CHAPTER OUTLINE
Genetic Alterations and Cancer
Classifications of Proto-Oncogenes
Proto-oncogenes and Inherited Cancer Syndromes
Epigenetics: DNA Methylation and Tumor Suppressors
High-Yield Terms
Carcinogen: any substance, such as a chemical, or any type of radiation that is directly involved in causing cancer
Neoplasm: any abnormal mass of tissue, medically synonymous with the term tumor
Malignancy: medically describes any condition that becomes progressively more severe, with respect to cancers the term characterizes invasiveness and the tendency to metastasize
Metastasis: refers to the spread of cancer from a site of origin to a new organ or another part of the body
Proto-oncogene: any normal gene that can become an oncogene due to gene mutation or abnormal expression
Oncogene: any gene that has the potential to cause cancer, in tumor cells, these genes are often mutated or expressed at high levels
Tumor suppressor: any gene that prevents the unregulated growth of cells, loss of function of this class of gene can result in cancer, also called antioncogene
Cancer Defined
Cancers, in the broadest terms, are a wide array of diseases typified by unregulated cell growth. The medical and biological term is neoplasm and defines an abnormal tissue that grows more rapidly than normal and continues to grow and proliferate in the absence of the originating growth signal. Neoplasms are transformed cells that can also harbor the characteristics of immortality. Transformation is a multistep process which results in the generation of the neoplastic cells. Immortalization refers to cells with unlimited life span but is not directly associated with aberrant growth or malignancy. Neoplasms can be benign or malignant. In the strictest sense, malignancy is defined as the ability to generate invasive tumors when cells are transplanted in vivo. However, medically this is not a useful definition due to the time required for the assessment of this capability. Therefore, it is more appropriate to define a malignant tumor, or mass, by its potential to worsen, to invade the surrounding tissues, and to exhibit the potential for metastases. Metastasis is the ability of cells from a tumor to break away from the original mass and spread to another location in the body. Although these terms broadly define all cancers, different types of cancers from different tissues exhibit a wide range of altered characteristics (Figure 52-1).

FIGURE 52-1: Some biochemical and genetic changes occurring in human cancer cells. Many changes are observed in cancer cells with many of the most significant diagrammed here. The roles of mutations in activating oncogenes and inactivating tumor suppressor genes are discussed in the text. Abnormalities of cell cycling and of chromosome structure, including aneuploidy, are common. Alterations of microRNA molecules that regulate gene activities have been reported, and the relationship of stem cells to cancer cells is a very active area of research. Telomerase activity is often detectable in cancer cells. Tumors sometimes synthesize certain fetal antigens, which may be measurable in the blood. Changes in plasma membrane constituents (eg, alteration of the sugar chains of various glycoproteins—some of which are cell adhesion molecules—and glycolipids) have been detected in many studies, and may be of importance in relation to decreased cell adhesion and metastasis. Various molecules can pass out of cancer cells and can be detected in the blood as tumor biomarkers. Angiogenic factors and various proteinases are also released by some tumors. Many changes in metabolism have been observed; for example, cancer cells often exhibit a high rate of aerobic glycolysis. (CAM, cell adhesion molecule; ECM, extracellular matrix.) Murray RK, Bender DA, Botham KM, Kennelly PJ, Rodwell VW, Weil PA. Harper’s Illustrated Biochemistry, 29th ed. New York, NY: McGraw-Hill; 2012.
Genetic Alterations and Cancer
The genetic damage present in a cancer cell can arise either spontaneously or the mutation can be inherited. Inherited mutations are classified as familial cancers such that the inheritance of the mutation predisposes an individual to cancer. This of course does not always mean that an individual acquiring an inherited mutation will develop cancer just that the individual has an increased likelihood relative to an individual who does not possess the mutant gene. Spontaneous mutations resulting in cancer are most often the result of exposure to some form of carcinogen. Carcinogens can be chemicals, radiant energies (such as ultraviolet or gamma irradiation), or oncogenic viruses. Chemical and radiant energy generally result in DNA damage that exceeds the capability of a cell to repair and thus, the damage is propagated in the resultant daughter cells. Often, individuals who harbor existing mutations in DNA repair process genes are highly susceptible to cancers caused by these types of carcinogens.
