14 Maya Davidovich-Pinhas and Havazelet Bianco-Peled Department of Chemical Engineering Technion – Israel Institute of Technology Israel The administration of medicines is a simple yet important clinical procedure. Different drug administration methods vary in their onset, intensity, ease of use and duration of pharmacological action. The use of direct injection of drugs into the blood is straightforward. However, it is clearly not convenient for the patients. Administration through the oral route is the most common approach being used, primarily due to its simplicity. However, it suffers from some limitations, such as risk of drug hydrolysis in the gastric tract and lack of solubility of some drugs leading to low bioavailability [1]. The limitations of these common administration methods have motivated studies seeking for an alternative drug delivery approach combining comfort of use with enhanced efficiency. One of the approaches that has been extensively studied in the last decade explores mucoadhesive polymers as a potential carrier for transmucosal drug release. Combining mucoadhesion ability with other advantages of polymeric drug vehicles, such as controlled drug release rate, protection of the drug from hydrolysis or other types of chemical degradation, protection from enzymatic degradation, reduction of drug toxicity and improvement of drug solubility and availability [2], allows the design of powerful drug delivery systems. The main focus of this chapter is a new family of mucoadhesive materials, termed acrylated polymers. These polymers are capable of forming covalent bonds with mucosal surfaces. Hence, they are characterized by remarkable adhesive capabilities. A thorough description of acrylated polymers is presented in this chapter following two short introductory sections, the first of which deals with mucoadhesion phenomenon and the second with various types of interactions involved in the mucoadhesion process. Mucoadhesion, defined as the ability of polymer dosage form to adhere to mucosa covered surfaces, was first introduced in the early 1980s as a new approach to improve drug release, targeting and absorption [3–5]. The mucosa gel layer is a secretion formed in specialized epithelial cells. It has a variety of roles depending on its physiology location. The epithelial tissue covers all organs that are exposed to the outer environment and yet not covered with skin. It is characterized by a high density of blood vessels and continuous blood flow, which makes it a powerful, easy and convenient target for noninvasive drug delivery. The epithelial tissue can be classified by the number of cell layers, single or multilayer surface and by the cell shape (squamous, cuboidal and columnar cells). Some epithelial tissues are characterized by membrane-bound vesicles called secretory granules embedded in between the epithelia cells. These granules store the secretion content in dehydrated form and are responsible for its release, either gradually or in response to specific stimulation. The secreted mucus gel layer has multiple functions, such as absorption, lubrication, entrapment and antibacterial activity [1]. Mucus is composed primarily of water (∼95%) but also contains small amounts of salts, lipids and proteins. The main components responsible for the elastic gel-like structure of the mucus are glycoproteins termed mucins. Mucins are high molecular weight extracellular glycoproteins that share many common features. Their structure is based on a polypeptide backbone with oligosaccharide side chains, which form an extended ‘bottle brush’ conformation, and cysteine-rich regions at both ends of the protein backbone. The cysteine-rich regions at the ends of the molecules are involved in the disulfide bond formation attributed to the gel-like structure. Due to the glycoprotein structure and characteristics they can form electrostatic, hydrophobic, disulfide and hydrogen bonding interactions with other substances, a process which potentially leads to mucoadhesion [6]. Many studies have illustrated the involvement of polymer chain penetration, entanglement and molecular interaction (covalent or/and noncovalent) in the mucoadhesion process. Therefore, mucoadhesion can be described using the diffusion and chemical bonding theories of adhesion. The diffusion theory of adhesion is based on the assumption that the adhesion strength of polymers to themselves (auto-adhesion) or to each other is due to mutual diffusion (interdiffusion) of macromolecules across the interphase. The chemical bonding theory of adhesion invokes the formation of interaction such as covalent, ionic or hydrogen bonds across the adhesive surface interphase [7–9]. The most common path of bioadhesion between polymers