Definitions
• Antibiotics—antimicrobials of microbial origin, most of which are produced by fungi or by bacteria of the genus Streptomyces.
• Antimicrobials—substances used in the treatment of infectious diseases. In the context of infectious diseases, it implies the agent is not an antibiotic in the strict sense of originating from a bacteria or fungus, but is still used in the treatment of infections.
• Bactericidal—antimicrobial activity that not only inhibits growth but is lethal to bacteria.
• Bacteriostatic—antimicrobial activity that inhibits growth but does not kill the organisms. The host defense mechanisms are ultimately responsible for eradication of infection.
• Minimal inhibitory concentration (MIC)—a laboratory term that defines the lowest concentration (μg/mL) able to inhibit growth of the microorganism in vitro.
• Resistant, nonsusceptible—term applied when organisms are not inhibited by clinically achievable concentrations of an antimicrobial agent.
• Sensitive, susceptible—term applied to microorganisms indicating that they will be inhibited by concentrations of the antimicrobial that can be achieved clinically.
• Spectrum—an expression of the categories of microorganisms against which an antimicrobial is typically active. A narrow-spectrum agent has activity against only a few organisms. A broad-spectrum agent has activity against organisms of diverse types (eg, Gram-positive and Gram-negative bacteria).
 Sources of Antimicrobial Agents
Sources of Antimicrobial Agents
There are three main sources of antimicrobial agents. First are antibiotics, which are molecules of biological origin. They probably play an important part in microbial ecology in the natural environment. Penicillin, for example, is produced by several molds of the genus Penicillium, and the first cephalosporin antibiotics were derived from other molds. Another source of naturally occurring antibiotics is the genus Streptomyces, which are Gram-positive, branching bacteria found in soil and freshwater sediments. Streptomycin, the tetracyclines, chloramphenicol, erythromycin, and many other antibiotics were discovered by screening large numbers of Streptomyces isolates from different parts of the world. Antibiotics are mass produced by techniques derived from the procedures of the fermentation industry.
Antibiotics are synthesized by molds or bacteria
Production in quantity is by industrial fermentation
Second are the chemically synthesized antimicrobial agents. These were initially discovered among compounds synthesized for other purposes and tested for their therapeutic effectiveness in animals. The sulfonamides, for example, were discovered as a result of routine screening of aniline dyes. More recently, active compounds have been synthesized with structures tailored to be effective inhibitors or competitors of known metabolic pathways. Trimethoprim, which inhibits dihydrofolate reductase, is an excellent example. “Structure-based drug design” involves the use of X-ray crystallography to understand the three-dimensional molecular conformation of potential drug targets, and then synthesizing small molecules to bind those targets. This technique holds great promise, although relatively few antimicrobials have yet been developed in this manner.
Chemicals with antibacterial activity are discovered by chance, as the result of screening programs, or via intentional molecular drug design
A third source of antimicrobials arises from the molecular manipulation of previously discovered antibiotics to broaden their range and degree of activity against microorganisms or to improve their pharmacologic characteristics. Examples include the development of penicillinase-resistant and broad-spectrum penicillins, as well as a large range of aminoglycosides and cephalosporins of increasing activity, spectrum, and resistance to inactivating enzymes.
Naturally occurring antibiotics can be chemically modified
 Spectrum of Action
Spectrum of Action
The spectrum of activity of each antimicrobial agent describes the genera and species against which it is typically active. See Table 23–1 for the most common antimicrobial agents and bacteria. Spectra overlap but are usually characteristic for each broad class of antimicrobial. Some antibacterial antimicrobials are known as narrow-spectrum agents; for example, benzyl penicillin is highly active against many Gram-positive and Gramnegative cocci but has little activity against enteric Gram-negative bacilli. The tetracyclines, the cephalosporins, and the carbapenems, on the other hand, are broad-spectrum agents that inhibit a wide range of Gram-positive and Gram-negative bacteria, including some obligate intracellular organisms.