The genetic damage found in cancer cells is of 2 types referred to as dominant or recessive. Dominant mutations are associated with genes defined as proto-oncogenes. The distinction between the terms proto-oncogene and oncogene relates to the activity of the protein product of the gene. A proto-oncogene is a gene whose protein product has the capacity to induce cellular transformation provided it sustains some genetic insult. An oncogene is a gene that has sustained some genetic damage and, therefore, produces a protein capable of cellular transformation. The process of activation of proto-oncogenes to oncogenes can include retroviral transduction or retroviral integration, point mutations, insertion mutations, gene amplification, chromosomal translocation, and/or protein-protein interactions. Proto-oncogenes can be classified into many different groups based upon their normal function within cells or based upon sequence homology to other known proteins. As predicted, proto-oncogenes have been identified at all levels of the various signal transduction cascades that control cell growth, proliferation and differentiation. Proto-oncogenes that were originally identified as resident in transforming retroviruses were initially designated with the letter c—indicative of the cellular origin as opposed to v—to signify original identification in retroviruses. Recessive mutations are associated with genes referred to as tumor suppressors, growth suppressors, recessive oncogenes, or antioncogenes.
High-Yield Concept
All cancer cells contain some form of genetic damage that appears to be the responsible event leading to tumorigenesis (the generation of cancer). The genetic damage present in a parental tumorigenic cell is maintained (ie, not correctable) such that it is a heritable trait of all cells of subsequent generations. This is referred to as clonal expansion such that all subsequent cells of a tumor contain the same genetic alteration that was present in the originating cell.
Types of Cancer
Sarcomas are cancers derived from transformed cells with a mesenchymal origin. These cancers include those affecting bone, muscle, connective tissue, fat, and the vasculature. Although hematopoietic cells (the cells of blood) are derived from mesenchymal stem cells and thus, hematopoietic cancers (leukemias and lymphomas) are strictly defined as sarcomas, they are more commonly separated into the separate category of liquid cancers.
Carcinomas are cancers derived from transformed epithelial cells that originate from the endoderm or ectoderm layer of the developing embryo. These cells are found in the lining of the mouth, throat, stomach and intestines, the mammary ducts, the pancreas, the prostate gland, and the lungs. Carcinomas represent the most common types of cancers in humans and can be divided into several distinct variants defined by histology (Table 52-1).

Aerobic Glycolysis and Cancer
Overall metabolism in cancer cells is accelerated relative to normal differentiated cells of the same tissue. This is due, in part, to the expression of growth-promoting genes. Cancer cells can accommodate high rates of metabolism because they are changed in such a way as to allow rapid influx of nutrients, often without the need for the extracellular signals (eg, hormones) required of normal cells. Within the blood, the carbohydrate glucose and the amino acid glutamine, account for the vast majority of metabolic carbon and nitrogen.
In cancer cells (as well as many proliferating cells) the fate of glucose depends not only on the proliferative state of the cell but also on the activities of the specific glycolytic enzymes that are expressed. Cancer cells only express the PKM2 form of pyruvate kinase but at low levels. Low PKM2 activity, in conjunction with increased glucose uptake and oxidation, facilitates the diversion of glucose carbons into the biomass needed for cancer cells to continue to grow at high rates. Also, in cells expressing PKM2 there is increased phosphorylation of the upstream glycolytic enzyme phosphoglycerate mutase (PGAM1) which increases its mutase activity. The phosphate donor is phosphoenolpyruvate (PEP) which is the substrate for pyruvate kinase. Phosphate transfer from PEP to PGAM1 yields pyruvate without concomitant generation of ATP (see Figure 10-5). This alternate pathway allows for a high rate of glycolysis that is needed to support the anabolic metabolism observed in many proliferating cells. Targeting PKM2 for the treatment of cancers has shown promise since recent work has demonstrated that small molecule PKM2-specific activators reduce the incorporation of glucose into lactate and lipids and result in decreased pools of nucleotide, amino acid, and lipid precursors accounting for the suppression of tumorigenesis observed with these drugs.