and mucosal surface uses noncovalent bonds such as hydrogen bonds, van der Waals forces, ionic interactions and/or chain entanglements [10]. Due to the negative surface charge of the mucus arising from the presence of oligosaccharide’s sialic acid terminal end groups, electrostatic interactions play an important role in the adhesion process [11,12]. Therefore, noncovalently binding mucoadhesive polymers are commonly classified according to their molecular charge into cationic, anionic, nonionic and ambiphilic polymers [3]. The adhesion of cationic polymers such as chitosan and polylysine is straightforward, due to ionic attraction between the positively charged amino groups carried by the polymer and the sialic acid end groups on the oligosaccharide side chains of the mucin. Adhesion of anionic polymers that carry –COOH groups, on the other hand, often arises from hydrogen bonds with the hydroxyl groups of the glycoprotein. This group includes, for example, polyacrylates, alginate and hyaluronic acid. The interaction of nonionic polymers is based on interpenetration of the polymer chains, also termed mucopenetration, followed by chain entanglement. Therefore, their ability to adhere is not influenced by the surrounding pH. Several studies have shown that poly(ethylene glycol) tends to penetrate and create entanglements with the mucosa surface in addition to its hydrophilic interaction ability [13–17]. Recent studies showed that nonionic polymers are, in most cases, less adhesive than anionic or cationic mucoadhesives. In this group can be found hydroxypropyl cellulose and poly(vinyl alcohol). Zwiterionic polymers benefit from both the cationic and anionic interactions; for example, ionic interaction with sialic end groups and hydrogen bonds with hydroxyl groups on the glycoprotein’s oligosaccharide side chain. Chitosan-EDTA and gelatine are good examples of such polymers [3]. An additional path for mucoadhesion involves specific noncovalent interactions often observed in cell recognition and adhesion which lead to the formation of strong interaction. These systems take advantage from known biological molecules, such as lectins and/or other adhesion molecules, to bind directly to receptors on the cell surface rather than to the mucus gel layer. Since specific binding to the cell surface is often followed by uptake and intracellular transport, new chances for drug delivery have evolved [18]. Other mucoadhesive systems found in the literature include dendrimers, boronic acid copolymers and synthetic glycopolymers [19]. Recently, attempts have been made to improve the mucoadhesive properties by modifications that enable formation of covalent bonds between the polymer and the mucosa surface through interactions with mucin-type glycoproteins. These modifications include polymers capable of forming disulfide bonds, termed thiomers [20–24], or acrylate–sulfide linking [25,26]. The adhesion of acrylated mucoadhesive polymers relies mostly on their ability to covalently associate with mucin-type glycoproteins through sulfide–acrylate interactions. Such interaction, also termed the Michael-type addition reaction, occurs between an electronegative vinyl end group, such as acrylate, and an electronegative neighbouring group, such as sulfide or amine, in physiological environment. The sulfide–acrylate interaction was previously used by Hubbell and co-workers [27,28] for conjugating sulfhydryl-containing biomolecules such as peptides or proteins to vinyl-carrying polymers. Their methodology was further developed for the development of many hydrogel systems based on poly(vinyl alcohol) [29], poly(ethylene glycol)-b-poly(lactic acid) [30], PEGylated fibrinogen [31] and other PEGylated proteins [31,32]. A proof of concept for the existence of a sulfide–glycoprotein interaction was demonstrated using a simple acrylated polymer, poly(ethylene glycol) diacrylate (PEG-DA), and a mixture of mucin-type glycoproteins extracted from fresh porcine intestine used as a model for mucosal surface content. PEG-DA is a linear poly(ethylene glycol) chain with varied molecular weight; it includes two acrylated terminal groups at its both ends (Figure 14.1). It can be synthesized from linear hydrophilic polyethylene glycol (PEG) [27,31]. Figure 14.1 The molecular structure of poly(ethylene glycol) diacrylate (PEG-DA). The ability of the acrylate end group to associate with mucin glycoproteins was monitored using proton nuclear magnetic resonance (1 H NMR) spectroscopy. The Michael-type addition reaction involves the coupling of an electronegative vinyl end group (e.g. acrylated end group) with other electronegative end group, such as thiols and amines. During this process the reactive electronegative double bond is opened