INFLUENZA VIRUS GROUP CHARACTERISTICS
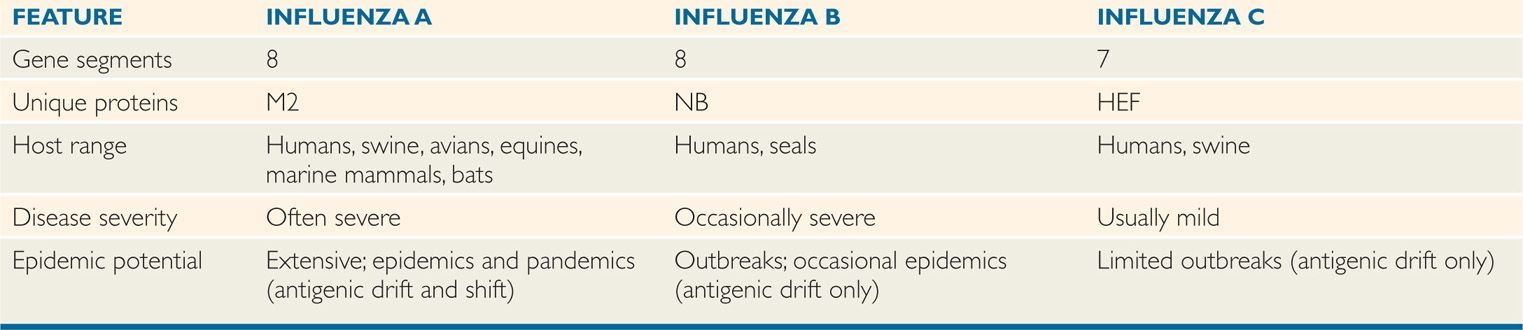
Influenza viruses are members of the orthomyxovirus group or family, which are enveloped, pleomorphic, single-stranded negative-sense segmented RNA viruses. They are classified into three major types, A, B, and C, on the basis of antigenic differences in their ribonucleoprotein (NP) and matrix (M) protein antigens. Influenza A viruses are the most extensively studied because of their predominance in epidemics, and much of the following discussion is based on knowledge of this type. They generally cause more severe disease and more extensive epidemics than the other types; naturally infect a wide variety of species, including mammals and birds; and have a great tendency to undergo significant antigenic changes (Table 9–1). Influenza B viruses are more antigenically stable, are known to infect humans and seals, and usually occur in more localized outbreaks. Influenza C viruses appear to be relatively minor causes of disease, affecting humans and pigs.
TABLE 9–1 Differences Among Influenza Viruses
Orthomyxoviruses divided into three types; A, B, and C
Type A has greatest virulence and predominance in epidemic spreads
Influenza A and B viruses each consist of a nucleocapsid containing eight segments of negative-sense, single-stranded RNA, which is enveloped in a lipid bilayer membrane derived from the host cell plasma membrane. The inner side of the envelope contains a layer of virus-specified matrix protein (M1). Two virus-specified glycoproteins, hemagglutinin (HA or H) and neuraminidase (NA or N), are embedded in the outer surface of the envelope and appear as “spikes” over the surface of the virion. The ratio of H to N is generally 4 or 5 to 1. There is another integral membrane protein in influenza A known as M2 ion channel protein. Figure 9–1 illustrates the makeup of influenza