CHAPTER OUTLINE
Platelet Activation and von Willebrand Factor (vWF)
The Kallikrein-Kinin System in the Intrinsic Pathway
Activation of Prothrombin to Thrombin
Thrombin Activation of Fibrin Clot Formation
Protein C: Control of Coagulation and Intravascular Inflammation
High-Yield Terms
Hemostasis: refers to the process of blood clotting and then the subsequent dissolution of the clot following repair of the injured tissue
Intrinsic pathway: pathway of coagulation initiated in response to contact of blood with a negatively charged surface; primarily represents a pathophysiological reaction
Extrinsic pathway: pathway of coagulation initiated in response to release of tissue factor in response to vascular injury; represents the normal physiological pathway of coagulation
Platelets: also called thrombocytes; are small, disk-shaped nonnucleated fragments released from megakaryocytes involved in initiating hemostasis
Platelet glycoproteins: refers to several types of platelet surface receptors (eg, GPIIb-GPIIIa) involved in the processes of platelet activation and aggregation
Kallikrein: any of several serine proteases that cleave kininogens to form kinins; includes plasma and tissue kallikreins
Kinin: any of a group of vasoactive polypeptides (eg, bradykinin) formed by kallikrein-catalyzed cleavage of kininogens causing vasodilation and altering vascular permeability
Tenase complex: the complex of calcium ions and factors VIIIa, IXa, and X on the surface of activated platelets
Prothrombinase complex: a complex composed of platelet phospholipids, calcium ions, factors Va and Xa, and prothrombin
Thrombus: refers to the blood clot which is the final product of the blood coagulation
von Willebrand factor, vWF: a complex multimeric glycoprotein that is produced by and stored in the α-granules of platelets and found associated with subendothelial connective tissue.
Factor V Leiden: an inherited disorder of blood clotting resulting from a variant factor V that causes a hypercoagulability disorder
Partial thromboplastin time (PTT): assay for defects in activities of the intrinsic pathway of coagulation
Prothrombin time (PT): assay for defects in activities of the extrinsic pathway of coagulation
Events of Hemostasis
The ability of the body to control the flow of blood following vascular injury is paramount to continued survival. The process of blood clotting and then the subsequent dissolution of the clot following repair of the injured tissue is termed hemostasis. Hemostasis proceeds through a series of 4 coordinated events required to respond to the loss of vascular integrity.
1. The initial phase of the process is vascular constriction. This limits the flow of blood to the area of injury.
2. Activation of platelets is absolutely required for hemostasis to proceed. Platelets become activated by thrombin and aggregate at the site of injury, forming a temporary, loose platelet plug. Platelets clump by binding to collagen that becomes exposed following rupture of the endothelial lining of vessels. Upon activation, platelets release contents of their granules that are critical for the coagulation cascade. In addition to induced secretion, activated platelets change their shape to accommodate the formation of the plug.
3. To ensure stability of the initially loose platelet plug, a fibrin mesh (also called the clot) forms and entraps the plug. If the plug contains only platelets and fibrin, it is termed a white thrombus; if red blood cells are present, it is called a red thrombus.
4. Finally, the clot must be dissolved in order for normal blood flow to resume following tissue repair. The dissolution of the clot occurs through the action of plasmin.
