CHAPTER OUTLINE
Steroids of the Adrenal Cortex
Regulation of Adrenal Steroid Synthesis
Functions of the Adrenal Steroid Hormones
Defects in Adrenal Steroidogenesis
High-Yield Terms
Androstanes: any 19-carbon steroid hormone, such as testosterone or androsterone, which controls the development and maintenance of male secondary sex characteristics
Estranes: any 18-carbon steroid hormone, such as estradiol and estrone, produced chiefly by the ovaries and responsible for promoting estrus and the development and maintenance of female secondary sex characteristics
Pregnanes: any 21-carbon steroid hormone, such as progesterone, responsible for changes associated with luteal phase of the menstrual cycle, differentiation factor for mammary glands
Cytochrome P450, CYP: any of a large number of enzymes produced from the cytochrome P450 genes, are involved in the synthesis and metabolism of various molecules and chemicals
Glucocorticoids: any of the group of corticosteroids predominantly involved in carbohydrate metabolism
Mineralocorticoids: steroid hormones characterized by their influence on salt and water balance
Hormone response elements, HREs: specific nucleotide sequences, residing upstream of steroid target genes, that are bound by steroid hormone receptors
The Steroid Hormones
The steroid hormones are all derived from cholesterol. The conversion of C27 cholesterol to the 18-, 19-, and 21-carbon steroid hormones (designated by the nomenclature C with a subscript number indicating the number of carbon atoms, eg, C19 for androstanes) involves the rate-limiting, irreversible cleavage of a 6-carbon residue from cholesterol, producing pregnenolone (C21).
Common names of the steroid hormones are widely recognized, but systematic nomenclature is gaining acceptance and familiarity. Steroids with 21 carbon atoms are known systematically as pregnanes, whereas those containing 18 and 19 carbon atoms are known as estranges and androstanes, respectively. The important mammalian steroid hormones are shown in Table 50-1 along with the structure of the precursor, pregnenolone.

All the steroid hormones exert their action by passing through the plasma membrane and binding to intracellular receptors. The mechanism of action of the thyroid hormones is similar; they interact with intracellular receptors. Both the steroid and thyroid hormone-receptor complexes exert their action by binding to specific nucleotide sequences in the DNA of responsive genes. These DNA sequences are identified as hormone response elements, HREs. The interaction of steroid-receptor complexes with DNA leads to altered rates of transcription of the associated genes.
Steroid Hormone Biosynthesis
The particular steroid hormone class synthesized by a given cell type depends upon its complement of peptide hormone receptors, its response to peptide hormone stimulation, and its genetically expressed complement of enzymes. Luteinizing Hormone (LH) stimulates the synthesis of progesterone and testosterone. Follicle-stimulating hormone (FSH) stimulates the synthesis of estradiol. Adrenocorticotropic hormone (ACTH) stimulates the synthesis of the glucocorticoids, principally cortisol. Angiotensin II stimulates the production of aldosterone.
Many of the enzymes of steroid hormone biosynthesis are members of the cytochrome P450 family of enzymes and they have common, preferred, and official nomenclatures. These enzymes are most correctly identified by the CYP acronym and include the family number, subfamily letter, and polypeptide number. Many of these enzymes are still identified by their historically common names or by the P450 nomenclature which includes a number designating the site of action of the particular enzyme (Table 50-2).