High-Yield Concept
The majority of the known proto-oncogenes in the human genome have been identified by either of 2 means: as the transforming genes (oncogenes) of transforming retroviruses or through transfection of DNA from tumor cell lines into nontransformed cell lines and screening for resultant tumorigenesis.
High-Yield Concept
Human cancers can be divided into 2 broad categories with one representing solid tumors and the second the liquid cancers. Solid cancers are the sarcomas and carcinomas. The liquid cancers are the blood-borne cancers, the hematopoietic cancers.
Metastases
A significant factor leading to mortality associated with cancer is the spread of the cancer from the site of its origination to another, or many other, locations. The process of cancer spreading is clinically referred to as metastasis. A simplified view of metastasis is diagrammed in Figure 52-2. In order for a cancer cell to metastasize it must first break away from the primary tumor and enter the blood stream. Many of the required processes of metastasis are outlined in Table 52-2. The process by which the cell enters the blood is called intravasation. The cell can then migrate to a new location usually where its migration is arrested in a small capillary. There the cell migrates into the local extracellular matrix (ECM) and begins to grow and divide. The process of migration into the ECM is referred to as extravasation. In order for a new tumor to form there must be an adequate blood supply to the site and this is accomplished via the secretion of angiogenic factors. Angiogenesis is the process of new vessel synthesis and this process is a current target of several anticancer therapies. One of the growth factors involved in stimulating angiogenesis is vascular endothelial growth factor (VEGF). The monoclonal antibody drug Avastin (bevacizumab) targets VEGF and in so doing prevents the growth of new vessels in various types of cancers.

FIGURE 52-2: Simplified scheme of metastasis. Schematic representation of the sequence of steps in metastasis, indicating some of the factors believed to be involved. Murray RK, Bender DA, Botham KM, Kennelly PJ, Rodwell VW, Weil PA. Harper’s Illustrated Biochemistry, 29th ed. New York, NY: McGraw-Hill; 2012.

High-Yield Concept
The rate of glycolysis is elevated in all cancer cells and in 1924 Otto Warburg made an observation that cancer cells metabolism glucose in a manner that was distinct from the glycolytic process occurring in normal cells. Warburg discovered that, unlike most normal tissues, cancer cells tended to “ferment” glucose into lactate even in the presence of sufficient oxygen to support mitochondrial oxidative phosphorylation. This observation became known as the Warburg effect (see Clinical Box 10-1).
In order for a cancer cell to break away, it must be released from the ECM of the primary tumor. Two mechanisms are involved in this process. One is a reduction in the expression of cell adhesion molecules. One cell adhesion molecule, particularly associated with metastatic cancers is E-cadherin. Cadherins are a class of calcium-dependent adhesion molecule. Reduction or loss of E-cadherin is highly correlated to the metastatic potential of several types of cancers. In particular, prostate cancers with reduced levels of E-cadherin are much more virulent than similar cancers with normal E-cadherin levels. Indeed, because of this association between E-cadherin levels and metastatic potential, E-cadherin is often considered to be a tumor suppressor. The other mechanism required for cancer cells to metastasize is that the ECM of the tumor must be locally degraded. One of the most important class of enzymes involved in ECM remodeling is the matrix metalloproteinases (MMPs). Many metastatic cancers express elevated levels of MMPs, particularly MMP-2 and MMP-9, leading to degradation of the basement membrane and ECM allowing the cells to escape to the blood.
Viruses and Cancer
Tumor cells can arise by the carcinogenic actions of specific tumor viruses (Table 52-3). Tumor viruses are of 2 distinct types. There are viruses with DNA genomes (eg, papilloma and adenoviruses) and those with RNA genomes called the retroviruses. It should be noted that not all DNA or RNA viruses cause cancer in animals, for example the influenza (Orthomyxoviridae family) and measles (Paramyxoviridae family) viruses, which are RNA viruses, are not tumor viruses.