TABLE 23–1 Characteristics of Antibacterial Drugs
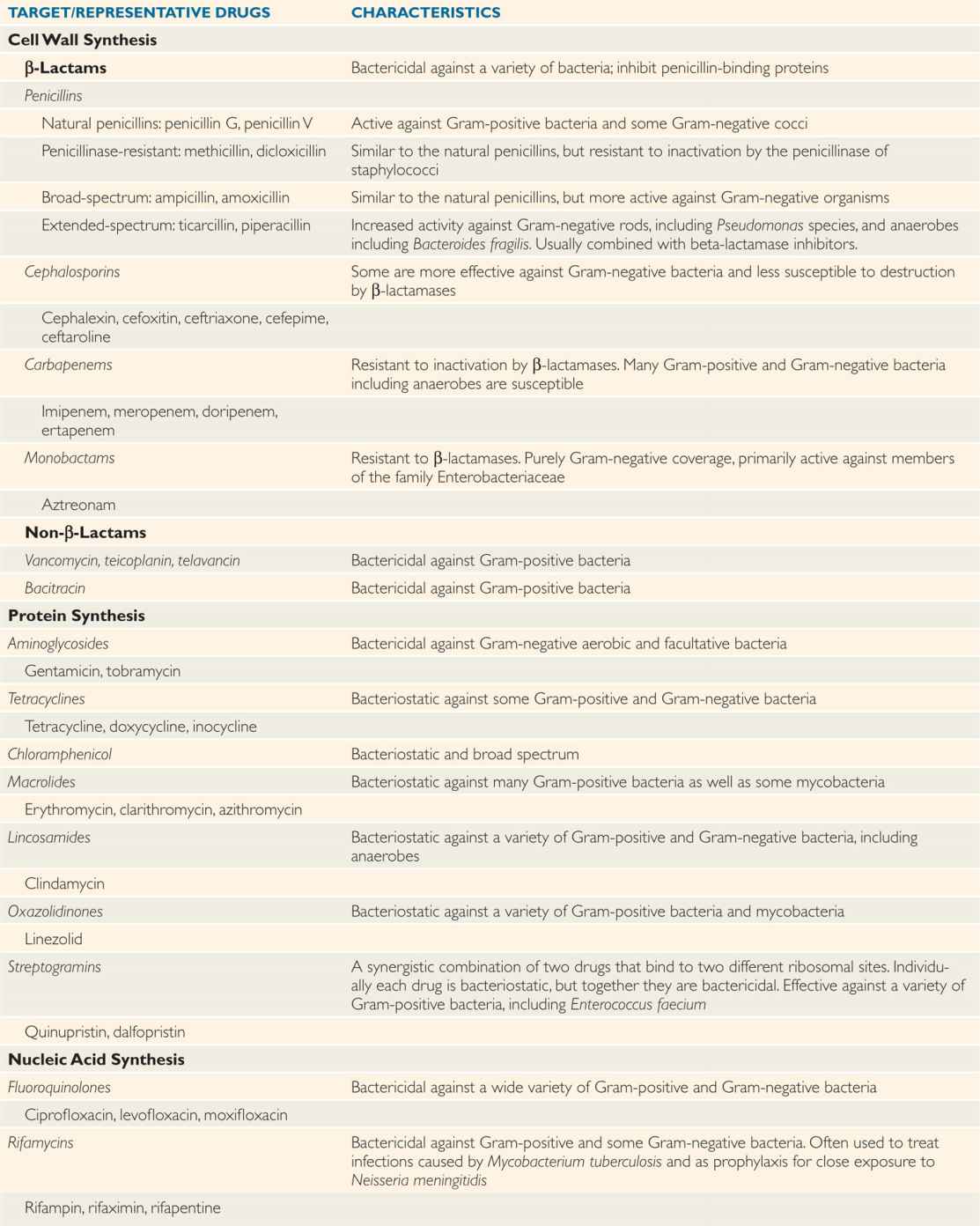

Spectrum is the range of bacteria against which the agent is typically active
Broad-spectrum agents inhibit both Gram-positive and Gram-negative species
SELECTED ANTIBACTERIAL AGENTS
The major antimicrobials are now considered in more detail, with emphasis on their modes of action and spectrum. Details on specific antimicrobial agent use, dosage, and toxicity should be sought in a specialized text or handbook written for that purpose.
 Antimicrobials That Act on Cell Wall Synthesis
Antimicrobials That Act on Cell Wall Synthesis
The peptidoglycan component of the bacterial cell wall provides its shape and rigidity. This giant molecule is formed by weaving the linear glycans N-acetylglucosamine and N-acetylmuramic acid into a basket-like structure. Mature peptidoglycan is held together by cross-linked short peptide side chains hanging off the long glycan molecules. This cross-linking process is the target of two of the most important groups of antimicrobials, the β-lactams and the glycopeptides (vancomycin and teicoplanin) (Figure 23–1). Peptidoglycan is unique to bacteria and its synthesis is described in more detail in Chapter 21.