to form a new covalent bond between the two components. Comparing the The ability of two polymers to interact was also monitored using rheology. As with the NMR measurements, comparing the solution viscosity of PEG-DA, mucin and their mixture provided a means to follow the interactions between the components. The underlying assumption was that viscosity increase is associated with molecular interactions due to the increase in the total molecular weight of the network [33]. Indeed, a viscosity increase upon mucin addition to PEG-DA solution was demonstrated (Figure 14.2). This result supported the suggestion that PEG-DA interacts with the mucin glycoproteins, and was in line with previous works in the field of mucoadhesive polymers that attributed viscosity enhancement after mucin addition to molecular interaction between the polymer and glycoproteins [20,34]. In order to overrule the possibility that the viscosity increase resulted from the addition of relatively high molecular weight glycoprotein to polymer solution, which might induce the formation of additional entanglements as a result of concentration increase, the experiment was repeated using PEG-OH with the same molecular weight. No pronounced changes in viscosity were observed upon mucin addition to PEG-OH solution. Thus, the viscosity increase can be attributed to Michael-type addition reaction between the PEG-DAs acrylate end groups and glycoprotein backbone, since this is the only possible interaction that cannot occur when PEG-OH chains are mixed with the mucin. Figure 14.2 Rate sweep experiment of PEG-OH 10 kDa 20 mg/ml ( The ability of acrylated functional groups to associate with glycoproteins present on fresh mucosal surface was further demonstrated by tensile measurements. The experiments were performed by cross-linking PEG-DA molecules with UV radiation on a fresh small intestine surface and then separating the two surfaces. The results were expressed as the maximum detachment force (MDF) needed to separate the surfaces [25]. The adhesion performance was compared to those demonstrated by a known covalently associated mucoadhesive polymer, alginate–thiol (Figure 14.3). A significant difference was observed between the results obtained from the 2% and 3% PEG-DA, whereas the difference between the 4% PEG-DA and the alginate–thiol was not significant. In addition, increase in the adhesion ability with increase in the PEG-DA concentration was demonstrated. The results could be an outcome of total increase in both covalent (acrylated) and noncovalent (penetration, hydrogen bonds, van der Waals etc.) interaction ability. Figure 14.3 Maximum detachment force (MDF) for various polymer samples from fresh mucus surface at 25 °C. Thiolated polymers have demonstrated similar adhesion ability to PEG-DA. For example, Bernkop-Schnürch et al. [35] characterized the adhesion of alginate and alginate–thiol to a commercial-grade crude porcine mucin. Maximum detachment forces of approximately 0.01 and 0.07 N were observed for alginate and alginate–thiol, respectively. In another study by the same group [36] maximum detachment forces of 0.027, 0.256 and 0.056 N were measured for low, medium and high molecular weight 2-iminothiolane conjugated chitosan (chitosan-TBA). It should be noted, however, that the exact set-up used for the adhesion measurements has a vast influence on the measured force, as described in detail in a previously published review [37]. In particular, the detachment experiments involving PEG-DA were performed using hydrated samples that were cross-linked on the mucus surface, whereas the above mentioned previous studies have used dry, compressed sample that did not contain any cross-linker. It is well known that during the swelling process, a dry sample’s polymer chains tend to penetrate the surface due to their swelling [38,39]. This process probably leads to an increase in the adhesion ability according to the diffusion theory of adhesion. Thus, the suggested acrylated mucoadhesive polymer displayed similar adhesion ability in hydrated environment in spite of the lack of swelling ability. Alginate is an anionic mucoadhesive polymer that is known for its ability to form hydrogen bonds with mucin-type glycoproteins through carboxyl–hydroxyl interactions [3]. This anionic biopolymer is used in many pharmaceutical and biotechnological applications [40]. Alginate is a linear, water-soluble polysaccharide of Enhancing the mucoadhesive properties of alginate was attempted by applying the acrylation approach. The resulting mucoadhesive polymer, termed alginate–poly(ethylene glycol) acrylate (alginate-PEGAc), was synthesized by the conjugation of PEG-DA molecules to alginate backbone. This polymer combines the strength, simplicity