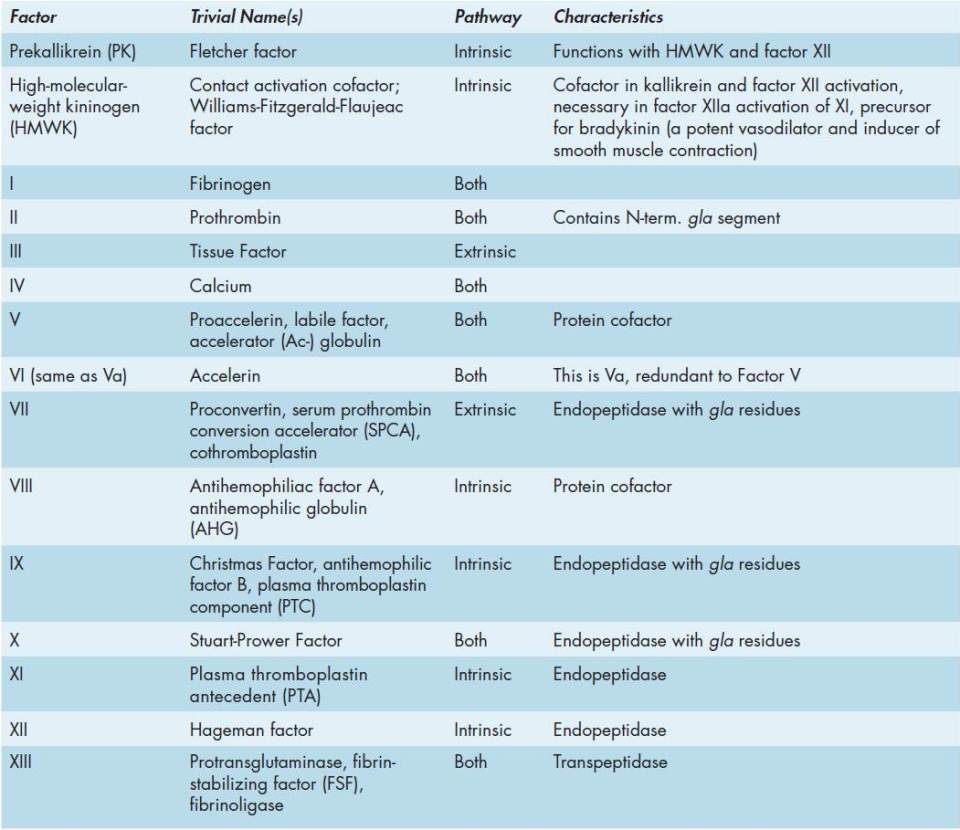
Two pathways lead to the formation of a fibrin clot: the intrinsic and extrinsic pathways (Figure 51-1). Although they are initiated by distinct mechanisms, the 2 converge on a common pathway that leads to clot formation. Both pathways are complex and involve numerous different proteins termed clotting factors (Tables 51-1 and 51-2). Fibrin clot formation in response to tissue injury is the most clinically relevant event of hemostasis under normal physiological conditions. This process is the result of the activation of the extrinsic pathway. The intrinsic pathway has low significance under normal physiological conditions. Most significant clinically is the activation of the intrinsic pathway by contact of the vessel wall with bacteria, lipoprotein particles, and other charged molecules.

FIGURE 51-1: The clotting cascades. The intrinsic cascade (which has less in vivo significance in normal physiological circumstances than the extrinsic cascade) is initiated when contact is made between blood and exposed negatively charged surfaces. The extrinsic pathway is initiated upon vascular injury which leads to exposure of tissue factor, TF (also identified as factor III), a subendothelial cell-surface glycoprotein that binds phospholipid. The green dotted arrow represents a point of crossover between the extrinsic and intrinsic pathways. The two pathways converge at the activation of factors X to Xa. Factor Xa has a role in the further activation of factors VII to VIIa as depicted by the green arrow. Active factor Xa hydrolyzes and activates prothrombin to thrombin. Thrombin can then activate factors XI, VIII, and V, furthering the cascade. Ultimately the role of thrombin is to convert fribrinogen to fibrin and to activate factors XIII to XIIIa. Factor XIIIa (also termed transglutaminase) cross-links fibrin polymers solidifying the clot. HMWK, high-molecular-weight kininogen; PK, prekallikrein; PL, phospholipid. Reproduced with permission of themedicalbiochemistrypage, LLC.

Platelet Activation and von Willebrand Factor (vWF)
Hemostasis requires that platelets adhere to exposed collagen, release the contents of their granules, and aggregate (Figure 51-2). Interaction of platelets with exposed collagen involves at least 2 collagen glycoprotein receptors identified as GPIa-IIa and GPVI. The GPIa-IIa receptor is a member for the integrin family of proteins and is, therefore, also identified as integrin α2β1. The GPVI receptor recognizes type III collagen.