A virus. Influenza B is somewhat similar but has a unique integral membrane protein, NB instead of M2 that is also believed to function as an ion channel. Influenza C differs from the others in that it possesses only seven RNA segments and has no neuraminidase, although it does possess other receptor-destroying capability (see further). In addition, the hemagglutinin of influenza C binds to a cell receptor different from that for types A and B.
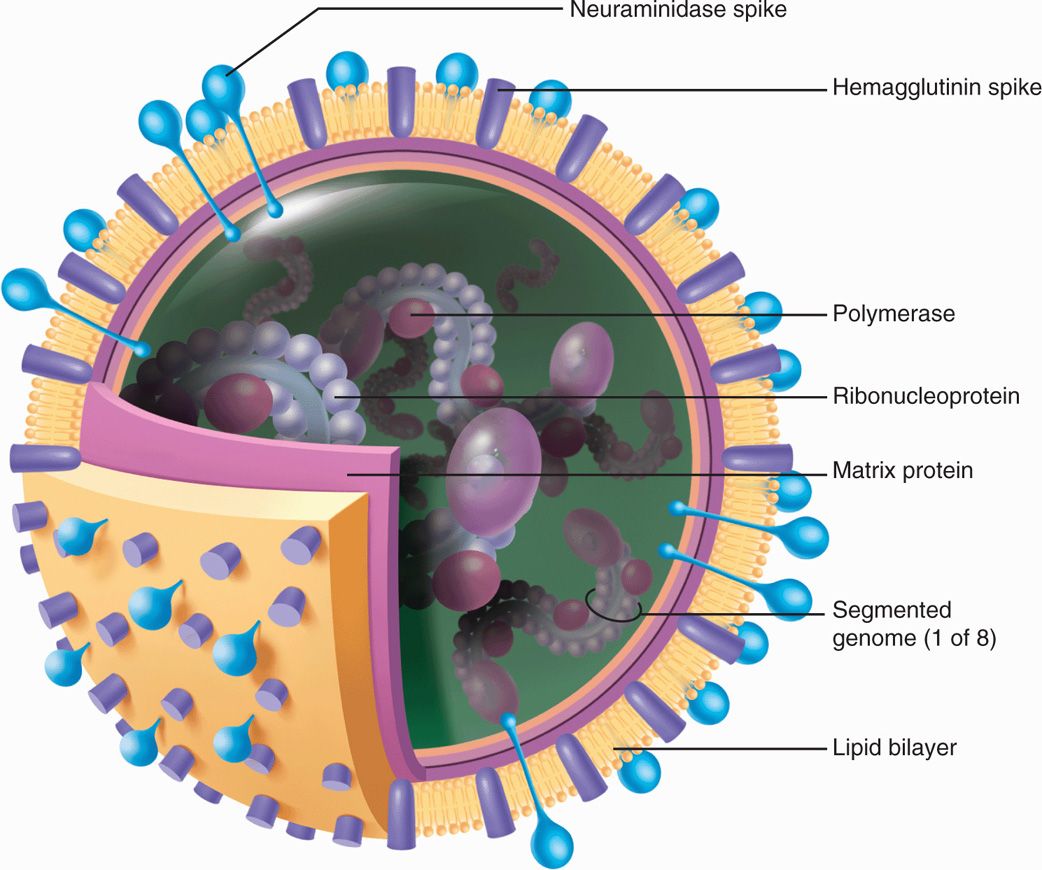
FIGURE 9–1. Diagrammatic view of influenza A virus. Three types of membrane proteins are inserted in the lipid bilayer: hemagglutinin (as trimer), neuraminidase (as tetramer), and M2 ion channel protein. The eight ribonucleoproteins segments each contain viral RNA surrounded by nucleoprotein and associated with RNA transcriptase. (Reproduced with permission from Willey JM: Prescott, Harley, & Klein’s Microbiology, 7th edition. McGraw-Hill, 2008.)
Enveloped RNA virus with segmented RNA genome
Virus-specific hemagglutinin (H) and neuraminidase (N) spikes expressed on envelope
Figure 9–2 illustrates the replication cycle of influenza virus. The virus-specific glycoproteins are antigenic and have special functional importance to the virus in pathogenesis and immunity. Hemagglutinin is so named because of its ability to agglutinate red blood cells from certain species (eg, chickens and guinea pigs) in vitro. Its major biologic function is to serve as a point of attachment to N-acetylneuraminic (sialic) acid-only containing gly-coprotein or glycolipid receptor sites on human respiratory cell surfaces, which is a critical first step in initiating infection of the cell.
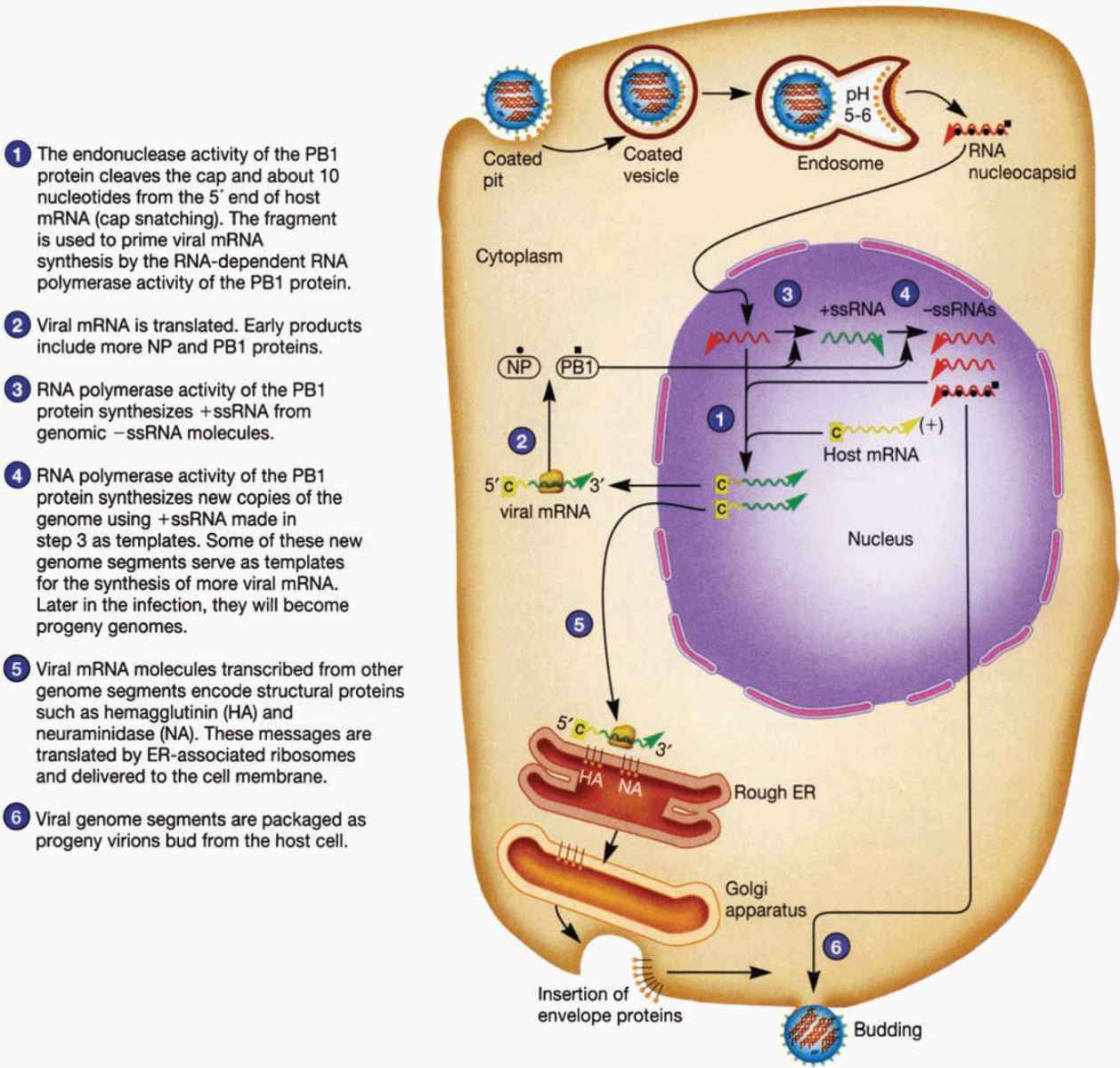
FIGURE 9–2. Diagrammatic view of influenza virus life cycle. (Reproduced with permission from Willey JM: Prescott, Harley, & Klein’s Microbiology, 7th edition. McGraw-Hill, 2008.)
