies reactive with steroidogenic enzymes and transcription factors that regulate the expression of these genes (166,167). Schröder et al. (168) reported the use of a monoclonal antibody (D11) that is reactive with normal and neoplastic cortical cells but nonreactive with normal adrenal medullary cells and pheochromocytomas. However, it reacted with carcinomas from other sites (169) and is no longer commercially available (119).
The monoclonal antibody A103, which reacts with melan-A, an antigen recognized by cytotoxic T cells and expressed in melanocytes, has also been used for the identification of adrenocortical (Fig. 14.32) and other steroid hormone–producing tumors (170). With the exception of melanoma, the only tumors that are consistently reactive with A103 are adrenocortical adenomas and carcinomas, testicular Leydig cell tumors, and ovarian Sertoli-Leydig cell tumors. Antibodies to inhibin A also provide an additional useful approach for the identification of steroid-producing cells (Fig. 14.32). Renshaw and Granter (171) have demonstrated that inhibin A and A103 are both useful for the identification of adrenocortical neoplasms and that A103 is marginally more specific and inhibin A is slightly more sensitive. Calretinin is also expressed in adrenal cortical neoplasms and is a useful adjunct in cases where stains for inhibin A are negative. Jorda et al. (172) demonstrated that 24 of 33 (73%) cortical neoplasms were positive for inhibin A; however, when calretinin was added, the numbers of tumors staining positively for the two markers increased to 94% (31 of 33 cases).
Some adrenocortical carcinomas exhibit evidence of neuroendocrine differentiation, manifested by the presence of immunoreactivity for synaptophysin (Fig. 14.32), neurofilament proteins, and neuron-specific enolase (173,174). The tumors are, however, typically negative for chromogranin/secretogranin proteins. Ultrastructural analysis of some cases showed clusters of membrane-bound secretory granules, which measured 150 to 300 nm in diameter and resembled neurosecretory granules. The tumor cells also contained synaptophysin messenger RNA (174). These observations have suggested that adrenocortical carcinomas with evidence of neuroendocrine differentiation could arise from stem cells, which are capable of multidirectional differentiation. In this regard, the studies of Bornstein et al. (42) have shown that hybrid corticomedullary cells exist normally in the rat adrenal cortex.
Browning et al. (175) reported that D2-40, a marker that is commonly used to highlight lymphatic endothelial cells, was highly specific and sensitive for differentiating adrenocortical tumors from both metastatic renal cell carcinomas and pheochromocytomas. In this series, D2-40 was strongly and diffusely positive in the cells of the neoplastic and nonneoplastic adrenal cortex but was negative in 13 cases of clear cell renal carcinomas. Normal and neoplastic medullary cells were negative for D2-40. Other markers of use for distinguishing adrenocortical carcinomas from other tumors are SF-1 and steroid receptor coactivator (119,176–178). As described earlier, SF-1 plays a key role in steroidogenic development and regulation of steroid biosynthesis. It is a nuclear stain (Fig. 14.32) that has been shown to be a highly sensitive and specific adrenocortical marker and is now considered by some as the best adrenocortical marker currently available (119).
Ultrastructurally, the mitochondria of cortical carcinomas may be round, ovoid, or elongated. Silva et al. (179) found isolated mitochondria with tubular cristae in virtually every case, but tumors containing a predominance of tubular mitochondria were found in only approximately half of the cases. Smooth endoplasmic reticulum is variable and glycogen deposits may be present in a smaller proportion of cases. El-Naggar et al. (180) have reported the presence of both dense matrix inclusions and crystalline inclusions within the mitochondria of a case of oncocytic adrenocortical carcinoma.