and gelation ability of alginate with the mucoadhesive properties arising from the PEGs characteristics and the acrylate functionality. It has the potential to be used in many biotechnology applications due to its unique characteristics. In particular, the ability to induce both physical cross-linking of the alginate backbone using divalent ions and/or chemical cross-linking of the PEGs acrylate end group using UV radiation, offers a new approach to control the polymer gel properties. The synthesis of alginate-PEGAc was designed as a two-step procedure where the synthesis of alginate–thiol is performed first, followed by the conjugation of PEG-DA to the alginate backbone [26] (Figure 14.4). In brief, the synthesis of alginate–thiol [47,52] was achieved by activating the alginate carboxylic groups by the carbodiimide functional group within the 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide hydrochloride (EDAC) intermediate reagent. Next, EDAC was replaced with L-cystein through its amine end group to form an amide bond. The thiol concentration in the thiolated product was measured using Ellman’s assay. The resulting thiolated alginate was dissolved in tris(2-carboxyethyl)phosphine hydrochloride (TCEP) solution in order to prevent intramolecular disulfide formation [52]. Finally, a Michael-type addition, involving a nucleophilic reaction of the thiols on the thiolated alginate with the vinyl group on the acrylate functionalized poly(ethylene glycol), was performed. In order to lower the probability of multiple attachments of a single PEG-DA molecule to the backbone, a large molar excess of PEG-DA was used. The resultant product includes PEG chains, still carrying one acrylate end group, linked to the alginate backbone through the cystein spacer molecule. The molecular structure of alginate, alginate–thiol and alginate-PEGAc were verified using Figure 14.4 Schematic illustration of the alginate-PEGAc synthesis where (a) native alginate, (b) thiolated alginate and (c) alginate-PEGAc. Alginate PEGylation was also performed by Laurienzo et al. [48], who conjugated PEG molecules to alginate through its hydroxyl end groups in order to maintain the gelation ability of alginate, which consumes the carboxylic end groups. The synthetic approach described above reduces the number of carboxylic groups on the alginate backbone. Yet, the product retained its gelation ability and gel microparticles could be formed by adding 1% PEGAc solution into 1% CaCl2 aqueous solution drop-wise [26]. The adhesion ability of alginate-PEGAc to mucosal surface was analysed by measuring the MDF needed to detach compressed polymer tablets from fresh small intestinal surface and compared to native alginate and thiolated alginate, a known covalently binding mucoadhesive polymer [26]. The MDF of two different alginate-PEGAc samples was significantly higher compared to both native alginate and thiolated alginate (Figure 14.5). However, no significant difference in adhesion properties was observed between the two native alginate samples. The adhesion of the thiolated alginate was higher than that of the native alginate in the case of alginate HF120 but not in the case alginate LF200 S. Moreover, the MDF values obtained from thiolated alginate HF120 were significantly higher compared to thiolated alginate LF200 S. This result was attributed to the higher thiol content of thiolated alginate HF120 (136.1 ± 6.6 μmol thiol/g polymer) compared to that of thiolated alginate LF200 S (56.7 ± 12 μmol thiol/g polymer), which is expected to increase the probability for disulfide interactions with mucin glycoproteins. A similar behaviour was also detected when comparing the two acrylated alginates. Alginate-PEGAc synthesized from HF120 demonstrated a significantly higher MDF compared to alginate-PEGAc prepared by modifying LF200 S. It can be assumed that the thiol content is strongly correlated with the PEGylation degree, since a larger thiol content increases the ability of PEG-DA to attach to the alginate, thus leading to a larger PEG content. An increase in PEG and acrylate content could be expected to increase the probability of chain entanglements, polymer noncovalent bonds such as hydrogen bonds and covalent bonds between the acrylate and mucin glycoproteins. Figure 14.5 Maximum detachment force (MDF) of a 13 mm dry uncross-linked compressed polymer samples to fresh small intestine surface at room temperature (n = 4). Characterization of the thermal stability of polymers intended for drug delivery applications is essential due to the high temperatures used in the accelerated stability tests that are required in order to determine the sample’s shelf-life [53]. Furthermore, the thermal behaviour of polymers is sensitive