Hemagglutinin binds to receptor on host cell for viral attachment
Neuraminidase is an antigenic hydrolytic enzyme that acts on the hemagglutinin receptors by splitting off their terminal neuraminic (sialic) acid. The result is destruction of receptor activity, which may help in preventing superinfection of the infected cell. Neuraminidase serves several functions. It may inactivate a free mucoprotein receptor substance in respiratory secretions that could otherwise bind to viral hemagglutinin and prevent access of the virus to the cell surface. Neuraminidase is important in fusion of the viral envelope with the host cell membrane as a prerequisite to viral entry. In addition, it aids in the release of newly formed virus particles from infected cells, thus making them available to infect other cells. Type-specific antibodies to neuraminidase appear to inhibit the spread of virus in the infected host and to limit the amount of virus released from host cells.
Neuraminidase has a role in viral envelope fusion and a major role in viral release
Neuraminidase promotes a smooth passage for the virus in the respiratory tract by inactivating mucoprotein receptors in respiratory secretions
Neuraminidase destroys viral receptor, thus preventing superinfection of infected cells
Nucleocapsid assembly takes place in the cell nucleus, but final virus assembly takes place at the plasma membrane. The ribonucleoproteins are enveloped by the plasma membrane, which by then contains hemagglutinin and neuraminidase. Virus “buds” are formed, and intact virions are released from the cell surface (Figure 9–2).
Nucleocapsid assembles in the nucleus and virus assembly occurs in the cytoplasm near the plasma membrane
Influenza A viruses were initially isolated in 1933 by intranasal inoculation of ferrets, which developed febrile respiratory illnesses. The viruses replicate in the amniotic sac of embryonated hen’s eggs, where their presence can be detected by the hemagglutination test. Most strains can also be readily isolated in cell culture systems, such as primary monkey kidney cells. Some cause cytopathic effects in culture.
Viral propagation and isolation in eggs or cell cultures
The most efficient method of detection is demonstration of hemadsorption by adherence of erythrocytes to infected cells expressing hemagglutinin or by agglutination of erythrocytes by virus already released into the extracellular fluid. The virus can then be identified specifically by inhibition of these properties by addition of antibody directed specifically against hemagglutinin. This method is called hemadsorption inhibition or hemagglutination inhibition (HI), depending on whether the test is conducted on infected cells or on extracellular virus, respectively. Because the hemagglutinin is antigenic, HI tests can also be used to detect antibodies in infected subjects. Research has shown that antibody directed against specific hemagglutinin is highly effective in neutralizing the infectivity of the virus.
Hemadsorption and hemagglutination inhibition used to detect presence of virus
Antihemagglutinin antibodies detectable in infected patients’ serum
 Influenza A
Influenza A
Influenza A is considered in detail because of its great clinical and epidemiologic importance.
The influenza A virion contains eight segments of negative-sense, single-stranded RNA with defined genetic responsibilities. These functions include coding for virus-specified proteins (Figure 9–1; Table 9–2). A unique aspect of influenza A viruses is their ability to develop a wide variety of subtypes through the processes of mutation and whole-gene “swapping” between strains, called reassortment. Recombination, which occurs when new genes are assembled from sections of other genes, is thought to occur rarely, if at all. These processes result in antigenic changes called drifts and shifts, which are discussed shortly.