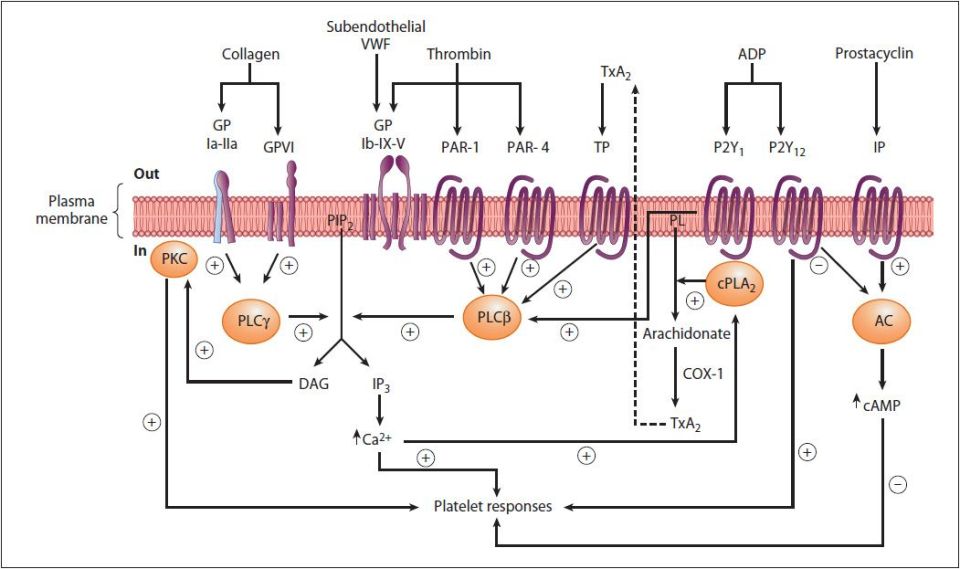
FIGURE 51-2: Diagrammatic representation of platelet activation by collagen, thrombin, thromboxane A2, and ADP, and inhibition by prostacyclin. The external environment, the plasma membrane, and the inside of a platelet are depicted from top to bottom. Platelet responses include, depending on the agonist, change of platelet shape, release of the contents of the storage granules, and aggregation. (AC, adenylyl cyclase; cAMP, cyclic AMP; COX-1, cyclooxgenase-1; cPLA2, cytosolic phospholipase A2; DAG, 1,2-diacylglycerol; GP, glycoprotein; IP, prostacyclin receptor; IP3, inositol 1,4,5-trisphosphate; P2Y1, P2Y12, purinoceptors; PAR, protease activated receptor; PIP2, phosphatidylinositol 4,5-bisphosphate; PKC, protein kinase C; PL, phospholipid; PLCβ, phospholipase Cβ; PLCγ, phospholipase Cγ; TP, thromboxane A2 receptor; TxA2, thromboxane A2; VWF, von Willebrand factor.) The G proteins that are involved are not shown. Murray RK, Bender DA, Botham KM, Kennelly PJ, Rodwell VW, Weil PA. Harper’s Illustrated Biochemistry. 29th ed. New York: McGraw-Hill; 2012.
Following binding to collagen the adhesion of platelets to the endothelial cell surface is mediated by von Willebrand factor (vWF). von Willebrand factor is a complex multimeric glycoprotein that is produced by and stored in the α-granules of platelets. It is also synthesized by megakaryocytes and found associated with subendothelial connective tissue. The function of vWF is to act as a bridge between a specific glycoprotein complex on the surface of platelets (GPIb-GPIX-GPV) and collagen fibrils. The GPIb part of the complex is composed of 2 proteins, GPIbα and GPIbβ, encoded by separate genes. Platelets also contain another collagen receptor.
Inherited deficiencies of vWF are the causes of von Willebrand disease, vWD (see Clinical Box 51-1). The importance of the interaction between vWF and the GPIb-GPIX-GPV complex of platelets is demonstrated by the inheritance of bleeding disorders caused by defects in 3 of the 4 proteins of the complex, the most common of which is Bernard-Soulier syndrome (also called giant platelet syndrome; see Clinical Box 51-2).
High-Yield Concept
In addition to its role as a bridge between platelets and exposed collagen on endothelial surfaces, vWF binds to and stabilizes coagulation factor VIII. Binding of factor VIII by vWF is required for normal survival of factor VIII in the circulation.
