CHAPTER OUTLINE
Nitrogen Distribution from Biosphere
The Glutamate Dehydrogenase Reaction
The Glutamine Synthetase Reaction
Removal of Nitrogen from Amino Acids
Nitrogen Homeostasis in the Brain
High-Yield Terms
Aminotransferase: any of a family of enzymes that catalyze the transfer of an amino group between a α-amino acid and an α-keto acid
AST and ALT: aspartate aminotransferase (AST; also called serum glutamate-oxaloacetate aminotransferase, [SGOT]) and alanine transaminase (ALT; also called serum glutamate-pyruvate aminotransferase [SGPT]), the two prevalent liver enzymes whose elevations in the blood have been used as clinical markers of tissue damage
Glucose-alanine cycle: mechanism for skeletal muscle to eliminate nitrogen while replenishing its glucose supply. Glucose oxidation produces pyruvate which can undergo transamination to alanine; the alanine then enters the blood stream and is transported to the liver where it is converted back to pyruvate, which is then a source of carbon atoms for gluconeogenesis
Urea: nitrogen compound composed of 2 amino groups (−NH2) joined by a carbonyl (−C=O) functional group; is the main nitrogen-containing substance in human urine
Kwashiorkor: an acute form of childhood protein-energy malnutrition with adequate caloric intake; characterized by edema, irritability, anorexia, ulcerating dermatoses, and an enlarged liver with fatty infiltrates
Nitrogen Distribution From Biosphere
Humans are totally dependent on other organisms for converting atmospheric nitrogen into forms available to the body. Nitrogen fixation is carried out by bacterial nitrogenases forming reduced nitrogen, NH4+, which can then be used by all organisms to form amino acids (Figure 29-1).

FIGURE 29-1: Overview of the flow of nitrogen in the biosphere. Nitrogen, nitrites, and nitrates are acted upon by bacteria (nitrogen fixation) and plants and we assimilate these compounds as protein in our diets. Ammonia incorporation in animals occurs through the actions of glutamate dehydrogenase and glutamine synthase. Glutamate plays the central role in mammalian nitrogen flow, serving as both a nitrogen donor and nitrogen acceptor. Reproduced with permission of themedicalbiochemistrypage, LLC.
Reduced nitrogen enters the human body as dietary free amino acids, protein, and the ammonia produced by intestinal tract bacteria. A pair of principal enzymes, glutamate dehydrogenase and glutamine synthetase, incorporates this ammonia into carbon skeletons generating the amino acids glutamate and glutamine, respectively. Amino and amide groups from these 2 amino acids are then freely transferred to other carbon skeletons by transamination (Figure 29-2) and transamidation reactions.

FIGURE 29-2: Transamination. The reaction is freely reversible with an equilibrium constant close to unity. Murray RK, Bender DA, Botham KM, Kennelly PJ, Rodwell VW, Weil PA. Harper’s Illustrated Biochemistry, 29th ed. New York: McGraw-Hill; 2012.
Aminotransferases exist for all amino acids except threonine and lysine. The most common compounds involved as a donor/acceptor pair in transamination reactions are glutamate and α-ketoglutarate (2-oxoglutarate), which participate in reactions with many different aminotransferases. Serum aminotransferases AST and ALT have been used as clinical markers of tissue damage, with increasing serum levels indicating an increased extent of damage. Alanine transaminase has an important function in the delivery of skeletal muscle carbon and nitrogen (in the form of alanine) to the liver. In skeletal muscle, pyruvate is transaminated to alanine, thus affording an additional route of nitrogen transport from muscle to liver. In the liver, alanine transaminase transfers the ammonia to α-ketoglutarate and regenerates pyruvate. The pyruvate can then be diverted into gluconeogenesis. This process is referred to as the glucose-alanine cycle (Chapter 13).