TABLE 9–2 Virus-Coded Proteins of Influenza A
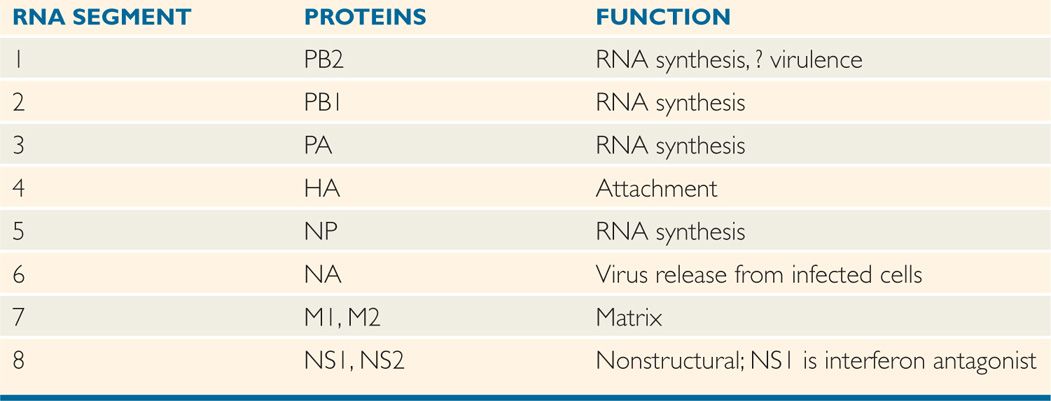
Influenza A genome divided into eight negative-sense RNA segments
Mutation and reassortment produce antigenic changes in the virus
The 17 recognized subtypes of hemagglutinin (H) and 10 neuraminidase (N) subtypes known to exist among influenza A viruses that circulate in birds and mammals represent a reservoir of viral genes that can undergo reassortment, or “mixing” with human strains. Although all 16 subtypes of hemagglutinins and nine subtypes of neuraminidases have been identified in aquatic birds, pigs are infected with two major hemagglutinins (H1 and H3) and neuraminidases (N1 and N2) and horses with two H (H3 and H7) and two N (N7 and N8). Three hemagglutinins (H1, H2, and H3) and two neuraminidases (N1 and N2) appear to be of greatest importance in human infections. A new subtype, H17N10 has been identified in bats. These subtypes are designated according to the H and N antigens on their surface (eg, H1N1, H3N2). There may also be more subtle, but sometimes important, antigenic differences (drifts) within each subtype. These differences are designated according to the major representative virus to which they are most closely related antigenically, using the place of initial isolation, number of the isolate, and year of detection. For example, two H3N2 strains that differ antigenically only slightly are A/Texas/1/77(H3N2) and A/Bangkok/1/79(H3N2).
Influenza A virus subtypes based on H and N antigens
Subtle changes known as antigenic drift (mutation) and drastic changes as antigenic shift (reassortment)
Antigenic drifts within major subtypes can involve either H or N antigens, as well as the genes encoding other structural and nonstructural proteins, and may result from as little as a single mutation in the viral RNA. These mutations are caused by viral RNA polymerase enzyme because it lacks proof reading ability. The mutant may come to predominate under selective immunologic pressures in the host population (Figure 9–3). Such drifts are common among influenza A viruses, occurring at least every few years and sometimes even during the course of a single epidemic. In addition, drifts can develop in influenza B viruses but considerably less frequently.
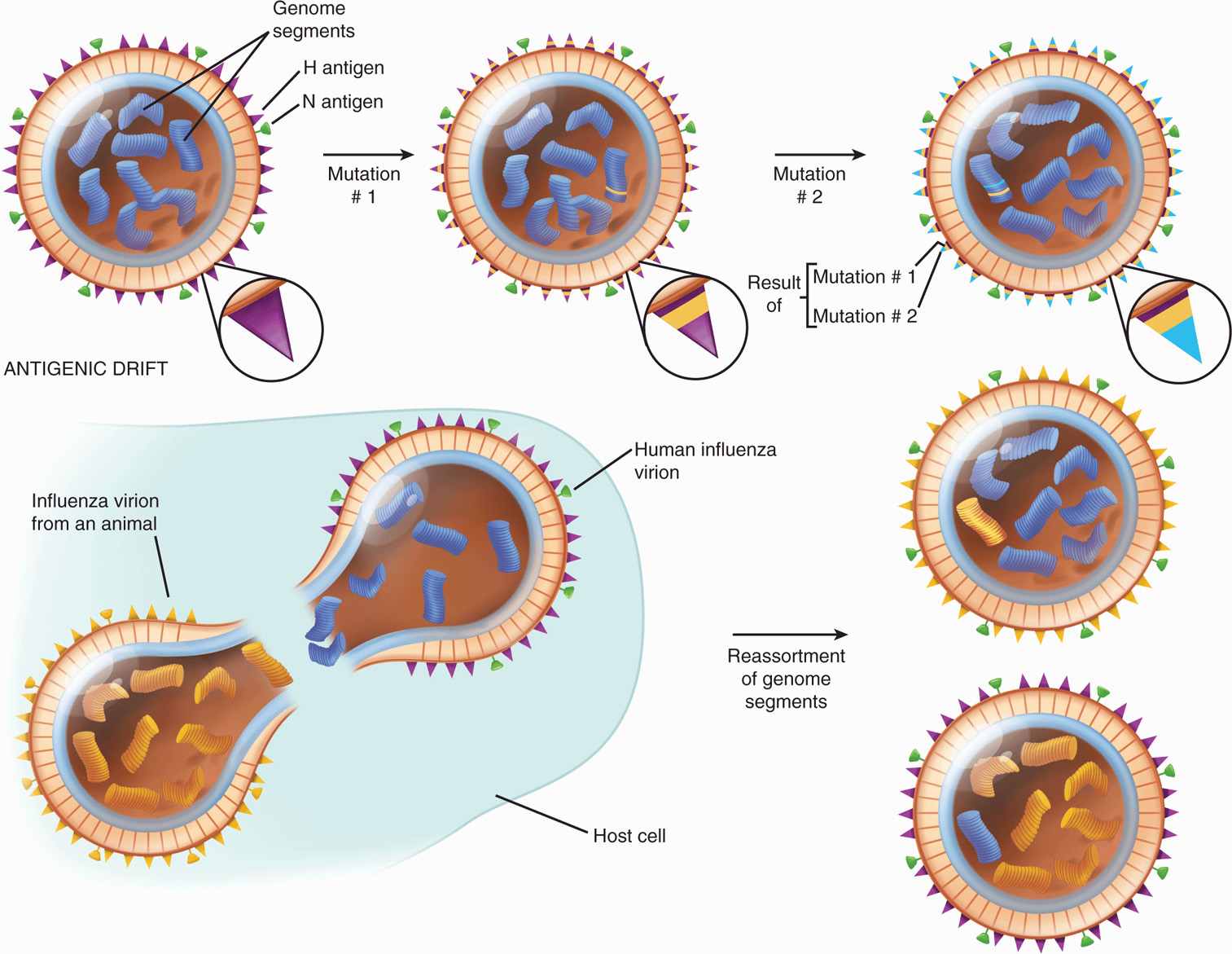
FIGURE 9–3. Influenza virus: antigenic drift and antigenic shift. With drift, repeated mutations cause a gradual change in the antigens composing hemagglutinin, such that antibody against the original virus becomes progressively less effective. With shift, there is an abrupt, major change in the hemagglutinin antigens because the virus acquires a new genome segment, which in this case codes for hemagglutinin. Changes in neuraminidase could occur by the same mechanism. (Reproduced with permission from Nester EW: Microbiology: A Human Perspective, 6th edition. 2009.)
Antigenic drift every year to few years with influenza A viruses
In contrast to the frequently occurring mutations that cause antigenic drift among influenza A strains, major changes (>50%) in the nucleotide sequences of the H or N genes can occur suddenly and unpredictably. These are referred to as antigenic shifts. (Figure 9–3 illustrates the difference between antigenic drifts and shifts.) They almost certainly result from reassortment that can be readily reproduced in the laboratory. Simultaneously infecting a cell with two influenza A subtypes yields progeny that contain antigens derived from either of the original viruses. For example, a cell infected simultaneously with influenza A (H3N2) and influenza A (H1N1) may produce a mixture of influenza viruses of the subtypes H3N2, H1N1, H1N2, and H3N1. When “new” epidemic strains emerge, they most likely have circulated into animal or avian reservoirs, where they have undergone genetic reassortment (and sometimes also mutation) and then are readapted and spread to human hosts when a sufficient proportion of the population has little or no immunity to the “new” subtypes. An example was the appearance of avian influenza A (H5N1) virus in Hong Kong in 1997 that caused infection in humans. The global spread of avian influenza (H5N1 and others) continued through 1997 and onward with several more cases every year. Studies indicated that all RNA segments were derived from an avian influenza A virus, but a single insert coding for several additional amino acids in the hemagglutinin protein facilitated cleavage by human cellular enzymes. In addition, a single amino acid substitution in the PB2 polymerase protein occurred. These two mutations together made the virus more virulent for humans; fortunately, human-to-human transmission was poor as discussed further. A recent example is the emergence of swine influenza virus (H1N1) in Mexico and the southwestern United States in 2009 that contained segments from avian, human, and swine influenza A viruses, and was easily transmitted to humans and caused a severe disease, mainly in young immune-competent adults, including deaths. In 2012, a new influenza virus strain, H3N8 has been identified in the autopsies of seals, which is closely related to a strain circulating in North American birds since 2002. As this strain has the ability to target the SAa-2,6 receptor found in the human respiratory tract, the new strain H3N8 poses risk to humans. In 2013, a new strain of avian flu (H7N9) infected humans in eastern China resulting in severe illness, including deaths. H7N9 has been found in chickens, ducks, and pigeons in live poultry markets in eastern China. H7N9 seems to be easily transmitted from poultry to humans compared with H5N1. Although there is no solid evidence of human-to-human transmission, a significant number of infected people had no contacts with poultry. Based on genetic analysis, H7N9 is responsive to neuraminidase inhibitors and that the virus has acquired some mutations that may allow it to infect mammals and humans.
Major antigenic shifts due to reassortment
Newly generated subtypes also develop mutations
H1N1 (human) and H5N1 (avian) target different regions of the respiratory tract
Additional molecular barriers limit human-to-human transmission of avian influenza virus (H5N1). One of the most important barriers is that avian and human influenza viruses target different regions of the human respiratory tract. Although the receptor for influenza viruses is sialic acid (SA) glycoprotein, there is a major difference in the sialic acid sugar positions with SA α 2,6 galactose for human influenza virus and SA α 2,3 galactose for avian influenza virus (H5N1). Human influenza virus receptor, SA α 2,6 galactose, is dominant on epithelial cells of nasal mucosa, paranasal sinuses, pharynx, trachea, and bronchi, whereas the H5N1 receptor SA α 2,3 galactose is mainly found on nonciliated bronchiolar cells at the junction between respiratory bronchioles and alveolus. It is interesting that A/Hong Kong/213/03 (H5N1) isolated from a patient recognized both SA α 2,6 galactose and SA α 2,3 galactose and is bound extensively to both bronchial and alveolar cells. More importantly, H1N1 swine influenza of 2009 was transmitted from human-to-human easily because it binds to the receptor SA α 2,6 galactose found in the upper respiratory tract, and caused greater severity because it infected the lower portion of the lungs by interacting with the receptor SA α 2,3 galactose.
