FIGURE 8–1. General scheme of antiviral action. The general sequence of viral replication, as in Figure 6–8, is shown with the points of action of selected antiviral agents.
Events in the cell unique to viral replication are the most desirable targets for antiviral therapy
In some cases, antiviral agents do not selectively inhibit a unique replicative event but inhibit DNA polymerase. Inhibitors of this enzyme take advantage of the fact that the virus is synthesizing nucleic acids more rapidly than the cell; therefore, there is relatively greater inhibition of viral than cellular DNA.
In many acute viral infections, especially respiratory ones, the bulk of viral replication has already occurred when symptoms are beginning to appear. Initiating antiviral therapy at this stage is unlikely to make a major impact on the illness. For these viruses, immuno- or chemoprophylaxis, rather than therapy, is a more logical approach. However, many other viral infections are characterized by ongoing viral replication and do benefit from viral inhibition, such as human immunodeficiency virus (HIV) infection and chronic hepatitis B and C.
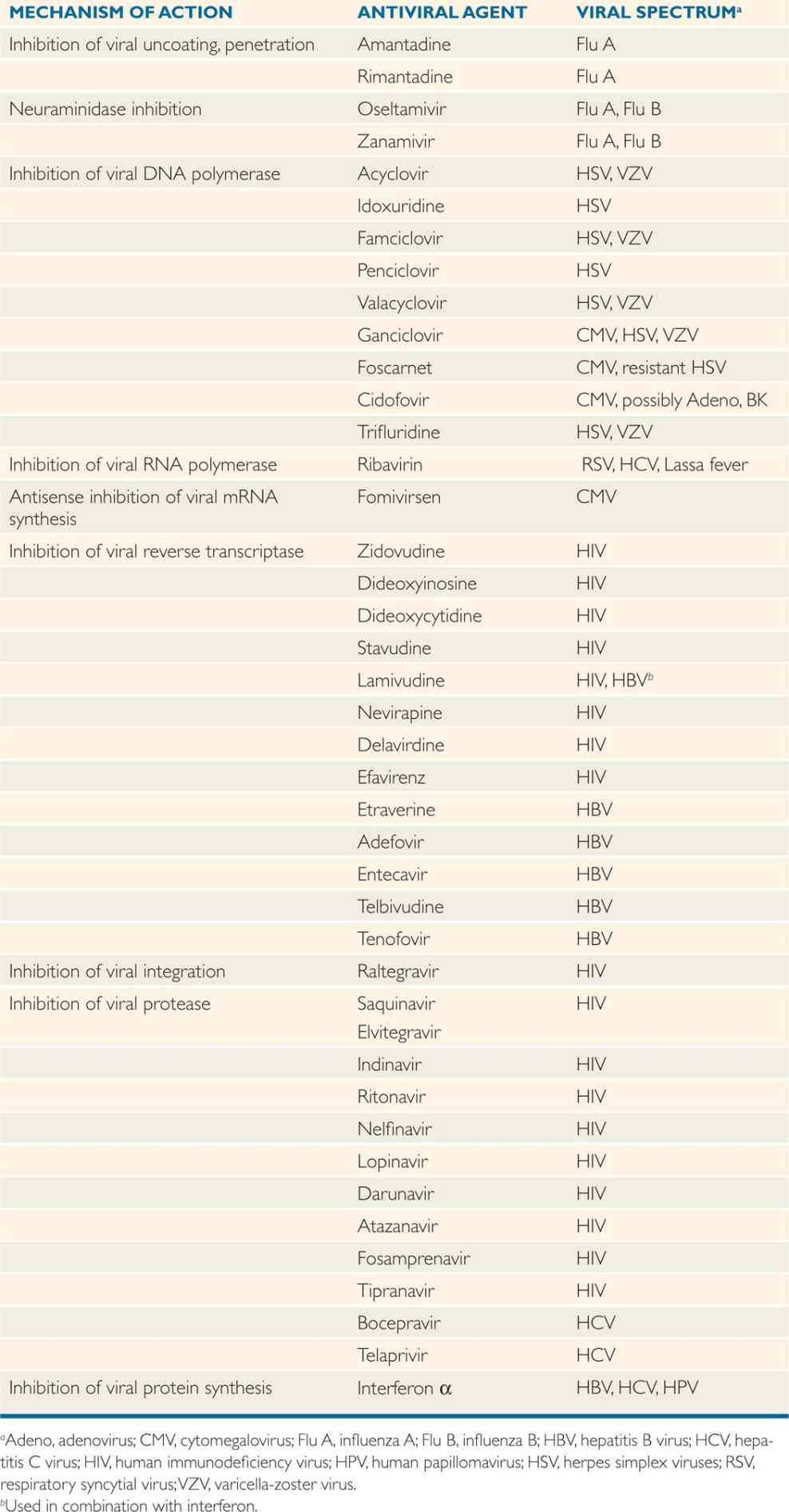
The principal antiviral agents in current use are discussed according to their modes of action. Their features are summarized in Table 8–1.