The Glutamate Dehydrogenase Reaction
Glutamate dehydrogenase is considered a gateway enzyme of energy and nitrogen homeostasis (Figure 29-3). The enzyme utilizes NAD+ in the direction of nitrogen liberation and NADP+ for nitrogen incorporation. In the forward reaction glutamate dehydrogenase is important in converting free ammonia and α-ketoglutarate to glutamate, forming one of the 20 amino acids required for protein synthesis. On the other hand, the reverse reaction is a key anapleurotic process linking amino acid metabolism with TCA cycle activity. In the reverse reaction, glutamate dehydrogenase provides an oxidizable carbon source used for the production of energy as well as a reduced electron carrier, NADH.

FIGURE 29-3: Glutamate dehydrogenase reaction. Reproduced with permission of themedicalbiochemistrypage, LLC.
As expected for a branch point enzyme with an important link to energy metabolism, glutamate dehydrogenase is regulated by the cell energy charge. ATP and GTP are positive allosteric effectors of the formation of glutamate, whereas ADP and GDP are positive allosteric effectors of the reverse reaction. Thus, when the level of ATP is high, conversion of glutamate to α-ketoglutarate is limited; when the cellular energy charge is low, glutamate is converted to ammonia and α-ketoglutarate which can then be oxidized in the TCA cycle. Glutamate is also a principal amino donor to other amino acids in subsequent transamination reactions.
The Glutamine Synthetase Reaction
The glutamine synthetase reaction (Figure 29-4) is also important to nitrogen homeostasis in several respects. First it produces glutamine, one of the 20 major amino acids. Second, in animals, glutamine is the major amino acid found in the circulatory system. Its role there is to carry ammonia to and from various tissues but principally from peripheral tissues to the kidney, where the amide nitrogen is hydrolyzed by the enzyme glutaminase (Figure 29-5); this process regenerates glutamate and free ammonium ion, which is excreted in the urine. Note that, in this function, ammonia arising in peripheral tissues is transported in the blood in a nonionizable form which has none of the neurotoxic or alkalosis-generating properties of free ammonia.

FIGURE 29-4: The glutamine synthase reaction strongly favors glutamine synthesis. Murray RK, Bender DA, Botham KM, Kennelly PJ, Rodwell VW, Weil PA. Harper’s Illustrated Biochemistry, 29th ed. New York: McGraw-Hill; 2012.

FIGURE 29-5: The glutaminase reaction proceeds essentially irreversibly in the direction of glutamate and NH4+ formation. Note that the amide nitrogen, not the α-amino nitrogen, is removed. Murray RK, Bender DA, Botham KM, Kennelly PJ, Rodwell VW, Weil PA. Harper’s Illustrated Biochemistry, 29th ed. New York: McGraw-Hill; 2012.
High-Yield Concept
The multiple roles of glutamate in nitrogen homeostasis make it a gateway between free ammonia and the amino groups of most amino acids.
High-Yield Concept
When acidosis occurs the body will divert more glutamine from the liver to the kidney. This allows for the conservation of bicarbonate ion since the incorporation of ammonia into urea requires bicarbonate. When glutamine enters the kidney, glutaminase releases 1 mole of ammonia generating glutamate and then glutamate dehydrogenase releases another mole of ammonia generating α-ketoglutarate. The ammonia will ionize to ammonium ion (NH4+), which is excreted. The net effect is a reduction in the concentration of hydrogen ion [H+], and thus an increase in the pH of the blood.
Liver contains both glutamine synthetase and glutaminase but the enzymes are localized in different cellular segments. This ensures that the liver is neither a net producer nor consumer of glutamine. The differences in cellular locations of these 2 enzymes allow the liver to scavenge ammonia that has not been incorporated into urea. The enzymes of the urea cycle are located in the same cells as those that contain glutaminase. The result of the differential distribution of these 2 hepatic enzymes makes it possible to control ammonia incorporation into urea or glutamine.
Digestive Tract Nitrogen
While glutamate, glutamine, and the remaining nonessential amino acids can be made by animals, the majority of the amino acids found in human tissues come from dietary sources. The details of protein digestion and amino acid uptake by the intestines are covered in Chapter 43. Since humans can neither synthesize the branched carbon chains found in branched chain amino acids or the ring systems found in phenylalanine and the aromatic amino acids nor incorporate sulfur into covalently bonded structures, there are 10 so-called essential amino acids (see Table 30-1) that must be supplied from the diet. However, depending on the composition of the diet and physiological state of an individual, one or another of the nonessential amino acids may also become a required dietary component. For example, arginine is only normally considered to be an essential amino acid during early childhood development because enough for adult needs is made by the urea cycle.