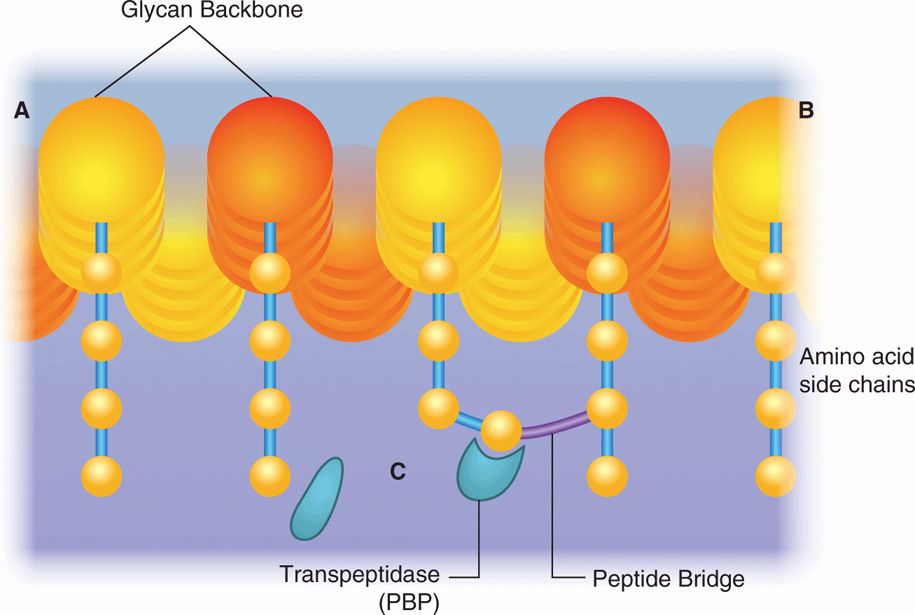
FIGURE 23–1. Action of antimicrobials on peptidoglycan synthesis. the glycan backbone and the amino acid side chains of peptidoglycan are shown. The transpeptidase enzyme catalyzes the cross-linking of the amino acid side chains. penicillin and other β-lactams bind to the transpeptidase, preventing it from carrying out its function. Vancomycin binds directly to the amino acids, preventing the binding of transpeptidase.
Cross-linking of peptidoglycan is the target of β-lactams and glycopeptides
β-Lactam Antimicrobials
The β-lactam antimicrobial agents comprise the penicillins, cephalosporins, carbapenems, and monobactams. They are named after the β-lactam ring in their structure; this ring is essential for antibacterial activity. Penicillin, the first member of this class, was derived from molds of the genus Penicillium. Later β-lactams were derived from both molds and bacteria of the genus Streptomyces. Today it is possible to synthesize β-lactams, but most are derived from semisynthetic processes involving chemical modification of the products of fermentation.
A β-lactam ring is part of the structure of all β-lactam antimicrobics
The β-lactam antibacterial agents interfere with the transpeptidation reactions that seal the peptide crosslinks between glycan chains. They do so by interference with the action of the transpeptidase enzymes which carry out this cross-linking. These targets of all the β-lactams are commonly called penicillin-binding proteins (PBPs). Several distinct PBPs occur in any one strain, are usually species specific, and vary in their avidity of binding to different β-lactam drugs.
β-Lactams interfere with peptidoglycan cross-linking by binding to transpeptidases called PBPs
The β-lactams are classified by chemical structure (Figure 23–2). They may have one β-lactam ring (monobactams), or a β-lactam ring fused to a five-member thiazolidine penem ring (penicillins, carbapenems) or a six-member dihydrothiazine cephem ring (cephalosporins). Within these major groups, differences in the side chain(s) attached to the single or double ring can have a significant effect on the drug’s pharmacologic properties and spectrum. These properties include resistance to gastric acid, which allows oral administration and their pattern of distribution into body compartments (eg, blood, cerebrospinal fluid, joints). The features that alter their spectrum include permeability into the bacterial cell, affinity for PBPs, and vulnerability to the various bacterial mechanisms of resistance.