TABLE 8–1 Summary of Antiviral Agents
SELECTED ANTIVIRAL AGENTS
 Inhibitors of Attachment
Inhibitors of Attachment
Attachment to a cell receptor is a virus-specific event. Antibodies can bind to the extracellular virus and prevent this attachment. However, although therapy with antibody is useful in prophylaxis, it has been minimally effective in treatment.
 Inhibitors of Cell Penetration and Uncoating
Inhibitors of Cell Penetration and Uncoating
Amantadine and rimantadine are symmetric amines, or acyclics, which are thought to inhibit viral uncoating as their primary antiviral effect.
Rimantadine differs from amantadine by the substitution of a methyl group for a hydrogen ion. They are extremely selective, with activity against only influenza A, where they act as inhibitors of the M2 protein. They have been used either as prophylaxis or for therapy. Unfortunately, since 2001, the rates of resistance to amantadine/rimantadine have increased so sharply (up to 100% for some strains) that they are no longer routinely recommended.
Effective only against influenza A viruses, but sharply rising resistance rates now preclude their routine use
Pharmacology and Toxicity
Both amantadine and rimantadine are available only as oral preparations. The pharmacokinetics of the two agents is quite different. Amantadine is excreted by the kidney without being metabolized, and its dose must be decreased in patients with impaired renal function. In contrast, rimantadine is metabolized by the liver, then excreted in the kidney, and dosage adjustment for renal failure is not necessary. The major toxicity is in the CNS—seizures, somnolence, etc.
Rimantadine is metabolized by the liver
Amantadine is excreted by the kidney
 Neuraminidase Inhibitors
Neuraminidase Inhibitors
Oseltamivir and zanamivir are antiviral agents that selectively inhibit the neuraminidase of influenza A and B viruses. The neuraminidase cleaves terminal sialic acid from glycoconjugates and plays a role in the release of virus from infected cells. Zanamivir was the first approved neuraminidase inhibitor. It is given by inhalation using a specially designed device. Oseltamivir phosphate is the oral prodrug of oseltamivir, a drug comparable with zanamivir in antineuraminidase activity.
Neuraminidase inhibitors are effective in treatment and prophylaxis of influenza A and B viruses
Treatment with either oseltamivir or zanamivir reduces influenza symptoms, shortens the course of illness by 0.5 to 1.5 days, and reduces the rate of complications. The activity of these compounds against both influenza A and B offers an advantage over amantadine and rimantadine, which are active only against influenza A.
 Inhibitors of Nucleic Acid Synthesis
Inhibitors of Nucleic Acid Synthesis
At present, most antiviral agents are nucleoside analogs that are active against virus-specific nucleic acid polymerases or reverse transcriptases and have much less activity against analogous host enzymes. Some of these agents serve as nucleic acid chain terminators after incorporation into nucleic acids.
Idoxuridine and Trifluorothymidine
Idoxuridine (5-iodo-2′-deoxyuridine, IUdR) is a halogenated pyrimidine that blocks nucleic acid synthesis by being incorporated into DNA in place of thymidine and producing a nonfunctional molecule (ie, by terminating synthesis of the nucleic acid chain). It is phosphorylated by cellular thymidine kinase to the active compound, which inhibits both viral and cellular DNA polymerase. The resulting host toxicity precludes systemic administration in humans. Idoxuridine can be used topically as effective treatment of herpetic infection of the cornea (keratitis). Trifluorothymidine, a related pyrimidine analog, is effective in treating herpetic corneal infections, including those that fail to respond to IUdR. Trifluorothymidine has largely replaced IUdR.