In addition to the amino acids obtained from the diet, many other nitrogenous compounds are found in the intestine. Most of these compounds are bacterial products of protein degradation with some exhibiting powerful pharmacological (vasopressor) effects (Table 29-1).

High-Yield Concept
Normal, healthy adults are generally in nitrogen balance, with intake and excretion being very well matched. Young growing children, adults recovering from major illness, and pregnant women are often in positive nitrogen balance. Their intake of nitrogen exceeds their loss as net protein synthesis proceeds. When more nitrogen is excreted than is incorporated into the body, an individual is in negative nitrogen balance. Insufficient quantities of even one essential amino acid are adequate to turn an otherwise normal individual into one with a negative nitrogen balance.
Nitrogen Balance
Unlike fats and carbohydrates, nitrogen has no designated storage depots in the body. Since the half-life of many proteins is short (on the order of hours), insufficient dietary quantities of even 1 amino acid can quickly limit the synthesis and lower the body levels of many essential proteins. The result of limited synthesis and normal rates of protein degradation is that the balance of nitrogen intake and nitrogen excretion is rapidly and significantly altered.
The biological value of dietary proteins is related to the extent to which they provide all the necessary amino acids. Proteins of animal origin generally have a high biological value; plant proteins have a wide range of values from almost none to quite high. In general, plant proteins are deficient in lysine, methionine, and tryptophan and are much less concentrated and less digestible than animal proteins. The absence of lysine in low-grade cereal proteins, used as a dietary mainstay in many underdeveloped countries, leads to an inability to synthesize protein (because of missing essential amino acids) and ultimately to a syndrome known as kwashiorkor, common among children in these countries.
Removal of Nitrogen from Amino Acids
The dominant reactions involved in removing amino acid nitrogen from the body are known as transaminations (see Figure 29-2). This class of reactions funnels nitrogen from all free amino acids into a small number of compounds; then, either they are oxidatively deaminated, producing ammonia, or their amine groups are converted to urea by the urea cycle. Transaminations involve moving a α-amino group from a donor α-amino acid to the keto carbon of an acceptor α-keto acid. These reversible reactions are catalyzed by a group of intracellular enzymes known as aminotransferases, which generally employ covalently bound pyridoxal phosphate as a cofactor. However, some aminotransferases employ pyruvate as a cofactor.
Aminotransferases exist for all amino acids except threonine and lysine. The most common compounds involved as a donor/acceptor pair in transamination reactions are glutamate and α-ketoglutarate, which participate in reactions with many different aminotransferases.
Alanine transaminase has an important function in the delivery of skeletal muscle carbon and nitrogen to the liver. In skeletal muscle, pyruvate is transaminated to alanine allowing for waste nitrogen from amino acid oxidation to be transported from muscle to liver. In the liver, alanine transaminase transfers the ammonia to α-ketoglutarate and regenerates pyruvate. The pyruvate can then be diverted into gluconeogenesis. This process is referred to as the glucose-alanine cycle (see Figure 13-2).
Because of the participation of α-ketoglutarate in numerous transaminations, glutamate is a prominent intermediate in nitrogen elimination as well as in anabolic pathways. Glutamate, formed in the course of nitrogen elimination, is either oxidatively deaminated by liver glutamate dehydrogenase forming ammonia, or converted to glutamine by glutamine synthetase and transported to kidney tubule cells. There the glutamine is sequentially deamidated by glutaminase and deaminated by kidney glutamate dehydrogenase. The ammonia produced in the latter two reactions is excreted as NH4+ in the urine, where it helps maintain urine pH in the normal range of pH4 to pH8. The extensive production of ammonia by peripheral tissue or hepatic glutamate dehydrogenase is not feasible because of the highly toxic effects of circulating ammonia. Normal serum ammonium concentrations are in the range of 20 to 40 μM, and an increase in circulating ammonia to about 400 μM causes alkalosis and neurotoxicity.