Molecular Pathology
Comparative genomic hybridization studies have revealed that genetic alterations are more common in malignant than in benign cortical tumors, whereas they occur rarely in hyperplastic lesions. Losses of 1p21-31, 2q, 3p, 3q, 6q, 9p, and 11q14-qter, and gains of 5q12, 9q32-qter, 12q, and 20q were the most frequent abnormalities present in carcinomas. Gains in 17q, 17p, and 9q32-qter were the most frequent abnormalities in adenomas, whereas gains in two of six cases of cortical hyperplasia involved 17 or 17q (181). Several groups have demonstrated LOH of 11q13 in a significant proportion of adrenal cortical carcinomas; however, none of the tumors demonstrated a mutation in the MEN1 gene (182,183). LOH of 17p13 and 11p15 has been demonstrated in approximately 80% of cortical carcinomas. Gicquel et al. (184) have further demonstrated that 17p13 LOH and histologic grade were independently associated with tumor recurrence.
Results of gene expression profiling have demonstrated that the most significantly upregulated genes in carcinomas include ubiquitin-specific protease 4 (USP4) and ubiquitin degradation 1-like (UFD1L), which are upregulated at least 40-fold compared with adenomas. Additional genes upregulated in carcinomas include members of the insulin-like growth factor (IGF) family such as IGF2, IGF2R, IGFBP3, and IGFBP6 (185). Giordano et al. (186) also demonstrated increased expression of IGF2 in adrenocortical carcinomas. Downregulated genes in carcinomas include chemokine (C-X-C motif) ligand 10 (CXCL10), retinoic acid receptor responder 2 (RARRES2), aldehyde dehydrogenase family member A1 (ALD1f1A1), cytochrome b reductase 1 (CYBRD1), and glutathione S-transferase A4 (GSTA4) (185).
West et al. (187) have demonstrated similar patterns of gene expression in pediatric adrenal cortical tumors. Interestingly, there was a consistent marked decrease in the expression of all major histocompatibility complex (MHC) class II genes in carcinomas compared with adenomas. These results parallel the observations by Marx et al. (188) that pre- and postnatal adrenals do not express MHC class II antigens in contrast to adult adrenals, which do express these antigens.
The use of microRNA expression in adrenocortical neoplasms has been reported to have prognostic a role by multiple investigators (119). Most recently, Wang et al. (189) reported that miR483-3p, which has been shown to be overexpressed in other carcinomas, was overexpressed in 68% of adrenocortical carcinomas (17 of 25) compared to 12% of adrenocortical adenomas (3 of 25) using in situ hybridization. They also compared miR483-3p expression to the immunohistochemical expression of IGF2 and Smad4. IGF2 expression correlated with that of miR483-3p, whereas Smad4 had an inverse relationship (decreased expression). The authors concluded that the combination of miR483-3p and Smad4 may be used in challenging cases to make the distinction between carcinoma and adenoma. Furthermore, they reported that there was a trend for elevated miR483-3p expression and negative/low Smad4 carcinomas to have a worse prognosis, but their results did not reach statistical significance in their study (189).
Altered DNA methylation in H19, IGF2, and RAS association domain family protein A (RASSF1A) genes has been reported in carcinomas. CpG island methylation status has also been reported to stratify carcinomas into prognostic subsets (119). Most recently, Assie et al. demonstrated recurrent alterations in p53, MEN1, and other previously reported and unreported genes by integrated genomic characterization of adrenocortical carcinomas (189a). Using this method they found that the most frequently (21% of cases) altered gene was the zinc finger and ring finger protein 3 (ZNRF3). ZNRF3 is a potential new tumor suppressor gene related to the beta-catenin pathway. It is still left to be seen how these and the previously described molecular data will be integrated into clinical practice.
Criteria for Malignancy in Adrenocortical Tumors
Conventional Adrenocortical Neoplasms. It may be extremely difficult to distinguish between benign and malignant cortical tumors, and different authors have used a variety of parameters to differentiate these groups of neoplasms (146,148,190–196). Hough et al. (148) examined a variety of histologic and clinical features to distinguish metastatic and nonmetastatic cortical tumors. These investigators found that the presence of necrosis (>2 hpfs in diameter) and broad fibrous bands were the most useful histologic discriminants. In addition, a diffuse pattern of growth, nuclear hyperchromasia, and vascular invasion were also helpful in making the distinction between benign and malignant cortical neoplasms.