Tumor-causing retroviruses are common in chickens, mice, and cats but rare in humans. The human T-cell leukemia viruses (HTLVs) and human immunodeficiency virus (HIV) are the most notable examples of RNA tumor viruses in humans. The HTLV belong to the Oncoviridae family of viruses and HIV belongs to the Lentiviridae family. Another human RNA virus that causes cancer is the hepatitis C virus (HCV) which belongs to the Flaviviridae family of viruses.
Retroviruses can induce the transformed state within the cells they infect by 2 mechanisms. Both of these mechanisms are related to the life cycle of these viruses. When a retrovirus infects a cell its RNA genome is converted into DNA by the viral encoded RNA-dependent DNA polymerase (reverse transcriptase). The DNA then integrates into the genome of the host cell where it can remain, being copied as the host genome is duplicated during the process of cellular division. Contained within the sequences at the ends of the retroviral genome are powerful transcriptional promoter sequences termed long terminal repeats (LTRs). The LTRs promote the transcription of the viral DNA leading to the production of new virus particles.
At some frequency the integration process leads to rearrangement of the viral genome and the consequent incorporation of a portion of the host genome into the viral genome. This process is termed transduction. Occasionally this transduction process leads to the virus acquiring a gene from the host that is normally involved in cellular growth control. Because of the alteration of the host gene during the transduction process as well as the gene being transcribed at a higher rate due to its association with the retroviral LTRs the transduced gene confers a growth advantage to subsequent infected cells. The end result of this process is unrestricted cellular proliferation leading to tumorigenesis. The transduced genes are termed oncogenes. The normal cellular gene in its unmodified, nontransduced form is termed a proto-oncogene.
The second mechanism by which retroviruses can transform cells relates to the powerful transcription promoting effect of the LTRs. When a retrovirus genome integrates into a host genome it does so randomly. At some frequency this integration process leads to the placement of the LTRs close to a gene that encodes a growth-regulating protein. If the protein is expressed at an abnormally elevated level it can result in cellular transformation. This is termed retroviral integration-induced transformation. It has been shown that HIV induces certain forms of cancers in infected individuals by this integration-induced transformation process.
Cellular transformation by DNA tumor viruses, in most cases, has been shown to be the result of protein-protein interaction. Proteins encoded by the DNA tumor viruses, termed tumor antigens or T antigens, can interact with cellular proteins. This interaction effectively sequesters the cellular proteins away from their normal functional locations within the cell. The predominant types of proteins that are sequestered by viral T antigens have been shown to be of the tumor suppressor type. It is the loss of their normal suppressor functions that results in cellular transformation.
Classifications of Proto-Oncogenes
Given the complexity of inducing and regulating cellular growth, proliferation and differentiation, it was suspected for many years that genetic damage to genes encoding growth factors, growth factor receptors, and/or the proteins of the various signal transduction cascades would lead to cellular transformation. This suspicion has proven true with the identification of numerous genes, whose products function in cellular signaling, that are involved in some way in the genesis of the tumorigenic state (Table 52-4).