In the peroxisomes of mammalian tissues, especially liver, there exists a minor enzymatic pathway for the removal of amino groups from amino acids. L-amino acid oxidase is FMN-linked and has broad specificity for the L-amino acids (Figure 29-6). A number of substances, including oxygen, can act as electron acceptors from the flavoproteins. If oxygen is the acceptor the product is hydrogen peroxide, which is then rapidly degraded by the catalases found in liver and other tissues. Missing or defective biogenesis of peroxisomes or L-amino acid oxidase is commonly associated with oculogyric crisis (OGC; spasmodic movements of the eyeballs that last several minutes to hours) and causes generalized hyperaminoacidemia and hyperaminoaciduria, generally leading to neurotoxicity and early death.
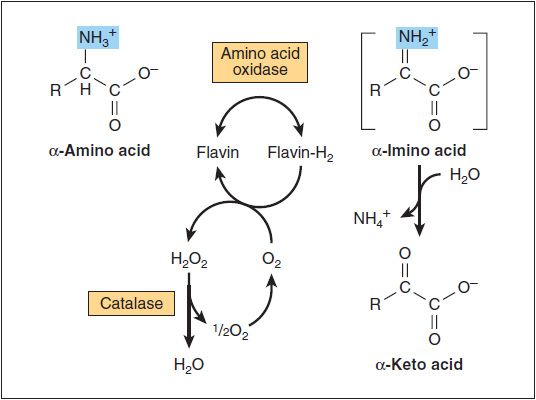
FIGURE 29-6: Oxidative deamination catalyzed by L-amino acid oxidase (L-α-amino acid: O2 oxidoreductase). The α-imino acid, shown in brackets, is not a stable intermediate. Murray RK, Bender DA, Botham KM, Kennelly PJ, Rodwell VW, Weil PA. Harper’s Illustrated Biochemistry, 29th ed. New York: McGraw-Hill; 2012.
High-Yield Concept
Serum aminotransferases such as aspartate aminotransferase (AST; also called serum glutamate-oxaloacetate aminotransferase [SGOT]) and alanine transaminase (ALT; also called serum glutamate-pyruvate aminotransferase [SGPT]) have been used as clinical markers of tissue damage, with increasing serum levels indicating an increased extent of damage.
The Urea Cycle
Although glutamine transport to the kidneys and the concerted actions of glutaminase and glutamate dehydrogenase can support a pathway for the elimination of waste nitrogen, about 80% of the excreted nitrogen is in the form of urea. The formation of urea takes place in the liver, in a series of reactions that are distributed between the mitochondrial matrix and the cytosol. The series of reactions that form urea is known as the urea cycle or the Krebs-Henseleit cycle (Figure 29-7).

FIGURE 29-7: Reactions and intermediates of urea biosynthesis. The nitrogen-containing groups that contribute to the formation of urea are shaded. Reactions ![]() and
and ![]() occur in the matrix of liver mitochondria and reactions
occur in the matrix of liver mitochondria and reactions ![]() ,
, ![]() , and
, and ![]() in liver cytosol. CO2 (as bicarbonate), ammonium ion, ornithine, and citrulline enter the mitochondrial matrix via specific carriers (see red dots) present in the inner membrane of liver mitochondria. Murray RK, Bender DA, Botham KM, Kennelly PJ, Rodwell VW, Weil PA. Harper’s Illustrated Biochemistry, 29th ed. New York: McGraw-Hill; 2012.
in liver cytosol. CO2 (as bicarbonate), ammonium ion, ornithine, and citrulline enter the mitochondrial matrix via specific carriers (see red dots) present in the inner membrane of liver mitochondria. Murray RK, Bender DA, Botham KM, Kennelly PJ, Rodwell VW, Weil PA. Harper’s Illustrated Biochemistry, 29th ed. New York: McGraw-Hill; 2012.