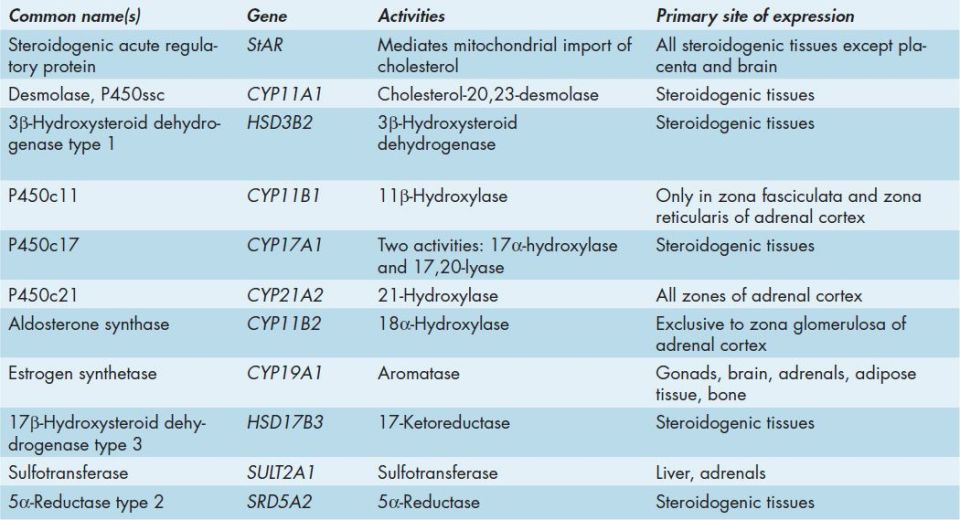
The first reaction in converting cholesterol to C18, C19, and C21 steroids involves the cleavage of a 6-carbon group from cholesterol-generating pregnenolone. This reaction is the principal committing and rate-limiting step in steroid biosynthesis. The enzyme system that catalyzes the cleavage reaction is historically referred to as desmolase. This enzyme is a cytochrome P450 enzyme and is also called P450-linked side chain cleaving enzyme (P450ssc) or more correctly CYP11A1. Other names include 20,22-desmolase or cholesterol desmolase. CYP11A1 is present in the mitochondria of steroid-producing cells, but not in significant quantities in other cells. In order for cholesterol to be converted to pregnenolone in the adrenal cortex it must be transported into the mitochondria where CYP11A1 resides. This transport process is mediated by steroidogenic acute regulatory protein (StAR). This transport process is the rate-limiting step in steroidogenesis.
Mitochondrial CYP11A1 is a complex enzyme system consisting of the cytochrome P450 and adrenodoxin (a P450 reductant). The activity of each of these components is increased by 2 principal PKA-dependent processes. First PKA phosphorylates a cholesteryl-ester esterase resulting in increased concentrations of cholesterol, the substrate for CYP11A1. Second, long-term regulation is effected at the level the CYP11A1 gene which contains a cAMP regulatory element (CRE) that binds cAMP resulting in increased transcription.
Steroids of the Adrenal Cortex
The adrenal cortex is responsible for production of 3 major classes of steroid hormones: glucocorticoids, which regulate carbohydrate metabolism; mineralocorticoids, which regulate the levels of sodium and potassium; and androgens, whose actions are similar to that of steroids produced by the male gonads. The adrenal cortex is composed of 3 main tissue regions: zona glomerulosa, zona fasciculata, and zona reticularis. Although the pathway to pregnenolone synthesis is the same in all zones of the cortex, the zones are histologically and enzymatically distinct, with the exact steroid hormone product dependent on the enzymes present in the cells of each zone (Figure 50-1).

FIGURE 50-1: Synthesis of the various adrenal steroid hormones from cholesterol. Only the terminal hormone structures are included. 3β-DH and Δ4,5-isomerase are the 2 activities of 3β-hydroxysteroid dehydrogenase type 1 (gene symbol HSD3B2), P450c11 is 11β-hydroxylase (CYP11B1), P450c17 is CYP17A1. CYP17A1 is a single microsomal enzyme that has 2 steroid biosynthetic activities: 17α-hydroxylase which converts pregnenolone to 17-hydroxypregnenolone (17-OH pregnenolone) and 17,20-lyase which converts 17-OH pregnenolone to DHEA. P450c21 is 21-hydroxylase (CYP21A2, also identified as CYP21 or CYP21B). Aldosterone synthase is also known as 18α-hydroxylase (CYP11B2). The gene symbol for sulfotransferase is SULT2A1. Reproduced with permission of themedicalbiochemistrypage, LLC.
Zona glomerulosa cells lack CYP17A1 that converts pregnenolone and progesterone to their C17 hydroxylated analogs. CYP17A1 is a single microsomal enzyme that has 2 steroid biosynthetic activities: 17α-hydroxylase which converts pregnenolone to 17-hydroxypregnenolone (17-OH pregnenolone) and 17,20-lyase which converts 17-OH pregnenolone to DHEA. Since zona glomerulosa cells lack CYP17A1, the pathways to the glucocorticoids (deoxycortisol and cortisol) and the androgens (dehydroepiandrosterone [DHEA] and androstenedione) are blocked in these cells. The next reaction of glomerulosa cell steroidogenesis is the conversion of pregnenolone to progesterone. This reaction requires the 2 enzyme activities of HSD3B2: the 3β-hydroxysteroid dehydrogenase and Δ4,5-isomerase activities.