Once adherent to the endothelium, further activation of platelets involves binding of thrombin as well as several factors released from platelet granules such as ADP and TXA2 (see Figure 51-2). Platelets contain 3 types of granules termed dense, alpha, and lysosomal. Dense granules contain serotonin, calcium ions, and ADP. Alpha granules contain fibronectin; vWF; coagulation factors I, V, and IX; protein S; high-molecular-weight kininogen (HMWK); platelet factor-4 (PF-4); thrombospondin-1; and plasminogen activator inhibitor-1 (PAI-1). Lysosomal granules contain acid hydrolases, β-galactosidase, and β-glucuronidase.
Thrombin binds to a class of GPCR on platelets called the protease activated receptors (PARs), specifically PAR-1, -3 and -4 (Figure 51-3). PARs utilize a unique mechanism to convert the result of extracellular proteolytic cleavage into an intracellular signaling event. PARs carry their own ligand which remains inactive until protease cleavage, such as by thrombin, “unmasks” the ligand. Following thrombin cleavage the unmasked ligand is still a part of the intact PAR but is now capable of interacting with the ligand-binding domain of the PAR resulting in the activation of numerous signaling cascades. Because the activation of PARs requires proteolytic cleavage the activation process is irreversible.

FIGURE 51-3: Mechanism of activation of protease-activated receptor 1 (PAR-1) by thrombin. Reproduced with permission of themedicalbiochemistrypage, LLC.
Platelet activation by collagen and thrombin leads to activation of members of the PLC family resulting in increased IP3 and DAG production and the consequent signal cascades induced by these second messengers. The activation-stimulated release of ADP from platelet granules triggers ADP-mediated receptor activation of PLA2. Active PLA2 hydrolyzes membrane phospholipids releasing arachidonic acid. The arachidonic acid serves as a substrate for production and release of thromboxane A2 (TXA2). TXA2 is a potent vasoconstrictor and inducer of platelet aggregation that functions by binding to receptors that function through the PLC pathway.
The calcium that is released from the ER in response to IP3 binding activates myosin light chain kinase (MLCK). Activated MLCK phosphorylates the light chain of myosin which then interacts with actin, resulting in altered platelet morphology and motility. This altered mobility allows enhanced adhesion and the shape change aids in the aggregation of platelets.
One of the most important substituents of platelet granules is ADP. The released ADP binds to members of the purinergic receptor family, specifically P2Y1 and P2Y12, the consequences of which are further stimulation of platelets. The important role of ADP in platelet activation can be appreciated from the use of ADP receptor antagonists (eg, clopidogrel), in the control of thrombosis (see below). ADP also modifies the platelet membranes leading to exposure of platelet glycoprotein receptor complex GPIIb-GPIIIa. GPIIb-GPIIIa constitutes a receptor for vWF and fibrinogen, resulting in fibrinogen-induced platelet aggregation. The GPIIb-GPIIIa complex is also a member of the integrin family; therefore, it is also called integrin αIIb-β3. The importance of the GPIIb-GPIIIa in platelet activation and coagulation is demonstrated by the fact that bleeding disorders result from inherited defects in this glycoprotein complex. The most commonly inherited platelet dysfunction is Glanzmann thrombasthenia, which results from defects in the GPIIb protein of this complex (see Clinical Box 51-3). In addition, the importance of this complex is the anticoagulant function of antibodies to this receptor (eg, abciximab: see below).
The Kallikrein-Kinin System in the Intrinsic Pathway
Activation of the intrinsic pathway of coagulation, in part, involves the contribution of the kallikrein-kinin system. This system comprises a complex of proteins that when activated leads to the release of vasoactive kinins. The kinins are released from both high-molecular-weight kininogen (HMWK) and low-molecular-weight kininogen (LMWK) as a result of activation of either tissue kallikrein or plasma kallikrein. The kinins are involved in many physiological and pathological processes including regulation of blood pressure and flow (via modulation of the renin-angiotensin pathway), blood coagulation, cellular proliferation and growth, angiogenesis, apoptosis, and inflammation. Kinin action on endothelial cells leads to vasodilation, increased vascular permeability, release of tissue plasminogen activator (tPA), production of nitric oxide (NO), and the mobilization of arachidonic acid, primarily resulting in prostacyclin (PGI2) production by endothelial cells. With respect to hemostasis, the most important kinin is bradykinin which is released from HMWK. Bradykinin is a 9-amino-acid vasoactive peptide that induces vasodilation and increases vascular permeability.