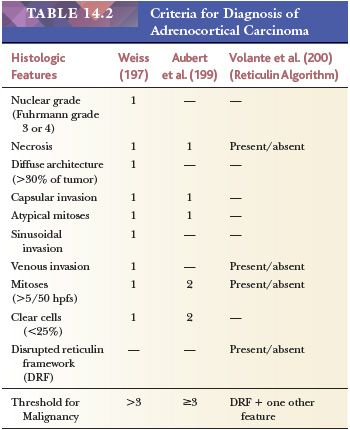
Weiss (146) proposed a system for the distinction of benign and malignant tumors based on the following nine parameters (Table 14.2): high nuclear grade (Fuhrman grade 3 or 4); greater than five mitoses per 50 hpfs; atypical mitoses; diffuse patterns of growth; necrosis; invasion of venous, sinusoidal, or capsular structures; and clear cells comprising less than 25% of the tumor (146). In a series of 43 cases, Weiss (146) found that tumors with fewer than two of these features never metastasized, whereas those with more than four almost invariably recurred or metastasized. Subsequently, Weiss et al. (197) lowered the threshold for malignancy from four to three parameters. Papotti et al. (119) and Duregon et al. (198) recently reported that the mitosis-specific antibody phosphohistone H3 was very useful in the screening for and counting of mitoses. Aubert et al. (199) confirmed the value of the Weiss system but have proposed a modification based on the most reproducible criteria, including mitotic rate (>5/50 hpfs), cytoplasm (<25% clear cells), abnormal mitoses, necrosis, and capsular invasion. The first two features were assigned values of 2, whereas each of the remainder was assigned values of 1. According to this system, each tumor could be given a score of 0 to 7. The threshold for malignancy remained a score of 3 or more.
From a clinicopathologic review of 92 adrenocortical carcinomas and 47 benign cortical tumors, Volante et al. (200) proposed an algorithm in which disruption of the reticulin network when combined with any one of the following three of Weiss criteria—(a) mitotic rate greater than five per 50 hpfs, (b) necrosis, and (c) vascular space invasion—resulted in successful classification of adrenocortical neoplasms as adrenocortical carcinomas with 100% sensitivity and specificity. Disruption of the reticulin network was assessed by silver histochemical staining, and basal lamina was assessed using immunohistochemical antibodies to laminin and collagen type IV. Disruption of the reticulin/basal lamina network was defined by loss of continuity of the reticular fibers (Fig. 14.33) or basal membrane network in 1 hpf (400× = 0.2 mm2) extended to at least one-third of the lesion.
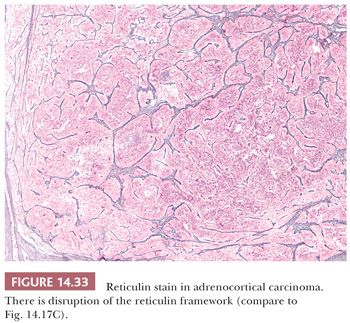
This group recently performed a multicenter validation study on their proposed reticulin algorithm on 245 adrenocortical tumors (61 adenomas and 184 carcinomas). After appropriate training, complete interobserver agreement (8/8 pathologists of variable experience) was reported in over 85% of cases (201). Of note, the authors adjusted their criteria for reticulin disruption to include qualitative disruption of the reticulin framework in the form of irregular thickening and fraying of reticulin fibers. Therefore, a subset of cases did not fulfill the quantitative requirements described earlier but demonstrated the latter qualitative requirement.