Zona glomerulosa cells are unique in the adrenal cortex in that they express the enzyme responsible for converting corticosterone to aldosterone, the principal and most potent mineralocorticoid. This enzyme is CYP11B2 (18α-hydroxylase or P450c18) which is historically called aldosterone synthase. The result is that the zona glomerulosa is mainly responsible for the conversion of cholesterol to the weak mineralocorticoid, corticosterone, and the principal mineralocorticoid, aldosterone.
Cells of the zona fasciculata and zona reticularis lack CYP11B2 so these tissues produce only the weak mineralocorticoid corticosterone. However, both these zones do contain CYP17A1 allowing them to synthesize the major glucocorticoid, cortisol. The presence of CYP17A1 in zona fasciculata and zona reticularis cells allows them to also produce the androgens, dehydroepiandrosterone (DHEA), and androstenedione. Thus, fasciculata and reticularis cells can make corticosteroids and the adrenal androgens, but not aldosterone.
Regulation of Adrenal Steroid Synthesis
Adrenocorticotropic hormone (ACTH) regulates steroid hormone production of the zona fasciculata and zona reticularis. The effect of ACTH on the production of cortisol is particularly important, with the result that a classic feedback loop is prominent in regulating the circulating levels of corticotropin-releasing hormone (CRH), ACTH, and cortisol.
Mineralocorticoid secretion from the zona glomerulosa is stimulated by angiotensins II and III through activation of CYP11A1 activity. In the kidney, aldosterone regulates sodium retention by stimulating expression of mRNA for the Na+/K+-ATPase responsible for the reaccumulation of sodium from the urine. The interplay between renin from the kidney and plasma angiotensinogen is important in regulating plasma aldosterone levels, sodium and potassium levels, and ultimately blood pressure. This feedback loop is closed by potassium, which is a potent stimulator of aldosterone secretion. Changes in plasma potassium of as little as 0.1 mM can cause wide fluctuations (±50%) in plasma levels of aldosterone. Potassium increases aldosterone secretion by depolarizing the plasma membrane of zona glomerulosa cells and opening a voltage-gated calcium channel, with a resultant increase in cytoplasmic calcium and the stimulation of calcium-dependent processes.
Although fasciculata and reticularis cells each have the capability of synthesizing androgens and glucocorticoids, the main pathway normally followed is that leading to glucocorticoid production. However, when genetic defects occur in the 3 enzyme complexes leading to glucocorticoid production, large amounts of the androgen, dehydroepiandrosterone (DHEA), are produced resulting in hirsutism and other masculinizing changes in secondary sex characteristics.
Functions of the Adrenal Steroid Hormones
Glucocorticoids
The glucocorticoids are a class of hormones so called because they are primarily responsible for modulating the metabolism of carbohydrates. Cortisol is the most important naturally occurring glucocorticoid. When released to the circulation, cortisol is almost entirely bound to protein. A small portion is bound to albumin with more than 70% being bound by a specific glycosylated α-globulin called transcortin or corticosteroid-binding globulin (CBG). Between 5% and 10% of circulating cortisol is free and biologically active.
Glucocorticoid function is exerted following cellular uptake and interaction with intracellular receptors. Cortisol inhibits uptake and utilization of glucose resulting in elevations in blood glucose levels. The effect of cortisol on blood glucose levels is further enhanced through the increased breakdown of skeletal muscle protein and adipose tissue triglycerides which provides energy and substrates for gluconeogenesis. Glucocorticoids also increase the synthesis of gluconeogenic enzymes. The increased rate of protein metabolism leads to increased urinary nitrogen excretion and the induction of urea cycle enzymes. Glucocorticoids also inhibit vitamin D–mediated intestinal calcium uptake, retard the rate of wound healing, and interfere with the rate of linear growth.