CLINICAL BOX 51-1: von WILLEBRAND DISEASE
Von Willebrand disease (vWD) is caused by deficiencies in the gene-encoding von Willebrand factor (vWF). vWD is the most commonly occurring bleeding disorder with clinically significant forms affecting approximately 1 in 1000 individuals. However, abnormalities in vWF have been estimated to occur with a frequency of 1 in 100 persons, thus a relatively small fraction of persons harboring mutations in vWF are actually symptomatic. There are both dominant and recessive forms of vWD. The characteristic feature of vWD is bleeding from mucocutaneous sites (typical skin and mucous membranes) rather than from deep tissues and joints. vWF is involved in 2 important reactions of blood coagulation. The first is the interaction of vWF with specific receptors on the surface of platelets and the subendothelial connective tissue. This interaction allows platelets to adhere to areas of vascular injury. The second role of vWF is to bind to and stabilize factor VIII allowing factor VIII to survive in the blood. As a consequence of these roles for vWF, deficiencies result in defective platelet adhesion and to secondary deficiencies in factor VIII. Since factor VIII deficiency results in hemophilia A, it is not surprising that vWF deficiencies can manifest with clinical symptoms that appear to be hemophilia A. The most common form of the vWD (type 1) is an autosomal dominant disorder resulting from quantitative deficiency in vWF. Recessive inheritance of a virtual lack of vWF protein characterizes the clinically severe form of the disease called type 3 vWD. The inheritance of qualitative defects in vWF (dysfunctional protein) is characteristic of type 2 vWD. The type 2 category is further subdivided into 4 variants that are based upon the functional characteristics of the mutant protein. In type 2A vWD the residual vWF has reduced function. In type 2B vWD the mutant vWF has an increased affinity for the GPIb protein of the platelet GPIb-IX-V complex. In type 2M vWD there is the presence of normal vWF multimeric complexes, but they exhibit reduced platelet-dependent function. In type 2N vWD there are factor VIII deficiencies that are the result of mutations in vWF that does not bind to factor VIII. In patients with type 1 and type 3 vWD the severity of the disease is related to the degree of functional deficiency in vWF. Clinically, type 1 and type 3 vWD patients can manifest from insignificant problems to life-threatening complications. Patients with type 2 vWD may exhibit symptoms whose severity may be greater than would be expected based upon a knowledge of the functional defect. Symptoms of vWD often present shortly after birth. The common symptoms are easy bruising, bleeding from the gums, cutaneous hematomas, and prolonged bleeding from cuts and abrasions. Individuals with severe forms of vWD can have bleeding complications because they either lack sufficient vWF to promote platelet adhesion at the site of vascular injury or they are factor VIII deficient due to the lack of the stabilizing action of vWF on factor VIII. Therefore, the therapeutic response to vWD requires an understanding of these distinct functions of vWF. Mucocutaneous and postsurgical bleeding can be treated by raising the level of factor VIII. In order to address the platelet adhesion deficit in vWD, it is necessary to increase the level of functional vWF. This can be accomplished using fresh frozen plasma or cryoprecipitates. The limitation to these approaches is that a large volume of frozen plasma is needed to deliver sufficient amount of vWF and cryoprecipitates are known to be associated with possible disease transmission. Platelet transfusions can be helpful in some patients with vWD, especially those who are refractory to factor VIII-vWF concentrates.