to the presence of molecular interactions or chemical modification [54,55]. Thus, thermal studies can improve the understanding of the polymer structure and behaviour on the molecular level. Thermal gravimetric analysis (TGA) of alginate revealed three thermal steps (Figure 14.6), in agreement with a previous publication by Saores et al. [56], who attributed the first one to a dehydration process and the other two to decomposition. Weight loss analysis revealed dehydration at temperatures of up to 200 °C with around 20% weight loss. The first decomposition step involved additional weight loss of around 30%; in the second decomposition step about 30% of the initial weight was lost. The total remaining ash is about 20%. It is evident from Figure 14.6 that native alginate and alginate–thiol share a similar dehydration behaviour, suggesting that they have similar hydrophilicity. Figure 14.6 Differential thermal gravimetry (DTG) thermogram obtained for alginate, alginate–thiol, PEG-DA and alginate-PEGAc under air flow of 100 ml/min at a heating rate of 20 °C/min. It is worthwhile mentioning that while several studies have identified two distinct decomposition temperatures for alginate and its derivatives, and the reported values for the location of the first decomposition peaks are similar, the location of the second decomposition peaks varies. For example, decomposition temperatures of 200 °C and 600 °C were reported for alginate by Saores et al. [56], while values of 240 °C and 380 °C were measured by Shah et al. [57]. A two-step decomposition process occurring at 250 °C and 320 °C was reported by Laurienzo et al. for 2,3-dioctyl amine alginate [48], and peaks at 200 °C and 400 °C were observed for alginic acids [56]. The first decomposition step at around 250 °C was also detected by Caykara et al. for alginate samples [58]. Notably, alginate and alginate–thiol share a similar first decomposition temperature (256 °C for alginate and 245 °C for alginate–thiol); however, they differ in the position of the second decomposition step (367 °C and 604 °C for alginate and alginate–thiol, respectively). This result is in line with the behaviour of other alginate derivatives as stated above. The shift of the second decomposition temperature of alginate–thiol to a higher value could result from the formation of inter- and intramolecular disulfide bonds, which allude to higher polymer thermal stability. It is recognized that intramolecular interactions increase the thermal stability of polymers, thus leading to a higher decomposition temperature [54,55]. Moreover, thermal studies of amino acids have shown that the presence of sulfide end groups has led to disulfide bridges, which shifted the thermal peaks to a higher temperature [59]. The thermogram of alginate-PEGAc is shown in Figure 14.6 along with the thermograms of the native alginate and PEG-DA. Due to the overlap between the decomposition peaks of alginate and PEG-DA full peak assignment is not straightforward, yet general trends can be realized. Firstly, the weight loss percentage of alginate-PEGAc and alginate at temperatures up to ∼400 °C are similar. Secondly, the total ash remaining for alginate-PEGAc is 10%, which is smaller than the value observed for both alginate and alginate–thiol from the TGA analysis [71], PEG-DA decomposes without any ashes left. Therefore, it can be concluded that the PEGAc chain attached to the alginate backbone will decompose in this temperature range, leaving ash from the alginate backbone alone. The sharp dehydration peak of about 15% weight loss was shifted to a higher temperature of around 170 °C, possibly due to hydrogen bond interactions between the water molecules and the hydrophilic PEG chains. Three additional small and broad peaks, located at 200, 249 and 378 °C are evident. The peaks at ∼250 and ∼370 °C are common to alginate and alginate-PEGAc, while conjugation of PEG has led to broadening of the alginate decomposition peaks and to the appearance of an additional small peak at ∼200 °C. Thus, this curve seems to reflect the thermal characteristics of both PEG-DA and alginate. Overall, modification of alginate by attachment of PEG chains has a significant impact on the thermal behaviour of the final product. This phenomenon is most likely due to the large molecular weight of the PEG and its chemical characteristics. Laurienzo et al. [48] synthesized alginate-PEG by a different methodology and also detected both broad peaks at a similar temperature range of 200–300 °C, which were attributed to the alginate backbone, and an additional peak at around 400 °C, which was attributed to the grafted PEG molecules. A TGA curve of thiolated chitosan cross-linked by PEG-DA molecules displayed a thermal peak