In addition to the metabolic effects of the glucocorticoids, these hormones are immunosuppressive and anti-inflammatory. The anti-inflammatory activity of the glucocorticoids is exerted, in part, through inhibition of phospholipase A2 (PLA2) activity with a consequent reduction in the release of arachidonic acid from membrane phospholipids (see Chapter 22). It is the anti-inflammatory activity of glucocorticoids that warrants the use of related drugs such as prednisone, in the acute treatment of inflammatory disorders.
Mineralocorticoids
The major circulating mineralocorticoid is aldosterone. As the name implies, the mineralocorticoids control the excretion of electrolytes. This occurs primarily through actions on the kidneys but also in the colon and sweat glands. The principal effect of aldosterone is to enhance sodium reabsorption in the cortical collecting duct of the kidneys. However, the action of aldosterone is exerted on sweat glands, stomach, and salivary glands to the same effect, that is, sodium reabsorption. This action is accompanied by the retention of chloride and water resulting in the expansion of extracellular volume. Aldosterone also enhances the excretion of potassium and hydrogen ions from the medullary collecting duct of the kidneys.
Androgens
The androgens, androstenedione and DHEA, circulate bound primarily to sex hormone–binding globulin (SHBG). Although some of the circulating androgen is metabolized in the liver, the majority of interconversion occurs in the gonads, skin, and adipose tissue. DHEA is rapidly converted to the sulfated form, DHEA-S, in the liver and adrenal cortex. The primary biologically active metabolites of the androgens are testosterone and dihydrotestosterone which function by binding intracellular receptors, thereby effecting changes in gene expression leading to the manifestation of the secondary sex characteristics.
Defects in Adrenal Steroidogenesis
Defective synthesis of the steroid hormones produced by the adrenal cortex can have profound effects on human development and homeostasis. Addison disease (Clinical Box 50-1) represents a disorder characterized by adrenal insufficiency that was first identified in a patient who presented with chronic adrenal insufficiency resulting from progressive lesions of the adrenal glands caused by tuberculosis. In addition to diseases that result from the total absence of adrenocortical function, there are syndromes that result from hypersecretion of adrenocortical hormones. Disorders that manifest with adrenocortical hyperplasia are referred to as Cushing syndrome (Clinical Box 50-2).
The CAHs are a group of inherited disorders that result from loss-of-function mutations in one of several genes involved in adrenal steroid hormone synthesis. In the virilizing forms of CAH the mutations result in impairment of cortisol production and the consequent accumulation of steroid intermediates proximal to the defective enzyme. The majority of CAH cases (90% to 95%) are the result of defects in CYP21A2 with a frequency of between 1 in 5000 and 1 in 15,000 (Clinical Box 50-3). However, defects in several additional genes result in the various other forms of CAH including CYP11B1, HSD3B2, and CYP19A1.
Gonadal Steroid Hormones
Although many steroids are produced by the testes and the ovaries, the 2 most important are testosterone and estradiol. These compounds are under tight biosynthetic control, with short and long negative feedback loops that regulate the secretion of follicle-stimulating hormone (FSH) and luteinizing hormone (LH) by the pituitary and gonadotropin-releasing hormone (GnRH) by the hypothalamus. Low levels of circulating sex hormone reduce feedback inhibition on GnRH synthesis (the long loop), leading to elevated FSH and LH. The latter peptide hormones bind to gonadal tissue and stimulate CYP11A1 activity, resulting in sex hormone production.