CLINICAL BOX 51-2: BERNARD-SOULIER SYNDROME
Bernard-Soulier syndrome (BSS) is the second most common inherited bleeding disorder that results as a consequence of defects in platelet function. It is characterized by, among other symptoms, the presence of giant platelets. Hence the disorder is also known as giant platelet syndrome. In addition to the presence of giant platelets, patients experience a prolonged bleeding time. Although platelets from these individuals will aggregate in response to the presence of epinephrine, collagen, and/or ADP, they will not aggregate in the presence of ristocetin. Ristocetin is an antibiotic isolated from Amycolatopsis lurida which was formerly used to treat staphylococcal infections. Ristocetin induces binding of vWF to platelet GPIb-IX-V and as a consequence of this activity the compound is used in a diagnostic assay for vWD. Bernard-Soulier syndrome is an autosomal recessive disorder in which most patients have either a decrease or absence in 3 of the 4 proteins of the platelet GPIb-IX-V complex. At least 18 different molecular defects have been identified resulting in Bernard-Soulier syndrome. Mutations in GPIbα result in type A BSS, mutations in GPIbβ result in type B BSS, and mutations in GPIX result in type C BSS. At least 6 missense mutations in the GPIX gene have been identified in BSS patients. No known mutations in GPV are associated with BSS. The clinical manifestations of BSS are similar to those in patients with dysfunctional platelets. These symptoms include mucocutaneous bleeding, gastrointestinal hemorrhage, and purpuric skin bleeding (bleeding from purplish patches on the skin). In female patients there is the additional complication of menorrhagia. Evaluation of BSS is done by analysis of platelet number and size. All patients will have some level of and platelets that are from 3 to 20 times normal size. Bleeding times are increased in BSS but the distinguishing abnormality is the failure of platelet aggregation (agglutination) in the presence of ristocetin. This aggregation defect cannot be corrected by the addition of normal plasma. Treatment of bleeding in BSS is accomplished by local application of pressure, topical thrombin, and platelet transfusion. In female patients hormonal management of menses is important.
CLINICAL BOX 51-3: GLANZMANN THROMBASTHENIA
Glanzmann thrombasthenia (weak platelets) represents a heterogenous group of bleeding disorders. The hallmark of the disease is the failure of platelets to bind fibrinogen and aggregate in the presence of a variety of physiological agonist such as ADP, thrombin, epinephrine, or collagen. Glanzmann thrombasthenia is an autosomal recessive bleeding disorder in which the underlying defect in all patients is an abnormality in the genes encoding either the GPIIb (αIIb) or GPIIIa (β3) proteins of the GPIIb-GPIIIa complex. The platelet GPIIb-GPIIIa complex is the most abundant platelet receptor representing approximately 15% of the total protein on the surface of platelets. Although Glanzmann thrombasthenia is a rare disorder, it is the most recognized inherited disorder of platelet function. The molecular abnormalities causing Glanzmann thrombasthenia range from major gene deletions and inversions to single point mutations. These mutations have been grouped according to the biochemical consequences and include transcriptional and protein functional defects. Among the functional defects there are protein stability mutants, glycoprotein complex maturation mutants, and ligand-binding mutants. The clinical manifestations of Glanzmann thrombasthenia are significant mucocutaneous bleeding that is evidenced at an early age. Lifelong repeated bleeding occurs at sites of minor trauma such as the gums (gingival) and the nose (epistaxis). In females heavy bleeding during menstruation may require therapeutic intervention. Severe intracranial or gastrointestinal hemorrhage can occur spontaneously (ie, unprovoked by injury) and is a significant cause of mortality in 5% to 10% of Glanzmann thrombasthenia patients. Treatment of the disorder is limited to local measures such as the application of pressure. Platelet transfusion may be useful but only on a limited basis as resistance will develop to the infused platelets.
These 2 kallikreins are serine proteases whose major substrates are HMWK (plasma kallikrein) and LMWK (tissue kallikrein). HMWK is considered an α-globulin and is composed of 6 functional domains. The protein circulates in the plasma as single-chain polypeptide. The heavy chain contains domains 1, 2, and 3 and the light chain comprises domains 5 and 6. The heavy and light chains are linked together through domain 4 which also contains the bradykinin sequence. Domain 1 contains a low-affinity calcium-binding site. Domains 2 and 3 inhibit cysteine proteases. Domain 3 also has platelet and endothelial cell–binding activity. Domain 5 has sequences for heparin binding and antiangiogenic properties. The binding of HMWK to negatively charged surfaces occurs through a histidine region of the light chain which is in domain 5. Domain 6 contains the prekallikrein and factor XI-binding sites. By being able to bind to charged surfaces via domain 5 and simultaneously binding factor XI and prekallikrein via domain 6, HMWK can serve as the cofactor in activation of the intrinsic pathway which is referred to as contact activation.