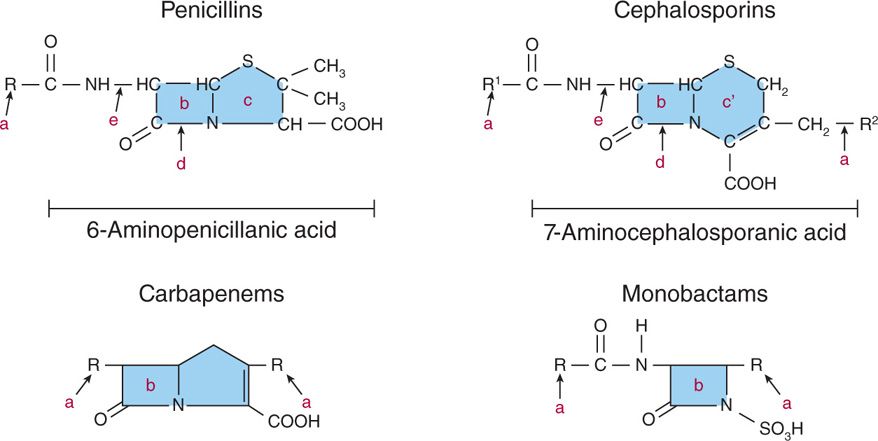
FIGURE 23–2. Structure of β-lactam antibiotics. a. Different side chains determine degree of activity, spectrum, pharmacologic properties, resistance to β-lactamases; b. β-lactam ring; c. thiazolidine ring; c’ dihydrothiazine ring; d. site of action of β-lactamases; e. site of action of amidase.
Penicillins, cephalosporins, monobactams, and carbapenems differ in terms of the structures fused to the β-lactam ring
β-Lactam antimicrobials are usually highly bactericidal, but only to growing bacteria synthesizing new cell walls. Killing involves attenuation and disruption of the developing peptidoglycan “corset,” liberation or activation of autolytic enzymes that further disrupt weakened areas of the wall, and finally osmotic lysis due to passage of water through the cytoplasmic membrane to the hypertonic interior of the cell. As might be anticipated, cell wall-deficient organisms, such as Mycoplasma, are not susceptible to β-lactam antimicrobials.
β-Lactam antimicrobics kill growing bacteria by lysing weakened cell walls
Penicillins. Penicillin G is the oldest penicillin. It remains active primarily against certain Gram-positive organisms, Gram-negative cocci, and some spirochetes, including Treponema pallidum, the cause of syphilis. It has little action against most Gram-negative bacilli, because their outer membrane prevents passage of these antibiotics to their sites of action on cell wall synthesis. Penicillin G is the least toxic of the penicillins. Its modification as penicillin V confers acid stability, so it can be given orally.
Penicillin’s penetration of outer membrane is often limited
Three major strategies in drug development have allowed penicillins to remain an important antibiotic class. First, semisynthetic penicillins were developed to cope with staphylococcal penicillinase. This penicillinase is one of a family of bacterial enzymes called β-lactamases that inactivate β-lactam antimicrobials. The penicillinase-resistant penicillins (methicillin, nafcillin, oxacillin) have narrow spectra, but are active against penicillinase-producing S aureus (although methicillin is no longer in use, these bacteria are still commonly referred to as “methicillin-susceptible S aureus, or “MSSA”).
Second, a group of broader spectrum penicillins was created, which owe their expanded activity to their ability to traverse the outer membrane of some Gram-negative bacteria, and in some cases to their resistance to hydrolysis by Gram-negative β-lactamases. Some, such as the aminopenicillins ampicillin and amoxicillin, have excellent activity against a range of Gram-negative pathogens but not against P aeruginosa, an important opportunistic pathogen. Others, such as the ureidopenicillins piperacillin and ticarcillin, are active against Pseudomonas when given in high dosage but are less active than ampicillin against some other Gram-negative organisms. These penicillins with a Gram-negative spectrum are slightly less active than penicillin G against Gram-positive organisms and are inactivated by staphylococcal penicillinase.
Resistance to staphylococcal and Gram-negative β-lactamases determines spectrum
Some penicillins are inactivated by staphylococcal penicillinase
Finally, in order to combat bacterial β-lactamases, penicillins are sometimes dosed with β-lactamase inhibitors.
Broad-spectrum penicillins penetrate the outer membrane of some Gram-negative bacteria
Cephalosporins. The structure of the cephalosporins confers resistance to hydrolysis by staphylococcal penicillinase and to varying degrees the β-lactamases of groups of Gram-negative bacilli. The cephalosporins are classified by generation—first, second, third, fourth, or fifth. The “generation” term relates to historical breakthroughs in expanding their spectrum through modification of the side chains. In general, a cephalosporin of a higher generation has a wider spectrum, and in some instances, more quantitative activity (lower MIC) against Gram-negative bacteria. As the Gram-negative spectrum increases, these agents typically lose some of their potency (higher MIC) against Gram-positive bacteria.