The essential features of the urea cycle reactions are as follows: Arginine from the diet or from protein breakdown is cleaved by the cytosolic enzyme arginase, generating urea and ornithine. In subsequent reactions of the urea cycle a new urea residue is built on the ornithine, regenerating arginine and perpetuating the cycle.
Ornithine, arising in the cytosol, is transported to the mitochondrial matrix via the action of ornithine translocase encoded by the ORNT1 gene. The ORNT1 transporter is a member of the solute carrier family of transporters and as such is also identified as SLC25A15. In the mitochondria ornithine transcabamoylase (OTC) catalyzes the condensation of ornithine with carbamoyl phosphate, producing citrulline. The energy for the reaction is provided by the high-energy anhydride of carbamoyl phosphate.
The carbamoyl phosphate is synthesized from bicarbonate (HCO3−) and ammonium (NH4+) ions via the action of carbamoyl phosphate synthetase I (CPS-I). This reaction is energetically expensive consuming 2 molar equivalents of ATP. Humans express 2 carbamoyl phosphate synthetases: a mitochondrial enzyme, CPS-I, which forms carbamoyl phosphate destined for inclusion in the urea cycle, and a cytosolic synthetase (CPS-II), which is involved in pyrimidine nucleotide biosynthesis.
Concomitant with ornithine transport into the mitochondria is the export of citrulline by SLC25A15 to the cytosol, where the remaining reactions of the cycle take place. Also important in the function of the urea cycle is the mitochondrial transporter called citrin (see Clinical Box 29-1). Citrin is involved in the mitochondrial uptake of glutamate and export of aspartate and as such functions in the malate-aspartate shuttle. Citrin is a Ca2+-dependent mitochondrial solute transporter that is also a member of the solute carrier family of transporters identified as SLC25A13.
CLINICAL BOX 29-1: CITRULLINEMIA
The major form of citrullinemia is the result of a urea cycle defect (Table 29-2). Another form of citrullinemia, with sudden onset between the ages of 11 and 79 years, is called citrullinemia type II (CTLN2). CTLN2 is caused by deficiency in the gene encoding the mitochondria transporter citrin (SLC25A13). Manifestations of CTLN2 are recurrent hyperammonemia with neuropsychiatric symptoms including nocturnal delirium, aggression, irritability, hyperactivity, delusions, disorientation, restlessness, drowsiness, loss of memory, flapping tremor, convulsive seizures, and coma. The symptoms of CTLN2 are often provoked by alcohol and sugar intake, as well as by certain medications, and/or surgery. The clinical course in adults with CTLN2 is milder than that of CTLN1, possibly distinguishing it from milder late-onset CTLN1. It is not known why CTLN2 is milder and later in onset than CTLN1 and distinguishing between the two forms of citrullinemia is difficult. Citrin deficiency can manifest in newborns as neonatal intrahepatic cholestasis caused by citrin deficiency (NICCD) and in older children as failure to thrive and dyslipidemia caused by citrin deficiency (FTTDCD). One of the most common characteristics of citrin deficiency is a fondness for protein-rich and/or lipid-rich foods and aversion to carbohydrate-rich foods. In NICCD children younger than 1 year exhibit growth retardation with transient intrahepatic cholestasis, hepatomegaly, diffuse fatty liver, and parenchymal cellular infiltration associated with hepatic fibrosis, variable liver dysfunction, hypoproteinemia, decreased coagulation factors, hemolytic anemia, and/or hypoglycemia. NICCD is generally not severe and symptoms may often resolve by 1 year of age provided appropriate treatment is given. However, some infants will die due to infection and liver cirrhosis. FTTDCD is usually observed in children around 1 to 2 years of age with the aforementioned food preferences being apparent. Some FTTDCD patients have growth retardation, hypoglycemia, hyperlipidemia, pancreatitis, fatty liver, hepatoma, and fatigue. In some cases of FTTDCD, the affected individual will manifest with characteristic CTLN2 symptoms later in life.