High-Yield Concept
In the presence of Ca2+, factor XIa activates factor IX to factor IXa. Active factor IXa cleaves factor X leading to its activation to factor Xa. The activation of factor Xa requires assemblage of the tenase complex (Ca2+ and factors VIIIa, IXa, and X) on the surface of activated platelets.
The plasma kinin forming system is composed of factor XII, and XI, prekallikrein, and HMWK. Factor XII, prekallikrein, and HMWK reversibly bind to endothelial cells, platelets, and granulocytes. When plasma makes contact with a negatively charged surface factor XII binds and is autoactivated to factor XIIa (the “a” signifies the activated factor). Factor XIIa then activates prekallikrein to kallikrein and kallikrein cleaves HMWK releasing bradykinin. There is also reciprocal activation of factor XII by kallikrein resulting in amplification of the system. Several physiologic substances serve as the charged surface to which the contact system binds. These substances include phospholipids, hematin (hydroxide of heme), fatty acids, sodium urate crystals, protoporphyrin, sulfatides, heparins, chondroitin sulfates, articular cartilage, endotoxin, L-homocysteine, and amyloid β-protein. Contact activation also occurs via lipoproteins, principally oxidized LDL (oxLDL). This is the basis of the role of hyperlipidemia in the promotion of a prothrombotic state and the development of atherosclerosis (see Chapter 48).
One of the responses of platelets to activation is the presentation of phosphatidylserine (PS) and phosphatidylinositol (PI) on their surfaces. The exposure of these phospholipids allows the tenase complex to form. The role of factor VIII in this process is to act as a receptor, in the form of factor VIIIa, for factors IXa and X. The activation of factor VIII to factor VIIIa (the actual receptor) occurs in the presence of minute quantities of thrombin. As the concentration of thrombin increases, factor VIIIa is ultimately cleaved by thrombin and inactivated. This dual action of thrombin, upon factor VIII, acts to limit the extent of tenase complex formation and thus the extent of the coagulation cascade.
Extrinsic Clotting Cascade
The extrinsic pathway is initiated at the site of injury in response to the release of tissue factor (factor III), and thus is also known as the tissue factor pathway. Tissue factor is a cofactor in the factor VIIa-catalyzed activation of factor X. Factor VIIa cleaves factor X to factor Xa in a manner identical to that of factor IXa of the intrinsic pathway. Indeed, activated factor Xa is the site at which the intrinsic and extrinsic coagulation cascades converge. The activation of factor VII occurs through the action of thrombin or factor Xa. The ability of factor Xa to activate factor VII creates a link between the intrinsic and extrinsic pathways. An additional link between the two pathways exists through the ability of tissue factor and factor VIIa to activate factor IX.
A major mechanism for the inhibition of the extrinsic pathway occurs at the tissue factor-factor VIIa-Ca2+-Xa complex. The protein lipoprotein-associated coagulation inhibitor (LACI) specifically binds to this complex. LACI is also referred to as extrinsic pathway inhibitor (EPI) or tissue factor pathway inhibitor (TFPI) and was formerly named anticonvertin.
Activation of Prothrombin to Thrombin
The common point in both pathways is the activation of factor X to factor Xa. Factor Xa activates prothrombin (factor II) to thrombin (factor IIa). Thrombin, in turn, converts fibrinogen to fibrin (see Figure 51-1).
Factor V is a cofactor in the formation of the prothrombinase complex, similar to the role of factor VIII in tenase complex formation. Like factor VIII activation, factor V is activated to factor Va by means of minute amounts and is inactivated by increased levels of thrombin. Factor Va binds to specific receptors on the surfaces of activated platelets and forms a complex with prothrombin and factor Xa. Factor Xa cleaves prothrombin to thrombin.
High-Yield Concept
The formation of a complex between factor VIIa and tissue factor is believed to be a principal step in the overall clotting cascade. Evidence for this stems from the fact that persons with hereditary deficiencies in the components of the contact phase of the intrinsic pathway do not exhibit clotting problems.