Cephalosporins are penicillinase resistant
Shifting between first- and third-generation cephalosporins gives a wider Gram-negative spectrum
The first-generation cephalosporins cefazolin and cephalexin have a spectrum of activity against Gram-positive organisms that resembles that of the penicillinase-resistant penicillins. In addition, they are active against some of the Enterobacteriaceae (see Table 13–1). These agents continue to have therapeutic value because of their high activity against Gram-positive organisms, because they are well tolerated, and because a broader spectrum is unnecessary in many infections due to MSSA and streptococci.
Second- and third-generation cephalosporins have less activity against Gram-positive bacteria
First-generation cephalosporins inhibit many Gram-positive bacteria and a few Enterobacteriaceae
Second-generation cephalosporins such as cefoxitin and cefaclor are resistant to β-lactamases of some Gram-negative organisms that inactivate first-generation compounds. Of particular importance is their expanded activity against Enterobacteriaceae species, although in theory this comes at the cost of reduced effectiveness against certain Gram positives.
Second-generation cephalospo-rins have improved coverage of Enterobacteriaceae
Third-generation cephalosporins, such as ceftriaxone, cefotaxime, and ceftazidime, have an even wider spectrum; they are active against Gram-negative organisms, often at MICs that are 10- to 100-fold lower than first-generation compounds. Of these three agents, only ceftazidime is active against P aeruginosa. The potency, broad spectrum, and low toxicity of the third-generation cephalosporins have made them preferred agents in life-threatening infections in which the causative organism has not yet been isolated. Selection depends on the clinical circumstances. For example, ceftriaxone or cefotaxime are preferred for meningitis because they have the highest activity against the three major causes, Neisseria meningitidis, Streptococcus pneumoniae, and Haemophilus influenzae. For a febrile stem cell transplant patient, ceftazidime might be chosen because of the possibility of P aeruginosa involvement.
Third-generation cephalosporins have increasing potency against Gram-negative organisms
Ceftriaxone and cefotaxime are preferred for meningitis
Ceftazidime is used for Pseudomonas
Fourth-generation cephalosporins have enhanced ability to cross the outer membrane of Gram-negative bacteria as well as resistance to many Gram-negative β-lactamases. Compounds such as cefepime have activity against a wider spectrum of Enterobacteriaceae as well as P aeruginosa. These cephalosporins retain the high affinity of third-generation drugs and activity against Neisseria and H influenzae. In effect, these drugs can be conceptualized as having the activity of ceftriaxone plus ceftazidime.
Fourth-generation cephalosporins have enhanced ability to penetrate outer membrane
Fifth-generation cephalosporins such as ceftaroline are defined by their unique ability to bind avidly to PBP-2A, the altered penicillin-binding protein that confers resistance to other β-lactam antibiotics in methicillin-resistant S aureus (MRSA). See Chapter 25 for information on MRSA. Fifth-generation cephalosporins retain some activity against Enterobacteriaceae, although they should be thought of primarily as anti-Gram-positive agents.
Fifth-generation cephalosporins have the unique ability to kill MRSA
Carbapenems. The carbapenems imipenem, meropenem, and doripemen have the broadest spectrum of all β-lactam antibiotics. This fact appears to be due to the combination of easy penetration of Gram-negative and Gram-positive bacterial cells and high level of resistance to β-lactamases. All three agents are active against streptococci, retain some antistaphylococcal activity, and are highly active against both β-lactamase-positive and -negative strains of gonococci and H influenzae. In addition, they are as active or more active then third-generation cephalosporins against Gram-negative rods. They are highly effective against obligate anaerobes such as Bacteroides fragilis. A closely related drug, ertapenem, is ineffective against Pseudomonas, but is otherwise similar. Imipenem is the carbapenem of choice against Gram-positive pathogens, but it is rapidly hydrolyzed by a renal tubular dehydropeptidase-1; therefore, it is administered together with an inhibitor of this enzyme (cilastatin), which greatly improves its urine levels and other pharmacokinetic characteristics. Meropenem, doripenem, and ertapenem are not significantly degraded by dehydropeptidase-1 and do not require coadministration of cilastatin.