Other methods that have been examined in an effort to distinguish benign from malignant adrenocortical neoplasms include DNA content, MIB-1 labeling index, and p53 status (192–194). Initially, carcinomas were shown to be aneuploid and adenomas were shown to be diploid, but subsequent studies demonstrated aneuploidy in some adenomas and diploidy in some carcinomas (195,196). Vargas et al. (202) demonstrated that the mean proliferative fraction, as determined by counting the proportion of MIB-1–positive cells, was 1.49% in adenomas, 20.8% in carcinomas, and 16.6% in recurrent or metastatic tumors. None of 20 benign lesions had an MIB-1 score that exceeded 8%, whereas only 1 of 20 malignancies had a score of less than 8%. Forty-five percent of the carcinomas were positive for p53, whereas none of 20 adenomas was p53 positive. Aubert et al. (199) also observed a statistically significant difference in MIB-1 labeling between benign (2.4% ± 1.3%) and malignant (21.2% ± 18.44%) tumors. In vitro analyses have suggested that any tumor with functional abnormalities, such as the secretion of precursor steroids and blunted or absent response to ACTH, should be considered potentially malignant (203). As described earlier in the section on molecular pathology, various molecular modalities have been reported that demonstrate differences between carcinomas and adenomas (119,181–189).
Oncocytic Adrenocortical Neoplasms. Bisceglia et al. (136) have also reviewed the criteria for the distinction of benign and malignant adrenocortical oncocytic tumors. According to these authors, major criteria for malignancy included high mitotic rate, atypical mitoses, and venous invasion, whereas minor criteria included large tumor size, necrosis, capsular invasion, and sinusoidal invasion. The presence of one major criterion was sufficient for the diagnosis of malignancy, whereas one to four minor criteria were sufficient for a diagnosis of tumors of uncertain malignant potential. The absence of all criteria indicated benignancy.
Duregon et al. (142) also applied the original reticulin algorithm to their series of oncocytic adrenocortical tumors and found that there tended to be a more heterogenous pattern of reticulin framework and that disruption was more likely to occur in nononcocytic areas, thus posing additional challenges. However, they concluded that its applicability was justified because it avoided underestimation of malignancy in this group of tumors. These authors noted that the oncocytic adrenocortical carcinomas usually behaved as low-grade malignant neoplasms (142).
Myxoid Adrenocortical Neoplasms. In the study of myxoid adrenocortical tumors by Papotti et al. (151), 8 of 10 cases with myxoid areas that ranged from 5% to 90% fulfilled Weiss criteria for malignancy (score >3), but two cases with 90% myxoid areas had a Weiss score of 1. In one of the latter tumors that measured 5 cm and weighed 55 g, there was local and peritoneal recurrence at 60 months after resection and the patient eventually died of disease at 68 months. In their study, they also cited four other cases with conventional features of adrenocortical carcinoma with focal myxoid degenerative changes. The authors concluded that myxoid adrenocortical tumors represent a rare but histologically and phenotypically distinct entity exhibiting malignant behavior (151). Weissferdt et al. (204) concluded that myxoid adrenocortical carcinomas have a more aggressive behavior than conventional adrenocortical carcinomas based on a clinicopathologic and immunohistochemical study of seven cases.
Pediatric Adrenocortical Neoplasms. The issue of malignancy is also complicated in the pediatric population in which the criteria of Weiss are less predictive of an aggressive course. As noted earlier, Cagle et al. (150) found tumor weight greater than 500 g to be the most useful determinant in predicting malignant behavior. Subsequently, Wieneke et al. (205) studied 83 cortical neoplasms in patients less than 20 years of age and found that 23 of 74 cases with a malignant histology based on adaptation of Weiss criteria had a clinically malignant course. From their analysis, features associated with an increased probability of malignant behavior included tumor weight greater than 400 g, size greater than 10.5 cm, vena cava invasion, capsular and/or vascular invasion, extension into periadrenal soft tissue, confluent necrosis, severe nuclear atypia, greater than 15 mitoses per 20 hpfs, and the presence of atypical mitotic figures. Of these, vena cava invasion, necrosis, and mitotic activity independently predicted malignant behavior in multivariate analyses. Because of the added uncertainty in pediatric tumors, Dehner (206) has suggested labeling tumors fulfilling Weiss criteria as “atypical adenomas.” More recently, Dehner and Hill (207) reviewed 39 adrenocortical neoplasms and proposed risk stratification of pediatric adrenocortical neoplasms based on their findings and review of the literature. They suggested that any cortical neoplasm confined to the adrenal gland and weighing less than 200 g should be assigned to the low-risk category. Those greater than 400 g with evidence of direct gross invasion into adjacent organs including the liver, spleen, or kidney, or with evidence of metastatic spread, to the high-risk category. They recommended that tumors 200 to 400 g confined to the adrenal gland or any tumor less than 400 g with microscopic invasion into the surrounding soft tissue but completely resected and with no evidence of metastatic spread should be placed in an intermediate-risk category (5,207).