Although receptors for H1N1 (human) are dominant in the upper part of the respiratory tract, H5N1 receptors are found in the lower portion of the lung in humans
H1N1 (swine) interacts with both receptors in the upper and lower respiratory tract
Major antigenic shifts, which occurred approximately every 8 to 10 years in the 20th century, often resulted in serious epidemics or pandemics among populations with little or no preexisting antibody to the new subtypes. Examples include the appearance of an H1N1 subtype in 1947, followed by an abrupt shift to an H2N2 strain in 1957, which caused the pandemic of Asian flu. A subsequent major shift in 1968 to an H3N2 subtype (the Hong Kong flu) led to another, but somewhat less severe epidemic. The Russian flu, which appeared in late 1977, was caused by an H1N1 subtype very similar to that which dominated between 1947 and 1957 (Table 9–3). The swine flu that appeared in April, 2009, in Mexico and southwestern United States was a previously unrecognized H1N1 strain, which caused a severe acute respiratory distress syndrome, including deaths, especially in young healthy immune-competent adults. Further analysis revealed that H1N1 swine influenza virus of 2009 was a reassortant that contained genetic components from four different flu viruses—North American swine influenza, North American avian influenza, human influenza, and swine influenza virus of Eurasian origin. Over the subsequent 3 months, this strain, designated swine-origin 2009 A (H1N1) rapidly spread globally. Fortunately, the pandemic tapered down in the following seasons. So, the key requirements for a pandemic influenza strain are: (1) generation of a new influenza A subtype, (2) causing a serious illness, and (3) easily transmitted from human-to-human. Although two of these three requirements were met in 2006 by H5N1, all these three prerequisites were fulfilled in 2009 by H1N1 swine. Each new human infection is an opportunity for the virus to change.
TABLE 9–3 Major Antigenic Shifts Associated With Influenza A Pandemics, 1947-1987

Major antigenic shifts correlate with epidemics
The concepts of antigenic shift and drift in human influenza A virus infections can be approximately summarized as follows. Periodic shifts in the major antigenic components appear, usually resulting in major epidemics in populations with little or no immunologic experience with the subtype. As the population of susceptible individuals is exhausted (ie, subtype-specific immunity is acquired by increasing numbers of people), the subtype continues to circulate for a time, undergoing mutations with subtle antigenic drifts from season to season. This allows some degree of virus transmission to continue. Infectivity persists because subtype-specific immunity is not entirely protective against drifting strains; for example, an individual may have antibodies reasonably protective against influenza A/Texas/77(H3N2), yet be susceptible in succeeding years to reinfection by influenza A/Bangkok/79(H3N2). Eventually, however, the overall immunity of the population becomes sufficient to minimize the epidemic potential of the major subtype and its drifting strains. Unfortunately, the battle is never entirely won; the scene is set for the sudden and usually unpredictable appearance of an entirely new subtype that may not have circulated among humans for 20 years or more. One example we saw in 2009 was when an H1N1 swine influenza virus appeared that had not been seen previously, and the existing population had no immunity to its components.
Minor antigenic drifts allow maintenance in population
Individual variation is significant
EPIDEMIOLOGY
Humans are the major hosts of the influenza viruses, and severe respiratory disease is the primary manifestation of infection. However, influenza A viruses closely related to those prevalent in humans circulate among many mammalian and avian species. As noted previously, some of these may undergo antigenic mutation or genetic recombination (reassortment) and emerge as new human epidemic strains.
Human, animal, and avian strains are similar
Characteristic influenza outbreaks have been described since the early 16th century, and outbreaks of varying severity have occurred nearly every year. Severe pandemics occurred in 1743, 1889-1890, 1918-1919 (the Spanish flu), 1957-1958 (the Asian flu), 1968-1969 (Hong Kong flu), 1977-1978 (Russian flu), and 2009-2010 (Swine flu). These episodes were associated with particularly high mortality rates; the Spanish flu was thought to have caused at least 30 to 50 million deaths, and some historians estimate the worldwide toll was closer to 100 million deaths. Usually, the elderly and persons of any age group with cardiac or pulmonary disease have the highest death rate. However, the severity in 2009 swine flu was mainly seen among the young healthy adult population.