attributed to the PEG residues at approximately 380 °C [60]. In accordance with the TGA results, differential scanning calorimetry (DSC) analysis of alginate and alginate–thiol revealed similar behaviour for the two polymers (Figure 14.7). Both curves exhibit one endothermic peak at around 90 °C (92 °C for alginate and 89 °C for alginate–thiol) followed by a second exothermic peak located around 255 °C (258 °C for alginate and 253 °C for alginate–thiol). Similar characteristic peaks at 103 °C and 260 °C were previously attributed to water dehydration and alginate exothermic decomposition, respectively [56]. These results are in line with the TGA analysis, where dehydration was observed at temperature of up to 200 °C and the first decomposition step was detected at around 250 °C for both samples, with the alginate decomposition temperature being slightly higher than that of alginate–thiol. It is noted that DSC analysis was limited to temperatures of up to 300 °C due to technical limitations; therefore, the second decomposition step could not be detected. Figure 14.7 DSC thermograms obtained from alginate, alginate–thiol, PEG-DA and alginate-PEGAc under nitrogen flow of 50 ml/min using a heating rate of 20 °C/min. The DSC curve of alginate-PEGAc displays a decomposition peak at 256 °C. It is located in a similar temperature range to that of alginate–thiol and alginate. However, its intensity is reduced, presumably due to the large molecular weight of the grafted PEG that reduces the alginate weight percentage in the sample. A second exothermic peak appears at 62 °C, which can be attributed to the PEG melting temperature in accordance with previously reported values of 59 [61], 67 [62], 60 [58] and 55 °C [48], depending on the details of the experimental protocol (heating rate, type of gas used and gas flow rate) [56]. The comparison of the thermograms obtained for the native alginate, PEG-DA and their conjugation product, suggests that the characteristic decomposition behaviour of alginate in the DSC temperature range is maintained after PEGAc grafting. The peak broadening and the small shoulder in the endothermic peak of alginate-PEGAc sample may result from dehydration that occurs in the same temperature range. In conclusion, a good correlation between the TGA and DSC results was observed for alginate, alginate–thiol and alginate-PEGAc. Moreover, the results show that the conjugation of PEG chains to the alginate backbone influences its thermal behaviour and increases its hydrophilicity. However, this modification does not decrease the thermal stability in a temperature range below about 400 °C. Alginate-PEGAc could be expected to undergo cross-linking (gelation) by two independent mechanisms: physical gelation though the alginate backbone and chemical gelation involving the acrylated end groups carried by the PEG side chains. The cross-linking schemes can potentially be used to cross-link the polymer chains in different ways, thus manipulating its characteristics and altering its properties. Alginate’s G and M monomers are organized in a block-wise pattern of homopolymeric regions of M and G interspersed with regions of an alternating structure of MG blocks [63]. Many of the physical properties of alginate depend on the proportion and distribution of these segments and their relative sequencing [64]. It is believed that the G units are responsible for stiff chain characteristics while the M units form a flexible chain structure, due to the differences in monomer conformation which was found to be
Acrylated Polymers
14.1 Introduction
14.2 Mucoadhesion
14.3 Types of Interactions Involved in the Mucoadhesion Process
14.4 Interactions Between Acrylate and Mucin Glycoprotein
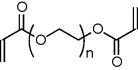
 NMR proton spectra of PEG-DA, mucin and their mixture provided a means to verify bond formation [25]. The spectrum obtained from native PEG-DA revealed several peaks ascribed to the vinyl end group protons (δ = 5.9–6.5 ppm) and to the protons of the methylene repeating unit (δ = 4.3 and δ = 3.6 ppm). While signals from the vinyl protons were also detected in the spectrum obtained from the mucin/PEG-DA mixture, their intensity decreased. The conjugation process is expected to lead to double bond opening, hence it should result in changes in the electron environment of the vinyl proton leading to a chemical shift. Therefore, the disappearance of the vinyl proton peaks, which are usually located around 6–8 ppm, is indicative to double bond opening due to reaction with glycoprotein. Moreover, new protons were found in the low ppm region where –CH2 groups are usually located, further supporting the hypothesis that PEG-DA formed intermolecular covalent bonds with mucin glycoproteins [25].