CLINICAL BOX 50-1: ADDISON DISEASE
Primary adrenal insufficiency is characterized by inadequate secretion of glucocorticoids and mineralocorticoids as a result of impaired function or destruction of the adrenal cortex. The disorder was originally associated with tuberculosis and referred to as Addison disease. Currently, 70% to 90% of cases of Addison disease are the result of autoimmune adrenalitis and frequently manifests in patients with other immunological and autoimmune disorders. Addison disease frequency is higher in females than males and is usually diagnosed between 30 and 50 years of age. The loss of cortisol and aldosterone synthesis and release from the adrenal cortex results in increased secretion of ACTH from pituitary corticotropes. This distinguishes primary adrenal insufficiency from secondary which is the result of ACTH deficiency. The characteristic clinical presentation of acute primary adrenal insufficiency includes orthostatic hypotension, agitation, confusion, circulatory collapse, abdominal pain, and fever. The clinical findings in chronic primary adrenal insufficiency include a longer history of malaise, fatigue, anorexia, weight loss, and joint and back pain. Darkening of the skin, especially in the sun-exposed areas, is a hallmark sign of primary adrenal insufficiency. Patients may also crave salt and develop unusual food preferences. Biochemical features for both presentations include hyponatremia, hypoglycemia, hyperkalemia, unexplained eosinophilia, and mild prerenal azotemia. Biochemical testing is needed to confirm the diagnosis of adrenal insufficiency. The disorder can be excluded by serum cortisol levels of more than 19 μg/dL (524 nmol/L) and is likely if the value is less than 3 μg/dL (83 nmol/L). Measurement of urine-free cortisol is not a useful diagnostic test. Measurement of plasma ACTH helps to determine if the disorder is primary (values above the normal range) or secondary (low values). Hypokalemia and elevated plasma renin activity identify mineralocorticoid deficiency which aids in the discrimination of primary adrenal insufficiency. Treatment of Addison disease involves physiological replacement of the deficient glucocorticoid and mineralocorticoid hormones. Patients with acute adrenal insufficiency should receive fluid resuscitation and medical care as appropriate, in addition to supraphysiological doses of hydrocortisone given intravenously. For chronic therapy, oral hydrocortisone provides glucocorticoid replacement that can be easily titrated and given as divided doses during the day to grossly recapitulate the normal diurnal pattern of cortisol secretion. Oral fludrocortisone replaces mineralocorticoids.
CLINICAL BOX 50-2: CUSHING SYNDROME
Cushing syndrome describes the clinical consequences of chronic exposure to excess glucocorticoid irrespective of the underlying cause. Endogenous causes of Cushing syndrome are rare and include a cortisol-producing adrenal tumor, excess secretion of ACTH from a pituitary tumor (Cushing disease), or an ectopic ACTH-producing tumor (ectopic Cushing syndrome). Cushing syndrome is most commonly associated with prolonged administration of supraphysiological glucocorticoid treatment (including tablets, inhalers, nasal sprays, and skin creams) and is referred to as iatrogenic Cushing syndrome. Cushing syndrome may mimic common conditions such as obesity, poorly controlled diabetes, and hypertension, which progress over time and often coexist in patients with metabolic syndrome. Untreated Cushing syndrome is associated with high rates of mortality predominantly from cardiovascular events (congestive cardiac failure or myocardial infarction) or infection. Cushing syndrome affects the musculature (causing myopathy and congestive cardiac failure), the bones (causing early osteoporosis), reproductive function (causing menstrual irregularity and infertility), and leads to mood disturbance. As a consequence, a delay in proper diagnosis can result in irreversible organ damage. The characteristic features of Cushing syndrome are psychiatric disturbances (depression, mania, and psychoses), central obesity, hypertension, diabetes, moon-shaped face, thin fragile skin, easy bruising, and purple striae (stretch marks). In addition, Cushing syndrome patients manifest with gonadal dysfunction that is characteristic of hyperandrogenism with excess body and facial hair (hirsutism) and acne. The most common symptoms that lead to a diagnosis of Cushing syndrome are weight gain, depression, subjective muscle weakness, and headache. Since these latter symptoms are not in and of themselves diagnostic of Cushing syndrome it is important to assess for any possible use of exogenous glucocorticoids, especially inhalers and skin creams. Management of Cushing syndrome depends on the underlying cause. Surgical resection of the pituitary, adrenal, or ACTH-producing tumor is the primary treatment of choice and is often curative. If the disease is secondary to exogenous glucocorticoid treatment then titration of the steroid dose as soon as clinically possible is indicated.