High-Yield Concept
The activation of thrombin occurs on the surface of activated platelets and requires formation of a prothrombinase complex (Figure 51-4). This complex is composed of platelet phospholipids, Ca2+, factors Va and Xa, and prothrombin.

FIGURE 51-4: Formation of the prothrombinase complex and release of active thrombin. Prothrombin (PT) binds to the complex of calcium ion, active factor V (Va), and active factor X (Xa) which then leads to the catalytic release of active thrombin. Reproduced with permission of themedicalbiochemistrypage, LLC.
Thrombin Activation of Fibrin Clot Formation
The major role of thrombin in hemostasis is the catalytic digestion of fibrinogen to fibrin. Fibrinogen (factor I) consists of 3 pairs of polypeptides ([Aα][Bβ][γ])2. The 6 chains are covalently linked near their N-terminals through disulfide bonds (Figure 51-5). The A and B portions of the Aα and Bβ chains comprise the fibrinopeptides A and B, respectively. Thrombin hydrolyses fibrinogen between the fibrinopeptide and the a and b portions of the protein releasing the fibrinopeptides.

FIGURE 51-5: Diagrammatic representation (not to scale) of fibrinogen showing pairs of Aα, Bβ, and γ chains linked by disulfide bonds. (FPA, fibrinopeptide A; FPB, fibrinopeptide B.) Murray RK, Bender DA, Botham KM, Kennelly PJ, Rodwell VW, Weil PA. Harper’s Illustrated Biochemistry. 29th ed. New York: McGraw-Hill; 2012.
The release of the fibrinopeptides generates fibrin monomers with a subunit structure (αβg)2. These monomers spontaneously aggregate in a regular array, forming a somewhat weak fibrin clot. In addition to fibrin activation, thrombin converts factor XIII to factor XIIIa, a highly specific transglutaminase that introduces cross-links between the fibrin monomers generating an insoluble fibrin clot (Figure 51-6).

FIGURE 51-6: Formation of a fibrin clot. (A) Thrombin-induced cleavage of Arg-Gly bonds of the Aα and Bβ chains of fibrinogen to produce fibrinopeptides (left-hand side) and the α and β chains of fibrin monomer (right-hand side). (B) Cross-linking of fibrin molecules by activated factor XIII (factor XIIIa). Murray RK, Bender DA, Botham KM, Kennelly PJ, Rodwell VW, Weil PA. Harper’s Illustrated Biochemistry. 29th ed. New York: McGraw-Hill; 2012.
Thrombin also activates thrombin-activatable fibrinolysis inhibitor (TAFI), thereby modulating the rate of fibrin clot dissolution (fibrinolysis). TAFI is also known as carboxypeptidase U (CPU) whose activity leads to removal of C-terminal lysines from partially degraded fibrin. This leads to an impairment of plasminogen activation, thereby reducing the rate of fibrinolysis.
Control of Thrombin Levels
There are 2 principal mechanisms by which thrombin activity is regulated. The predominant mechanism is the regulated conversion of prothrombin to thrombin. The activation of thrombin is also regulated by specific thrombin inhibitors. Antithrombin III is the most important since it can also inhibit the activities of factors IXa, Xa, XIa, and XIIa; plasmin; and kallikrein. The activity of antithrombin III is potentiated in the presence of heparin. Heparin binds to a specific site on antithrombin III, producing an altered conformation of the protein, and the new conformation has a higher affinity for thrombin as well as its other substrates. This effect of heparin is the basis for its clinical use as an anticoagulant. The naturally occurring heparin activator of antithrombin III is present as heparan and heparan sulfate on the surface of vessel endothelial cells. It is this feature that controls the activation of the intrinsic coagulation cascade.
High-Yield Concept
Factor V Leiden is the most common inherited thrombophilia in Caucasian populations of European descent. Overall, 5% of the world population harbors the factor V Leiden mutation. The symptoms of factor V Leiden are deep vein thrombosis (DVT) and pulmonary embolism, both of which can be fatal. In fact, it is estimated that as many as 30% of patients who experience DVT and/or pulmonary embolisms are carriers of the factor V Leiden mutation.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