Pandemic influenza may have high mortality
Direct droplet spread is the most common mode of transmission. Influenza infections in temperate climates tend to occur most frequently during midwinter months. Major epidemics of influenza A usually occur at 2- to 3-year intervals, and influenza B epidemics occur irregularly, usually every 4 to 5 years. The typical epidemic develops over a period of 3 to 6 weeks, and can involve 10% of the population. Illness rates may exceed 30% among school-aged children, residents of closed institutions, and industrial groups. One major indicator of influenza virus activity is an abrupt rise in school or industrial absenteeism. In severe influenza A epidemics, the number of deaths reported in a given area of the country often exceeds the number expected for that period. This significant increase, referred to as excess mortality, is another indicator of severe, widespread illness. Influenza B rarely causes such severe epidemics. In general, human influenza viruses are not stable in the environment and are sensitive to heat, acid pH, and solvents. In contrast, avian influenza viruses (H5N1 and others) retain infectivity for several weeks outside the host. The avian virus is shed in respiratory secretions and feces, and the virus survives in the feces for a long time.
Seasonality favors winter months
Epidemic intervals usually a few years
Excess mortality or increased absenteeism are indicators of epidemics
PATHOGENESIS
Influenza viruses have a predilection for the respiratory tract because of the presence of their receptors. They multiply in ciliated respiratory epithelial cells, leading to functional and structural ciliary abnormalities and viremia is rarely detected. This is accompanied by a switch-off of protein and nucleic acid synthesis in the affected cells, the release of lysosomal hydrolytic enzymes, and desquamation of both ciliated and mucus-producing epithelial cells. Thus, there is substantial interference with the mechanical clearance mechanism of the respiratory tract. The process of programmed cell death (apoptosis) results in the cleavage of complement components, leading to localized inflammation. Early in infection, the primary chemotactic stimulus is directed toward mononuclear leukocytes, which constitute the major cellular inflammatory component. The respiratory epithelium may not be restored to normal for 2 to 10 weeks after the initial insult.
Virus multiplies in respiratory epithelium
Synthetic blocks cause cilial damage and cell desquamation
Clearance mechanisms are compromised
The virus particles are also toxic to tissues. This toxicity can be demonstrated by inoculating high concentrations of inactivated virions into mice, which produces acute inflammatory changes in the absence of viral penetration or replication within cells. Other host cell functions are also severely impaired, particularly during the acute phase of infection. These functions include chemotactic, phagocytic, and intracellular killing functions of polymorphonuclear leukocytes and, perhaps, of alveolar macrophage activity.
Viral toxicity causes inflammation
Phagocytic host defenses compromised
The net result of these effects is that, on entry into the respiratory tract, the viruses cause cell damage, especially in the respiratory epithelium, which elicits an acute inflammatory response and impairs mechanical and cellular host responses. This damage renders the host highly susceptible to invasive bacterial superinfection. In vitro studies also suggest that bacterial pathogens such as staphylococci can more readily adhere to the surfaces of influenza virus-infected cells. Recovery from infection begins with interferon (α/β) production, which limits further virus replication, and with rapid generation of natural killer cells. Shortly thereafter, class I major histocompatibility complex (MHC)-restricted cytotoxic T cells appear in large numbers to participate in the lysis of virus-infected cells and, thus, in initial control of the infection. This is followed by the appearance of local and humoral antibody together with an evolving, more durable cellular immunity. Finally, there is repair of tissue damage.
Damage creates susceptibility to bacterial invasion
Interferon and cytotoxic T-cell responses associated with recovery
IMMUNITY
Although cell-mediated immune responses are undoubtedly important in influenza virus infections, humoral immunity has been investigated more extensively. Typically, patients respond to infection within a few days by producing antibodies directed toward the group ribonucleoprotein antigen, the hemagglutinin, and the neuraminidase. Peak antibody titer levels are usually reached within 2 weeks of onset and then gradually wane over the following months to varying low levels. Antibody to the ribonucleoprotein appears to confer little or no protection against reinfection because it is an internal protein of the virus particle that cannot be recognized by circulating antibody. Antihemagglutinin antibody is considered the most protective; it has the ability to neutralize virus on reexposure because it is a surface protein of the virus easily recognized by the antibody. However, such immunity is relative, and quantitative differences in responsiveness exist among individuals. Furthermore, antigenic shifts and drifts often allow the virus to subvert the antibody response on subsequent exposures. Antibody to neuraminidase antigen is not as protective as antihemagglutinin antibody, but plays a role in limiting virus spread within the host.
Antihemagglutinin antibody has protective effect
Antineuraminidase may limit viral spread
 CLINICAL ASPECTS
CLINICAL ASPECTS
MANIFESTATIONS
Influenza A and B viruses tend to cause the most severe illnesses, whereas influenza C seems to occur infrequently and generally causes milder disease. The typical acute influenzal syndrome is described here.
The incubation period is brief, lasting an average of 2 days. Onset is usually abrupt, with symptoms developing over a few hours. These include fever, myalgia, headache, and occasionally shaking chills. Within 6 to 12 hours, the illness reaches its maximum severity, and a dry, nonproductive cough develops. The acute findings persist, sometimes with worsening cough, for 3 to 5 days, followed by gradual improvement. By about 1 week after onset, patients feel significantly better. However, fatigue, nonspecific weakness, and cough can remain frustrating lingering problems for an additional 2 to 6 weeks.
Short incubation period followed by acute disease with dry cough
Occasionally, patients develop a progressive infection that involves the tracheobronchial tree and lungs. In these situations, pneumonia, which can be lethal, is the result. Other unusual acute manifestations of influenza include central nervous system dysfunction, myositis, and myocarditis. In infants and children, a serious complication known as Reye syndrome may develop 2 to 12 days after onset of the infection. It is characterized by severe fatty infiltration of the liver and by cerebral edema. This syndrome is associated not only with influenza viruses but with a wide variety of systemic viral illnesses. The risk is greatly enhanced by exposure to salicylates, such as aspirin.