CLINICAL BOX 50-3: CONGENITAL ADRENAL HYPERPLASIAS
Congenital adrenal hyperplasia resulting from deficiencies in the CYP21A2 gene represent the most commonly occurring forms (>95%) of the disease. There are two CYP21 genes but one is a pseudogene (CYP21P). The active CYP21 gene is identified as CYP21A2, CYP21, CYP21B, and 21-hydroxylase. Older nomenclature identifies the CYP21P gene as the 21-hydroxylase A gene (or CYP21A1) and CYP21A2 as the 21-hydroxylase B gene. All 21-hydroxylase activity is synthesized from the mRNA encoded by the CYP21A2 gene. The majority of the mutations in CYP21A2 that result in CAH have been identified and the severity of the disorder can be correlated to specific mutations and the consequent effect of the mutation on enzyme activity. Deficiencies in CYP21A2 result in decreased secretion and plasma concentration of cortisol. The reduced levels of cortisol result in a reduction in the negative feedback exerted by this hormone on the hypothalamic-pituitary axis. The reduced negative feedback leads to increased secretion of corticotropin-releasing hormone (CRH) and ACTH. The resultant high plasma concentrations of ACTH are responsible for the adrenocortical hyperplasia characteristic of this disorder. CAH resulting from CYP21A2 deficiencies is divided into 3 distinct clinical forms. The most severe enzyme impairment mutations result in the salt-losing form. Females with this form of the disease present at birth due to ambiguity in the external genitalia. Males with the salt-losing form present with acute adrenal crisis shortly after birth or in early infancy. Females who present with no acute adrenal crisis and only mild masculinization of the external genitalia and males who present with virilism early in life are considered to be suffering from the simple virilizing form of CAH. The final clinical form of CAH manifests only in females at puberty or shortly thereafter with symptoms of mild excess androgen development resulting in hirsutism, amenorrhea, and infertility. This form of the disorder is referred to as the attenuated form or the late onset or nonclassic form.
Salt-losing form: In the salt-losing form of this disorder the degree of loss of enzyme activity is severe to complete. As a result of the level of enzyme deficiency the synthesis of cortisol is negligible. Because CYP21A2 is also needed for the synthesis of aldosterone, which is a major hormone involved in Na+ retention by the kidney, there is excessive salt loss leading to hyponatremic dehydration which can be fatal if not treated. Although there is some aldosterone made, the level of salt loss exceeds the ability of the adrenal cortex to make sufficient aldosterone to compensate. The net result is an acute adrenal crisis. The near-complete, or complete, loss in cortisol production results in maximal activity of the CRH-ACTH axis leading to maximal adrenal androgen secretion with the result being masculinization of the female genitalia. The first sign of CAH due to CYP21A2 deficiency is, in fact, the ambiguous female genitalia in neonates. The masculinization can be so extreme as to result in the fusion of the labia and the formation of a penile urethra. Diagnosis of the salt-losing form of this disorder is made in females born with masculinized genitalia, who are of normal 46,XX karyotype, with marked elevation in plasma 17α-hydroxyprogesterone and androstenedione, as well as serum chemistry showing hyponatremia, hypochloremia, and hyperkalemic acidosis.
Simple virilizing form: In the simple virilizing form of the disorder the deficiency in CYP21A2 is not complete or as severe as in the salt-losing form. As a result the adrenal cortex can compensate for salt loss with increased aldosterone synthesis and release. In addition, the increased ACTH release result in normal plasma cortisol levels and thus, there is no glucocorticoid deficit. However, as in the salt-losing form the CRHACTH axis is hyperactive leading to excessive adrenal androgen synthesis with consequent masculinization of the female genitalia.
Attenuated form: As the name of this form of disease implies, patients with the attenuated form exhibit only mild reductions in CYP21A2 activity. Symptoms associated with this form of the disorder manifest in females at puberty. There are no signs of masculinization of the genitalia in females with this form of the disease. Although females have relatively normal breast development, the androgen excess results in excessive body hair (hirsutism), amenorrhea, and the development of small ovarian cysts.
High-Yield Concept
Despite the characterizations of adrenal insufficiency and adrenal hyperplasia, there remained uncertainty about the relationship between adrenocortical hyperfunction and virilism (premature development of male secondary sex characteristics) referred to as adrenogenital syndromes. These adrenogenital syndromes are now more commonly referred to as congenital adrenal hyperplasias (CAHs).
The biosynthetic pathway to sex hormones in male and female gonadal tissue includes the production of the androgens, androstenedione, and dehydroepiandrosterone. Testes and ovaries contain an additional enzyme, 17β-hydroxysteroid dehydrogenase (HSD17B3), that enables androgens to be converted to testosterone.