Proto-Oncogenes and Inherited Cancer Syndromes
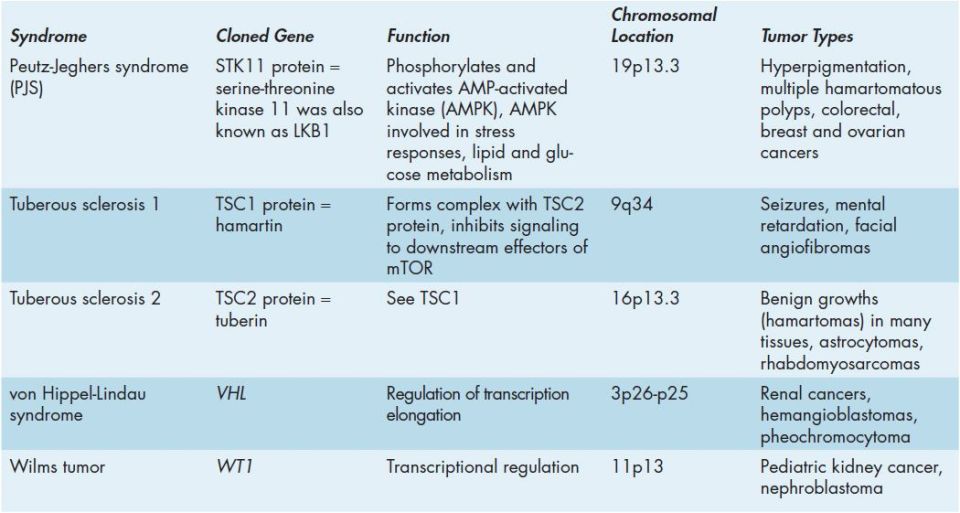
Hereditary cancer syndromes have their origin in random genetic mutations that occur in the germ cells and, thus are passed on in the germ line of an individual with typical mendelian patterns of inheritance. Specific hereditary susceptibility syndromes have been characterized that increase the risk of malignancies of the breast, ovary, colon, endometrium, and endocrine organs (Table 52-5). The most prevalent hereditary cancer syndromes are hereditary nonpolyposis colorectal cancer (HNPCC), familial adenomatous polyposis (FAP, Clinical Box 52-3), hereditary breast and ovarian cancer (HBOC), and multiple endocrine neoplasia type 2 (MEN2). The identification of hereditary cancer susceptibility syndromes, at the level of the gene, has significant medical implications. One major benefit of being able to test for cancer susceptibility genes is that individuals in high-risk families, who did not themselves inherit the susceptibility gene, can avoid unnecessary medical interventions.

Tumor Suppressors
Tumor suppressor genes are genes that regulate the growth of cells and when these genes are functioning properly, they can prevent and inhibit the growth of tumors. Tumor suppressor genes were first identified by making cell hybrids between tumor and normal cells. On some occasions a chromosome from the normal cell reverted the transformed phenotype of the tumor cell. When a tumor suppressor gene is altered, for example due to sustaining a mutation, the protein product of the gene may lose the ability to properly controlling cell growth. Under these conditions the cell can grow and proliferate in an uncontrolled way leading to development of cancer. Several familial cancers have been shown to be associated with the loss of function of a tumor suppressor gene (Table 52-6). There are 3 main classifications of tumor suppressor genes. These include genes that encode proteins that tell cells to stop growing and dividing. Another class encodes proteins responsible for repairing DNA damage prior to allowing cells to complete the cell cycle. The third class encodes proteins that are involved in regulating cell death processes, called apoptosis.

Epigenetics: DNA Methylation and Tumor Suppressors
The term epigenetics is used to define the mechanism by which changes in the pattern of inherited gene expression occur in the absence of alterations or changes in the nucleotide composition of a given gene. This is simply defined as the phenotypic consequences that can be observed due to alterations “on” the genes and not by alterations “to” the genes. Several different types of epigenetic events have been identified and include DNA methylation and histone modifications (see Chapter 35). DNA methylation is likely to be the most important epigenetic event controlling and maintaining the pattern of gene expression during development.
Whereas, epigenesis plays a vital role in the regulation, control, and maintenance of gene expression leading to the many differentiation states of cells in an organism, recent evidence has identified a linkage between epigenetic processes and disease. Most significant is the link between epigenesis at the level of DNA methylation and cancer. Alterations in DNA methylation have been suggested to be a contributing factor in nearly half of all human cancers. In most cases the higher the level of DNA methylation the more transcriptionally repressed is the methylated gene. The best characterized epigenetic “lesions” in malignant cells are the promoter CpG island hypermethylations that lead to transcriptional repression of tumor suppressor genes. These changes have been observed in numerous cancers and were first fully characterized in retinoblastomas (the RB gene), breast cancers (the BRCA1 gene), renal carcinomas (the VHL gene), and numerous types of cancers involving hypermethylation of the cyclin-dependent kinase (CDK) inhibitory gene, p16. Hypermethylation of the p16 gene has been observed in nearly 20% of all primary human neoplasms.