Carbapenems have very broad spectra
Monobactams. Aztreonam, the first monobactam licensed in the United States, has a spectrum limited to aerobic and facultatively anaerobic Gram-negative bacteria, including Enterobacteriaceae, P aeruginosa, Haemophilus, and Neisseria. Monobactams have poor affinity for the PBPs of Gram-positive organisms and strict anaerobes and thus demonstrate little activity against them. However, they are highly resistant to hydrolysis by β-lactamases of Gram-negative bacilli. Anaerobic superinfections and major distortions of the bowel flora are less common with aztreonam therapy than with other broad-spectrum β-lactam antimicrobials, presumably because aztreonam does not produce a general suppression of gut anaerobes.
Monobactam activity is exclusively against Gram-negatives
β-Lactamase Inhibitors. A number of β-lactams with little or no antimicrobial activity are capable of binding irreversibly to β-lactamase enzymes and, in the process, rendering them inactive. Three such compounds, clavulanic acid, sulbactam, and tazobactam, are referred to as suicide inhibitors, because they must first be hydrolyzed by a β-lactamase before becoming effective inactivators of the enzyme. They are highly effective against staphylococcal penicillinases and broad-spectrum β-lactamases; however, their ability to inhibit cephalosporinases is significantly less. Combinations of one of these inhibitors with an appropriate β-lactam antimicrobial agent protects the therapeutic agent from destruction by many β-lactamases and significantly enhances its spectrum. Four such combinations are now available in the United States: amoxicillin/clavulanate, ticarcillin/clavulanate, ampicillin/sulbactam, and piperacillin/tazobactam. Bacteria that produce chromosomally encoded inducible cephalosporinases are not susceptible to these combinations. Whether these combinations offer therapeutic or economic advantages compared with the β-lactamase-stable antibiotics now available remains to be determined. See the following section on enzymatic inactivation as a resistance mechanism for more information on β-lactam resistance.
β-Lactamase inhibitors are β-lactams that bind β-lactamases
Penicillin activity is enhanced in the presence of β-lactamase inhibitors
Clinical Use. The β-lactam antibiotics are usually the drugs of choice for infections caused by susceptible organisms because of their bactericidal action and low toxicity. They also have great value in the prevention of many infections, such as surgical site infections. Most are excreted by the kidney and achieve high urinary levels. Penicillins reach the cerebrospinal fluid when the meninges are inflamed and are effective in the treatment of meningitis, although first- and second-generation cephalosporins are not. In contrast, the third-generation cephalosporins penetrate the blood-brain barrier much better and have become the agents of choice in the treatment of undiagnosed meningitis and meningitis caused by most Gram-negative organisms.
Low toxicity favors use of β-lactams
Glycopeptide Antimicrobials
Two agents, vancomycin and teicoplanin, belong to this group. Each of these antimicrobials inhibits assembly of the linear peptidoglycan molecule by binding directly to the terminal amino acids of the peptide side chains. The effect is the same as with β-lactams: prevention of peptidoglycan cross-linking. Both agents are bactericidal, but are primarily active only against Gram-positive bacteria. Their main use has been against multiresistant Gram-positive infections including those caused by strains of staphylococci that are resistant to the penicillinase-resistant penicillins and cephalosporins, especially MRSA. Neither agent is absorbed by mouth; this feature allows these medications to be given orally to treat Clostridium difficile infections of the bowel (see Chapter 29). A related drug, telavancin, was created by adding a lipid tail onto a glycopeptide backbone, thus giving it the theoretical advantage of cell membrane activity and cell wall activity; its clinical usefulness remains to be firmly established.