NMR proton spectra of PEG-DA, mucin and their mixture provided a means to verify bond formation [25]. The spectrum obtained from native PEG-DA revealed several peaks ascribed to the vinyl end group protons (δ = 5.9–6.5 ppm) and to the protons of the methylene repeating unit (δ = 4.3 and δ = 3.6 ppm). While signals from the vinyl protons were also detected in the spectrum obtained from the mucin/PEG-DA mixture, their intensity decreased. The conjugation process is expected to lead to double bond opening, hence it should result in changes in the electron environment of the vinyl proton leading to a chemical shift. Therefore, the disappearance of the vinyl proton peaks, which are usually located around 6–8 ppm, is indicative to double bond opening due to reaction with glycoprotein. Moreover, new protons were found in the low ppm region where –CH2 groups are usually located, further supporting the hypothesis that PEG-DA formed intermolecular covalent bonds with mucin glycoproteins [25].
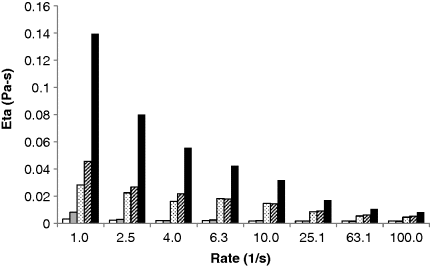
 ), PEG-Da 10 kDa 20 mg/ml (
), PEG-Da 10 kDa 20 mg/ml ( ), mucin 20 mg/ml (
), mucin 20 mg/ml ( ), mucin 20 mg/ml + PEG-OH 10 kDa 20 mg/ml (
), mucin 20 mg/ml + PEG-OH 10 kDa 20 mg/ml ( ), and mucin 20 mg/ml + PEG-Da 10 kDa 20 mg/ml (
), and mucin 20 mg/ml + PEG-Da 10 kDa 20 mg/ml ( ) in distilled water at 25 °C.
) in distilled water at 25 °C.
14.5 Acrylated Alginate (Alginate-PEGAc)
 linked α-L-guluronic acid (G) and β-D-mannuronic acid (M) [41–43]. Gelation of alginate is based on its affinity toward certain multivalent cations, such as Ca+2, and its ability to bind those ions selectively and cooperatively [42], a process which leads to the formation of ionically or physically cross-linked alginate gel [41]. Over the years several approaches have been developed in order to improve alginate characteristics by conjugating various molecules, [44] such as acrylic acid [45,46], cystein [47] and PEG [48], to its backbone. Alginate was also used in combination with PEG molecules by physical blending of the polymers followed by alginate cross-linking. This approach has led to the formation of alginate hydrogel with larger pore sizes that can also be used for cell encapsulation [49–51].
linked α-L-guluronic acid (G) and β-D-mannuronic acid (M) [41–43]. Gelation of alginate is based on its affinity toward certain multivalent cations, such as Ca+2, and its ability to bind those ions selectively and cooperatively [42], a process which leads to the formation of ionically or physically cross-linked alginate gel [41]. Over the years several approaches have been developed in order to improve alginate characteristics by conjugating various molecules, [44] such as acrylic acid [45,46], cystein [47] and PEG [48], to its backbone. Alginate was also used in combination with PEG molecules by physical blending of the polymers followed by alginate cross-linking. This approach has led to the formation of alginate hydrogel with larger pore sizes that can also be used for cell encapsulation [49–51].
14.5.1 Synthesis of Alginate-PEGAc
 NMR experiments, where both methylene and vinyl protons of the PEG-acrylate chain were detected in the alginate-PEGAc product [26]. Lack of cytotoxicity was demonstrated using in vitro cell assay [26], where alginate-PEGAc samples prepared from two types of alginate were cultured with human foreskin fibroblasts (HFFs) cells for 24 hours and assayed for the live/dead cells using fluorescent calcien and ethidium homodimer labelling followed by fluorescent microscopy imaging. None of the alginate-PEGAc caused cytotoxic effects in HFFs cells.