CLINICAL BOX 52-1: RETINOBLASTOMA
Retinoblastoma (RB) is a rare type of eye cancer that forms in immature cells of the retina and most often manifest in early childhood prior to the age of 5. Diagnosis of retinoblastoma after the age of 6 is extremely rare due to the terminal differentiation of retinal epithelial cells. In most children with retinoblastoma, the disease affects only one eye. However, in about 30% of children with retinoblastoma the cancer develops in both eyes. The most common first sign of retinoblastoma is a visible whiteness in the pupil referred to as leukocoria. This is also called amaurotic cat’s eye reflex. Additional symptoms of retinoblastoma include crossed eyes or eyes that do not point in the same direction (strabismus), persistent eye pain, redness, or irritation; and blindness or poor vision in the affected eye(s). Retinoblastoma is often curable when it is diagnosed early. If not treated promptly, the cancer can spread to other parts of the body leading to life-threatening metastasis. When retinoblastoma is associated with a gene mutation that occurs in all of the body’s cells, it is known as germinal retinoblastoma. Individuals with this form of retinoblastoma also have an increased risk of developing several other cancers outside the eye. Specifically, they are more likely to develop a cancer of the pineal gland in the brain (pinealoma), a type of bone cancer known as osteosarcoma, cancers of soft tissues such as muscle, and an aggressive form of skin cancer called melanoma. In the familial form of this disease individuals inherit a mutant, loss-of-function allele from an affected parent. A subsequent later somatic mutational event inactivates the normal allele resulting in retinoblastoma development. This leads to an apparently dominant mode of inheritance. The requirement for an additional somatic mutational event at the unaffected allele means that penetration of the defect is not always complete. There are also sporadic forms of retinoblastoma which requires 2 independent somatic mutational events to occur and is, thus extremely rare. The gene responsible for retinoblastoma is called the retinoblastoma susceptibility gene (RB). The RB RNA encodes a 110 kDa protein (pRB) of 928 amino acids. pRB is a nuclear localized phosphoprotein. Detectable levels of pRB can be found in most proliferating cells but the protein is not detectable in any retinoblastoma cells, typical of the case for many cancers involving tumor suppressor genes. The largest percentage (30%) of retinoblastomas contains large scale deletions in the RB gene. The germline mutations at RB occur predominantly during spermatogenesis as opposed to oogenesis. However, the somatic mutations occur with equal frequency at the paternal or maternal locus. In contrast, somatic mutations at RB in sporadic osteosarcomas occur preferentially at the paternal locus. This may be the result of genomic imprinting. The major function of pRB is in the regulation of cell cycle progression. Its ability to regulate the cell cycle correlates to the state of phosphorylation of pRB. Phosphorylation is maximal at the start of S phase and lowest after mitosis and entry into G1. Stimulation of quiescent cells with mitogen induces phosphorylation of pRB, while in contrast, differentiation induces hypophosphorylation of pRB. It is, therefore, the hypophosphorylated form of pRB that suppresses cell proliferation. One of the most significant substrates for phosphorylation by the G1 cyclin-CDK complexes that regulate progression through the cell cycle is pRB. pRB forms a complex with the E2F family of transcription factors, a result of which renders E2F inactive (Figure 52-3). When pRB is phosphorylated by G1 cyclin-CDK complexes it is released from E2F allowing E2F to transcriptionally activate genes. In the context of the cell cycle, E2F increases the transcription of the S-phase cyclins as well as leads to increases in its own transcription. Transformation by the DNA tumor viruses, SV40, adeno, polyoma, and human papilloma, is accomplished by binding of the transforming proteins (T antigens) of these viruses to pRB when pRB is in the hypophosphorylated, proliferation inhibitory, state.