Glycopeptide antimicrobics bind directly to amino acid side chains
 Inhibitors of Protein Synthesis (Figure 23–3)
Inhibitors of Protein Synthesis (Figure 23–3)
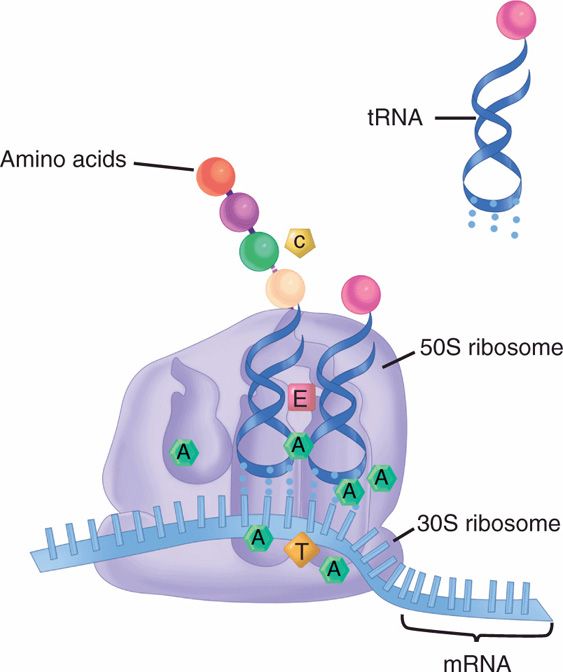
FIGURE 23–3. Action of antimicrobials on protein synthesis. Aminoglycosides (A) bind to multiple sites on both the 30S and 50S ribosomes in a manner that prevents tRNA from forming initiation complexes. Tetracyclines (T) act in a similar manner, binding only to the 30S ribosomes. Chloramphenicol (C) blocks formation of the peptide bond between the amino acids. Erythromycin (E) and macrolides block the translocation of tRNA from the acceptor to the donor side on the ribosome.
Aminoglycosides
All members of the aminoglycoside group of antibacterial agents have a six-member aminocyclitol ring with attached amino sugars. The individual agents differ in terms of the exact ring structure and the number and nature of the amino sugar residues. Aminoglyco-sides are active against a wide range of bacteria, but only those organisms that are able to transport them into the cell by a mechanism that involves oxidative phosphorylation. Thus, they have little or no activity against strict anaerobes or facultative organisms that metabolize only fermentatively (eg, streptococci). It appears highly probable that aminoglycoside activity against facultative organisms is similarly reduced in vivo when the oxidation–reduction potential is low, as in abscesses.
Aminoglycosides must be transported into cell by oxidative metabolism
Not active against anaerobes
Once inside bacterial cells, aminoglycosides inhibit protein synthesis by binding to the bacterial ribosomes either directly or by involving other proteins. This binding destabilizes the ribosomes and blocks initiation complexes, thus preventing the elongation of poly-peptide chains. The agents may also cause distortion of the site of attachment of mRNA, mistranslation of codons, and failure to produce the correct amino acid sequence in proteins. The first aminoglycoside, streptomycin, binds to the 30S ribosomal subunit, but the newer and more active aminoglycosides bind to multiple sites on both 30S and 50S subunits. This gives the newer agents broader spectra and less susceptibility to resistance due to binding site mutation.
Ribosome binding disrupts initiation complexes
Newer agents bind to multiple sites on the ribosome
Eukaryotic ribosomes are resistant to aminoglycosides, and the antimicrobials are not actively transported into eukaryotic cells. These properties account for their selective toxicity and also explain their ineffectiveness against intracellular bacteria such as Rickettsia and Chlamydia.
No entry into human cells
Gentamicin and tobramycin are the major aminoglycosides; they have an extended spectrum, which includes Enterobacteriaceae, and of particular importance, P aeruginosa. They are sometimes beneficial in treating serious infections caused by Gram-positive pathogens such as S aureus and enterococci, although only when used in combination with other drugs. Streptomycin and amikacin are now primarily used in combination with other antimicrobial agents in the therapy of tuberculosis and other mycobacterial diseases. Neomycin, the most toxic aminoglycoside, is used in topical preparations and as an oral preparation before certain types of intestinal surgery, because it is poorly absorbed.
Spectrum includes P aeruginosa
All of the aminoglycosides are toxic to the vestibular and auditory branches of the eighth cranial nerve to varying degrees; this damage can lead to complete and irreversible loss of hearing and balance. These agents may also be toxic to the kidneys. It is often essential to monitor blood levels during therapy to ensure adequate yet nontoxic doses, especially when renal impairment diminishes excretion of the drug.
Renal and vestibular toxicity must be monitored
The clinical value of the aminoglycosides is a consequence of their rapid bactericidal effect, their broad spectrum, and the slow development of bacterial resistance, including retained action against Pseudomonas strains that resist many other drugs. They cause fewer disturbances of the resident microbiota than most other broad-spectrum antimicrobials, probably because of their lack of activity against the predominantly anaerobic flora of the bowel, and because they are only used parenterally for systemic infections. The β-lactam antibiotics often act synergistically with the aminoglycosides, most likely because their action on the cell wall facilitates aminoglycoside penetration into the bacterial cell. This effect is most pronounced with organisms such as streptococci and enterococci, which lack the metabolic pathways required to transport aminoglycosides to their interior.