NMR experiments, where both methylene and vinyl protons of the PEG-acrylate chain were detected in the alginate-PEGAc product [26]. Lack of cytotoxicity was demonstrated using in vitro cell assay [26], where alginate-PEGAc samples prepared from two types of alginate were cultured with human foreskin fibroblasts (HFFs) cells for 24 hours and assayed for the live/dead cells using fluorescent calcien and ethidium homodimer labelling followed by fluorescent microscopy imaging. None of the alginate-PEGAc caused cytotoxic effects in HFFs cells.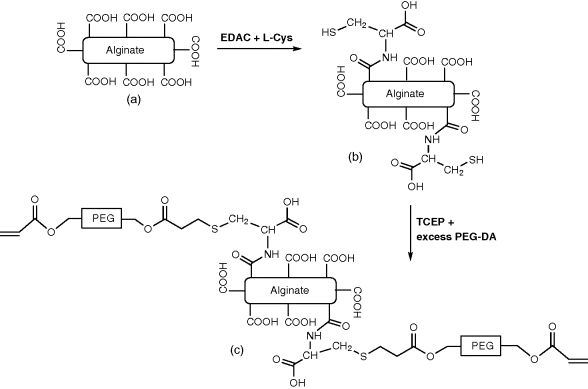
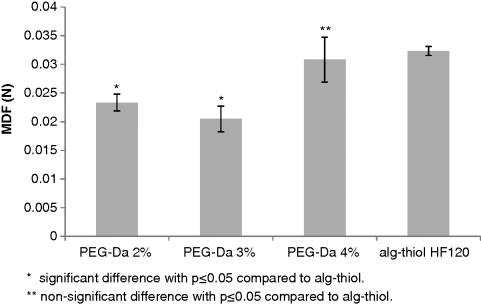
14.5.2 Mucoadhesion Ability
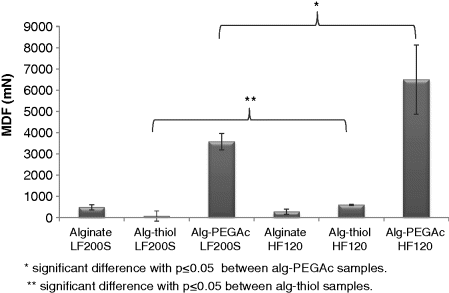
14.5.3 Thermal Properties of Alginate-PEGAc
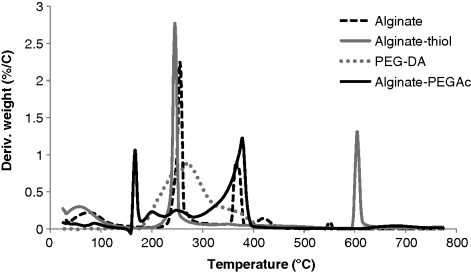
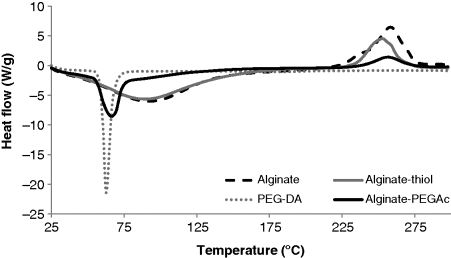
14.5.4 Gelation of Acrylated Alginate
 and
and  chair conformation for the guluronate and mannuronate residues, respectively. Thus, the overall conformation of the chain backbone is assumed to be a combination of stiff G blocks connected by flexible M blocks. Small angle X-ray scattering (SAXS) from such chains was previously described in terms of the ‘broken rod linked with flexible chain’ model [40,65]
chair conformation for the guluronate and mannuronate residues, respectively. Thus, the overall conformation of the chain backbone is assumed to be a combination of stiff G blocks connected by flexible M blocks. Small angle X-ray scattering (SAXS) from such chains was previously described in terms of the ‘broken rod linked with flexible chain’ model [40,65]
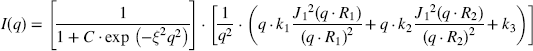
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