Idoxuridine and trifluorothymidine block DNA synthesis
Acyclovir
This antiviral agent differs from the nucleoside guanosine by having an acyclic (hydroxyethoxymethyl) side chain. It must be phosphorylated by viral thymidine kinase to be active. Therefore, the compound is essentially nontoxic because it is not phosphorylated or activated in uninfected host cells. Viral thymidine kinase catalyzes the phosphorylation of acyclovir to a monophosphate. From this point, host cell enzymes complete the progression to the diphosphate and, finally, the triphosphate. Acyclovir triphosphate inhibits viral replication by competing with guanosine triphosphate and inhibiting the function of the virally encoded DNA polymerase. The selectivity and minimal toxicity of acyclovir are aided by its 100-fold or greater affinity for viral DNA polymerase than for cellular DNA polymerase. A second mechanism of viral inhibition results from incorporation of acyclovir triphosphate into the growing viral DNA chain. This causes termination of chain growth because there is no 3′-hydroxy group on the acyclovir molecule to provide attachment sites for additional nucleotides.
Activity of acyclovir against herpesviruses directly correlates with the capacity of the virus to induce a thymidine kinase. Susceptible strains of herpes simplex virus types 1 and 2 (HSV-1 and -2) are the most active thymidine kinase inducers and are the most readily inhibited by acyclovir. Cytomegalovirus (CMV) induces little or no thymidine kinase and is not inhibited. Varicella-zoster and Epstein-Barr viruses are between these two extremes in terms of both thymidine kinase induction and acyclovir susceptibility.
Acyclovir is effective against the herpesviruses, which induce thymidine kinase
Resistant strains of HSV have been recovered from immunocompromised patients, including patients with acquired immunodeficiency syndrome (AIDS); in most instances, resistance results from mutations in the viral thymidine kinase gene, rendering it inactive in phosphorylation. Resistance may also result from mutations in the viral DNA polymerase. Resistant virus has rarely been recovered from immunocompetent patients, even after years of drug exposure.
Acyclovir inhibits viral DNA poly-merase and terminates viral DNA chain growth
Pharmacology and Toxicity Acyclovir is available in three forms: topical, oral, and parenteral. Topical acyclovir is rarely used. The oral form has low bioavailability (~10%), but achieves concentrations in blood that inhibit HSV and, to a lesser extent, varicellazoster virus (VZV). Intravenous acyclovir is used for serious HSV infection (eg, congenital, encephalitis) as well as for VZV infection in immunocompromised patients. Because acyclovir is excreted by the kidney, the dosage must be reduced in patients with renal failure. Central nervous system toxicity and renal toxicity have been reported in patients treated with prolonged high intravenous doses. Acyclovir is remarkably free of bone marrow toxicity, even in patients with hematopoietic disorders.
Intravenous acyclovir used in serious HSV infections
Treatment and Prophylaxis Acyclovir is effective in the treatment of primary HSV mucocutaneous infections or for severe recurrences in immunocompromised patients. The agent is useful in neonatal herpes and encephalitis, infection in immunocompromised patients and for varicella in older children or adults. Acyclovir is beneficial against herpes zoster in elderly patients or any patient with eye involvement. In patients with frequent severe genital herpes, the oral form is effective in preventing recurrences. Because it does not eliminate the virus from the host, it must be taken daily to be effective. Acyclovir is minimally effective in the treatment of recurrent genital or labial herpes in otherwise healthy individuals.
Valacyclovir, Famciclovir, and Penciclovir
Valacyclovir is a prodrug of acyclovir that is better absorbed and, therefore, can be used in lower and less frequent dosage (bioavailability ~60%). When absorbed, it becomes acyclovir. It is currently approved for use in HSV and VZV infections. Dosage adjustment is necessary in patients with impaired renal function.
Famciclovir is similar to acyclovir in its structure and requirement for phosphorylation, but differs slightly in its mode of action. After absorption, the agent is converted to penciclovir, the active moiety, which inhibits viral DNA polymerase. However, it does not irreversibly terminate DNA replication. Famciclovir is currently approved for treatment of HSV and VZV infections. Penciclovir is approved for topical treatment of recurrent herpes labialis.
Agents that are similar to or become acyclovir after absorption are available
Ganciclovir
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