In a 2-step reaction, catalyzed by cytosolic argininosuccinate synthetase, citrulline and aspartate are condensed to form argininosuccinate. The reaction involves the addition of AMP (from ATP) to the amido carbonyl of citrulline, forming an activated intermediate and the subsequent addition of aspartate to form argininosuccinate. Arginine and fumarate are produced from argininosuccinate by the cytosolic enzyme argininosuccinate lyase (also called argininosuccinase). In the final step of the cycle arginase cleaves urea from arginine, regenerating cytosolic ornithine, which can be transported to the mitochondrial matrix for another round of urea synthesis. The fumarate, generated via the action of arginiosuccinate lyase, is reconverted to aspartate for use in the argininosuccinate synthetase reaction. This occurs through the actions of cytosolic versions of the TCA cycle enzymes, fumarase (which yields malate) and malate dehydrogenase (which yields oxaloacetate). The oxaloacetate is then transaminated to aspartate by AST, which can then be used in another round of the urea cycle.
Beginning and ending with ornithine, the reactions of the cycle consume 3 equivalents of ATP and a total of 4 high-energy nucleotide phosphates. Urea is the only new compound generated by the cycle; all other intermediates and reactants are recycled. The energy consumed in the production of urea is more than recovered by the release of energy formed during the synthesis of the urea cycle intermediates. Ammonia released during the glutamate dehydrogenase reaction is coupled to the formation of NADH. In addition, when fumarate is converted back to aspartate, the malate dehydrogenase reaction used to convert malate to oxaloacetate generates a mole of NADH. These two moles of NADH are oxidized in the mitochondria yielding 4 to 6 moles of ATP depending upon the shuttle mechanism employed for NADH transport.
Regulation of the Urea Cycle
The urea cycle operates only to eliminate excess nitrogen. On high-protein diets the carbon skeletons of the amino acids are oxidized for energy or stored as fat and glycogen, but the amino nitrogen must be excreted. To facilitate this process, enzymes of the urea cycle are controlled at the gene level. With long-term changes in the quantity of dietary protein, changes of 20 fold or greater in the concentration of cycle enzymes are observed. When dietary proteins increase significantly, enzyme concentrations rise. On return to a balanced diet, enzyme levels decline. Under conditions of starvation, enzyme levels rise as proteins are degraded and amino acid carbon skeletons are used to provide energy, thus increasing the quantity of nitrogen that must be excreted.
Short-term regulation of the cycle occurs principally at CPS-I, which is relatively inactive in the absence of its allosteric activator N-acetylglutamate. The steady-state concentration of N-acetylglutamate is set by the concentration of its components acetyl-CoA and glutamate and by arginine, which is a positive allosteric effector of N-acetylglutamate synthase (Figure 29-8).

FIGURE 29-8: N-acetylglutamate synthetase reaction. Reproduced with permission of themedicalbiochemistrypage, LLC.
Urea Cycle Disorders
A complete lack of any one of the enzymes of the urea cycle will result in death shortly after birth. However, deficiencies in each of the enzymes of the urea cycle, including N-acetylglutamate synthase, have been identified. These disorders are referred to as urea cycle disorders (UCDs). A common thread to most UCD is hyperammonemia leading to ammonia intoxication. Blood chemistry will also show elevations in glutamine. In addition to hyperammonemia, UCD all present with encephalopathy and respiratory alkalosis. The most dramatic presentation of UCD symptoms occurs in neonates between 24 and 48 hours after birth. Afflicted infants exhibit progressively deteriorating symptoms due to the elevated ammonium levels. Deficiencies in arginase do not lead to symptomatic hyperammonemia as severe or as commonly as in the other UCD. Deficiencies in CPS-I (CPSD), OTC (OTCD), argininosuccinate synthetase (ASD), and argininosuccinate lyase (ALD) comprise the common neonatal UCD.
Clinical symptoms are most severe when the UCD is at the level of CPS-I. Symptoms of the UCD usually arise at birth and encompass ataxia, convulsions, lethargy, poor feeding, and, eventually, coma and death if not recognized and treated properly. In fact, the mortality rate is 100% for UCD that are left undiagnosed. Several UCD manifest with late-onset and in these cases the symptoms are hyperactivity, hepatomegaly, and an avoidance of high-protein foods (Table 29-2).

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