Differential Diagnosis
Adrenocortical carcinomas must be distinguished from a variety of secondary tumors involving the adrenal gland, including renal cell carcinoma, hepatocellular carcinoma, metastatic carcinoma, and liposarcoma. Immunohistochemistry may be of particular value in discriminating cortical carcinomas from these tumor types (22,165,170–179,208–210), as summarized in (Fig. 14.32).
Grading
Grading of adrenocortical carcinomas is difficult, and there are no widely accepted criteria for the grading of cortical carcinomas. In an analysis of 42 carcinomas, the only parameter that had a strong statistical association with patient outcome was mitotic rate (197). Patients with tumors containing more than 20 mitoses per 50 hpfs had a mean survival of 14 months, whereas patients with tumors with less than 20 mitoses had a mean survival of 58 months. Atypical mitoses, capsular invasion, tumor weight in excess of 250 g, and size in excess of 10 cm each had a marginal association with survival. Other features, including nuclear grade, presence of necrosis, venous or sinusoidal invasion, character of the tumor cell cytoplasm, and architectural pattern, had no impact on survival.
Volante et al. (200) stratified adrenocortical carcinomas in their proposed algorithm into low-, intermediate-, and high-risk groups. Low risk: stage I or II and mitoses less than or equal to nine per 50 hpfs; intermediate risk: stage I or II and greater than nine mitoses per 50 hpfs or stage III or IV and less than or equal to nine mitoses per 50 hpfs; and high risk: stage III or IV and greater than nine mitoses per 50 hpfs. Mean disease-free survivals were reported to be 61.9, 17.3, and 12.2 months in the low-, intermediate-, and high-risk groups, respectively.
Natural History
Didolkar et al. (145) have studied the natural history of a large series of patients with adrenocortical carcinoma. The mean duration of symptoms in patients with or without hormonal manifestations was 6 months. Fifty-two percent of patients had distant metastases at the time of diagnosis, 41% had locally advanced disease, and 7% had tumor confined to the adrenal gland. The overall median survival was 14 months, and the 5-year survival rate was 24%. The median survival of patients with functional tumors was somewhat longer than that of patients with nonfunctional tumors. The most common sites of metastasis were the lungs, followed by retroperitoneal lymph nodes, liver, and bone. The staging of adrenocortical carcinoma is summarized in Table 14.3.
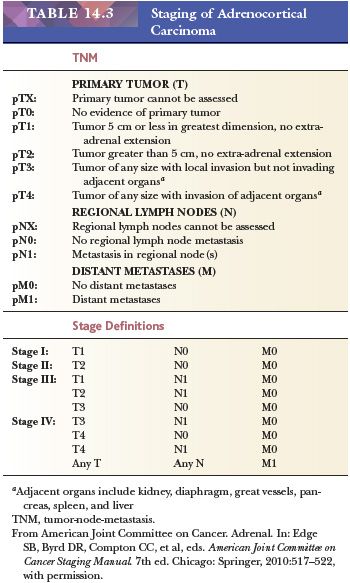
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