Progressive respiratory infection and pneumonia may be lethal
Reye syndrome may follow
The most common and important complication of influenza virus infection is bacterial superinfection. Such infections usually involve the lung, but bacteremia with secondary seeding of distant sites can also occur. The superinfection, which can develop at any time in the acute or convalescent phase of the disease, is often heralded by an abrupt worsening of the patient’s condition after initial stabilization. The bacteria most commonly involved include Streptococcus pneumoniae, Haemophilus influenzae, and Staphylococcus aureus.
Sudden worsening suggests bacterial superinfection
In summation, there are essentially three ways in which influenza may cause death:
Underlying disease with decompensation. Individuals with limited cardiovascular or pulmonary reserves can be further compromised by any respiratory infection. Thus, the elderly and those of any age with underlying chronic cardiac or pulmonary disease are at particular risk.
Superinfection. Superinfection can lead to bacterial pneumonia and, occasionally, disseminated bacterial infection.
Direct rapid progression. Less commonly, progression of the viral infection can lead to overwhelming viral pneumonia with asphyxia. This phenomenon has been seen most commonly in severe pandemics; for example, the Spanish flu in 1918-1919 often produced fulminant death in healthy young soldiers.
Clinical manifestations of avian flu (H5N1) and swine flu (H1N1) varied with high fever, respiratory symptoms, neurologic symptoms, lymphopenia, and diarrhea. The virus replicated in the lower portion of the lung via interacting with the SA α 2,3 galactose receptor resulting in primary viral pneumonia in the absence of any secondary bacterial infection, including deaths, especially in healthy young adults. The cause of death was believed to be related to systemic dissemination, alveolar flooding, Na+ channel blockage, and cytokine storm (see Figure 7–5).
DIAGNOSIS
During the acute phase of illness, influenza viruses can be readily isolated from respiratory tract specimens, such as nasopharyngeal and throat swabs. Most strains grow in primary monkey kidney cell cultures, and they can be detected by hemadsorption or hemagglutination. Rapid diagnosis of infection is possible by direct immunofluorescence or immunoenzymatic detection of viral antigen in epithelial cells or secretions from the respiratory tract and by polymerase chain reaction (PCR). Serologic diagnosis is of considerable help epidemiologically and is usually made by demonstrating a fourfold or greater increase in HI antibody titers in acute and convalescent specimens collected 10 to 14 days apart. For details about the HI assay, see Chapter 4.
Virus isolation detects virus
Rapid detection of antigen or of viral genome by PCR often used
Serodiagnosis is useful epidemiologically
TREATMENT
The two basic approaches to management of influenza disease are symptomatic care and anticipation of potential complications, particularly bacterial superinfection. After the diagnosis has been made, rest, adequate fluid intake, conservative use of analgesics for myalgia and headache, and antitussives for severe cough are commonly prescribed. It must be emphasized that nonprescription drugs must be used with caution. This applies particularly to drugs containing salicylates (aspirin) given to children, because the risk of Reye syndrome must be considered.
Supportive therapy indicated
Bacterial superinfection is often suggested by a rapid worsening of clinical symptoms after patients have initially stabilized. Antibiotic prophylaxis has not been shown to enhance or diminish the likelihood of superinfection, but can increase the risk of acquisition of more resistant bacterial flora in the respiratory tract and make the superinfection more difficult to treat. Ideally, physicians should instruct patients regarding the natural history of the influenza virus infection and be prepared to respond quickly to bacterial complications, if they occur, with specific diagnosis and therapy.
Antibiotic prophylaxis does not prevent bacterial superinfection
Two classes of antiviral agents are available for use against influenza viruses—neuraminidase (N) inhibitors and viral protein M2 inhibitors (Table 9–4). Neuraminidase inhibitors include oseltamivir (Tamiflu) and zanamivir (relenza) that were approved in 1999 and block the function of neuraminidase enzyme of both influenza A and B viruses, which is required for viral release, spread, and infectivity. The mechanism of action of these neuraminidase inhibitors is to competitively inhibit the function of the viral neuraminidase enzyme. As neuraminidase removes sialic acid from the glycoprotein receptors, the inhibitors do not cleave sialic acid residues on the surfaces of host cells and influenza viral envelopes. Therefore, viral hemagglutinin (H) binds to the uncleaved sialic acid residues, resulting in viral aggregation at the surface of the host cell and inhibition of virus release and reinfection of uninfected cells. These drugs are effective in reducing the severity of influenza virus if taken within 48 hours of onset of illness. Oseltamivir is recommended for treatment in subjects 2 weeks and older, and chemoprophylaxis in 1 year and older. Zanamivir is recommended for treatment in subjects 7 years and older, and chemoprophylaxis in 5 years and older. Zanamivir that is administered as oral inhalation is not recommended for people with underlying respiratory disease. Viral resistance has now been demonstrated for some strains of influenza A and is currently low, but this might change in the future.
TABLE 9–4 Comparison of Antiviral Drugs for Influenza
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