In males, LH binds to Leydig cells, stimulating production of the principal Leydig cell hormone, testosterone. Testosterone is secreted to the plasma and also carried to Sertoli cells by androgen-binding protein (ABP). In Sertoli cells the Δ4 double bond of testosterone is reduced, producing dihydrotestosterone. Testosterone and dihydrotestosterone are carried in the plasma, and delivered to target tissue, by a specific gonadal-steroid binding globulin (GBG). In a number of target tissues, testosterone can be converted to dihydrotestosterone (DHT). DHT is the most potent of the male steroid hormones, with an activity that is 10 times that of testosterone. Because of its relatively lower potency, testosterone is sometimes considered to be a prohormone (Figure 50-2).

FIGURE 50-2: Synthesis of the male sex hormones in Leydig cells of the testis. P450ssc, 3β-DH, and P450c17 are the same enzymes as those needed for adrenal steroid hormone synthesis. 17,20-lyase is the same activity of CYP17A1 described earlier for adrenal hormone synthesis. Aromatase (also called estrogen synthetase) is CYP19A1. 17-Ketoreductase is also called 17β-hydroxysteroid dehydrogenase type 3 (gene symbol HSD17B3). The full name for 5α-reductase is 5α-reductase type 2 (gene symbol SRD5A2). Reproduced with permission of themedicalbiochemistrypage, LLC.
Testosterone is also produced by Sertoli cells, but in these cells it is regulated by FSH. In addition, FSH stimulates Sertoli cells to secrete androgen-binding protein (ABP), which transports testosterone and DHT from Leydig cells to sites of spermatogenesis. There, testosterone acts to stimulate protein synthesis and sperm development.
In females, LH binds to thecal cells of the ovary, where it stimulates the synthesis of androstenedione and testosterone. An additional enzyme complex known as aromatase (CYP19A1, also called estrogen synthetase) is responsible for the final conversion of the latter 2 molecules into the estrogens. Aromatase is a complex enzyme present in the ER of the ovary and in numerous other tissues in both males and females. Its action involves hydroxylations and dehydrations that culminate in aromatization of the A ring of the androgens (Figure 50-3).

FIGURE 50-3: Synthesis of the major female sex hormones in the ovary. Synthesis of testosterone and androstenedione from cholesterol occurs by the same pathways as indicated for synthesis of the male sex hormones. Aromatase (also called estrogen synthetase) is CYP19A1. Reproduced with permission of themedicalbiochemistrypage, LLC.
Aromatase activity is also found in granulosa cells, but in these cells the activity is stimulated by FSH. Normally, thecal cell androgens produced in response to LH diffuse to granulosa cells, where granulosa cell aromatase converts these androgens to estrogens. As granulosa cells mature they develop competent large numbers of LH receptors in the plasma membrane and become increasingly responsive to LH, increasing the quantity of estrogen produced from these cells. Granulosa cell estrogens are largely, if not all, secreted into follicular fluid. Thecal cell estrogens are secreted largely into the circulation, where they are delivered to target tissue by the same globulin (GBG) used to transport testosterone.
Thyroid Hormones
The thyroid hormones are triiodothyronine (T3) and thyroxine (T4) (Figure 50-4). These hormones are derived from the thyroid hormone precursor, thyroglobulin. Thyroglobulin is glycosylated and contains more than 115 tyrosine residues, which become iodinated and are used for the synthesis of both T3 and T4. Thyroglobulin is exocytosed through the apical membrane into the closed lumen of thyroid follicles, where it accumulates as the major protein of the thyroid and where maturation takes place. Briefly, a Na+/K+-ATPase–driven pump concentrates iodide (I–) in thyroid cells, and the iodide is transported to the follicle lumen. There it is oxidized to I+ by a thyroperoxidase found only in thyroid tissue. The addition of I+ to tyrosine residues of thyroglobulin is catalyzed by the same enzyme, leading to the production of thyroglobulin containing monoiodotyrosyl (MIT) and diiodotyrosyl (DIT) residues. The thyronines, T3 and T4, are formed by combining MIT and DIT residues on thyroglobulin (Figure 50-4).

FIGURE 50-4: Structures of T3 and T4.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