FIGURE 52-3: pRB regulation of the E2F transcription factor. When hypophosphorylated, pRB binds to E2F preventing it from migrating to the nucleus and therefore, when bound to pRB the E2F protein is incapable of activating transcription. When pRB is phosphorylated, primarily at low levels by CDK2, it releases E2F which enters the nucleus and activates its own transcription as well as the cyclin E gene. Production of cyclin E gene results in a dramatic increase in CDK2 kinase activity. This results in further phosphorylation of pRB keeping E2F in the transcriptionally active state allowing for progression into S phase of the cell cycle. Reproduced with permission of themedicalbiochemistrypage, LLC.
CLINICAL BOX 52-2: LI-FRAUMENI SYNDROME
Li-Fraumeni syndrome (LFS) is a rare autosomal dominant disorder that greatly increases the risk of developing several types of cancer, particularly in children and young adults. The disorder is named for the 2 physicians who first recognized and described the syndrome: Frederick Pei Li and Joseph F. Fraumeni, Jr. The cancers most often associated with Li-Fraumeni syndrome include breast cancer, a form of bone cancer called osteosarcoma, and cancers of soft tissues (such as muscle) called soft tissue sarcomas. Other cancers commonly seen in this syndrome include brain tumors, cancers of blood-forming tissues (leukemias), and a cancer called adrenocortical carcinoma that affects the outer layer of the adrenal glands. LFS is linked to germline mutations in the tumor suppressor gene, p53. The p53 protein is involved in the blocking cell cycle progression while DNA repair takes place or if the DNA damage is severe enough p53 initiates a program of apoptosis. It was first thought that p53 was a dominant oncogene since cDNA clones isolated from tumor lines were able to cooperate with the RAS oncogene in transformation assays. This proved to be misleading since the cDNA clones used in all these studies were mutated forms of wild-type p53 and cDNAs from normal tissue were later shown to be incapable of RAS cotransformation. The mutant p53 proteins were shown to be altered in stability and conformation as well as binding to the chaperone, hsp70. Subsequent analyses showed that the p53 locus was lost by either insertions or deletions on both alleles in certain leukemias. This suggested that wild-type p53 may be a tumor suppressor not a dominant proto-oncogene. Direct confirmation came when it was shown that wild-type p53 could suppress transformation in oncogene cooperation assays with mutant p53 and RAS. It has now been demonstrated that mutant p53 involvement in neoplasia is more frequent than any other known tumor suppressor or dominant proto-oncogene. The protein encoded by p53 is a transcription factor whose activity is regulated by its state of phosphorylation. The p53 protein binds to target DNA sequences as a tetramer which explains the fact that mutant p53 proteins act in a dominant manner. They are present in complexes with wild type p53 and alter the function of the normal tetramer. The stability of p53 complexes is regulated by interaction with the ubiquitin ligase, MDM2. Phosphorylation of p53 protects it from MDM2-mediated ubiquitination. The level of p53 is low after mitosis but increases during G1. During S phase the protein becomes phosphorylated by the M-phase cyclin-CDK complex of the cell cycle and also by casein kinase II (CKII). Genes that are targets for transcriptional regulation by p53 are involved in the control of cell cycle progression and the suppression of cell growth. One major cell cycle–regulating gene that is a target for p53 is the CDK inhibitory protein (CIP), p21CIP. Activation of p53 by DNA damage-mediated phosphorylation (via the ATM kinase) results in increased expression of p21CIP with a resultant arrest in the G1 and G2 phases of the cell cycle (Figure 52-4). If the damaged DNA is unable to be repaired, p53 triggers activation of apoptosis.

FIGURE 52-4: DNA damage induction of p53 and activation of p21CIP. Ionizing radiation, or other causes of DNA damage, activate the ataxia-telangiectasia mutated (ATM) kinase. ATM phosphorylates p53 which stabilizes the protein and releases it from MDM2. The phosphorylated p53 then migrates to the nucleus where it activates transcription of various target genes. One target of p53 is the gene encoding CDK inhibitory protein 21, p21CIP. Increased expression of p21CIP results in inhibition of G1-S- and S-cyclin-CDK complexes ensuring that cells do not progress through S phase of the cell cycle until DNA damage is repaired. Reproduced with permission of themedicalbiochemistrypage, LLC.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