Broad spectrum and slow development of resistance enhance use
Often combined with β-lactam antimicrobics
Tetracyclines
Tetracyclines are composed of four fused benzene rings. Substitutions on these rings provide differences in pharmacologic features of the major members of the group, tetracycline and doxycycline. The tetracyclines inhibit protein synthesis by binding to the 30S ribosomal subunit at a point that blocks attachment of aminoacyl-tRNA to the acceptor site on the mRNA ribosome complex. Unlike the aminoglycosides, their effect is reversible; they are bacteriostatic rather than bactericidal. A recently developed drug in the related class of glycylcyclines is tigecycline, which covers anaerobes aggressively and thus may be used for treating polymicrobial intraabdominal infections and other complicated deep-tissue infections. Because it is poorly tolerated from a gastrointestinal standpoint, and because of concerns for clinical failure when used for bloodstream infections, this drug’s most useful role may be in the treatment of nontuberculous mycobacterial infections.
Tetracyclines block tRNA attachment
Activity is bacteriostatic
The tetracyclines are broad-spectrum agents with a range of activity that encompass most common pathogenic species, including Gram-positive and Gram-negative rods and cocci and both aerobes and anaerobes. They are active against cell wall-deficient organisms, such as Mycoplasma, and against some obligate intracellular bacteria, including members of the genera Rickettsia and Chlamydia. Differences in spectrum of activity between members of the group are relatively minor. Acquired resistance to one generally confers resistance to all; however, tigecycline appears to overcome the two major resistance mechanisms to other tetracyclines, and thus may be a useful alternative in select cases.
Broad spectrum includes some intracellular bacteria
The tetracyclines are absorbed orally. In practice, they are divided into those agents that generate blood levels for only a few hours (tetracycline) and those that are longer acting (minocycline and doxycycline), which can be administered less often. The tetracyclines are chelated by divalent cations, which reduce their absorption and activity. Thus, they should not be taken with dairy products or many antacid preparations. Tetracyclines are excreted in the bile and urine in active form.
Orally absorbed but chelated by some calcium-rich foods
Tetracycline has a strong affinity for developing bone and teeth, to which it gives a yellowish color and enamel damage, and they are avoided in children up to 8 years of age. This may be less of a significant problem with doxycycline, however. Common complications of tetracycline therapy include photosensitivity, nausea, and esophagitis.
Dental staining and enamel damage to permanent teeth limits use of tetracyclines in children
Chloramphenicol
Chloramphenicol has a simple nitrobenzene ring structure that can be mass produced by chemical synthesis. It influences protein synthesis by binding to the 50S ribosomal subunit and blocking the action of peptidyl transferase, which prevents formation of the peptide bond essential for extension of the peptide chain. Its action is reversible in most susceptible species; thus, it is bacteriostatic. It has little effect on eukaryotic ribosomes, which explains its selective toxicity.
Chloramphenicol blocks peptidyl transferase
Like tetracycline, chloramphenicol is a broad-spectrum antibiotic with a wide range of activity against both aerobic and anaerobic species (see Table 13–1). Chloramphenicol is readily absorbed from the upper gastrointestinal tract and diffuses readily into most body compartments, including the cerebrospinal fluid. It also permeates readily into mammalian cells and is active against obligate intracellular pathogens such as Rickettsia and Chlamydia. It is poorly concentrated in urine.
Diffusion into body fluid compartments occurs readily
The major drawback to this inexpensive, broad-spectrum antimicrobial with almost ideal pharmacologic features is a rare but serious toxicity. Between 1 in 100,000 and 1 in one million patients treated with even low doses of chloramphenicol have an idiosyncratic reaction that results in aplastic anemia. The condition is irreversible and, before the advent of stem cell transplantation, it was universally fatal. In high doses, chloramphenicol also causes a reversible depression of the bone marrow and, in neonates, abdominal, circulatory, and respiratory dysfunction. The inability of the immature infant liver to conjugate and excrete chloramphenicol aggravates this latter condition.
Marrow suppression and aplastic anemia are serious toxicities
In the United States, chloramphenicol use is now restricted to the treatment of rickettsial or ehrlichial infections in which tetracyclines cannot be used because of hypersensitivity or pregnancy. Its central nervous system (CNS) penetration and activity against anaerobes continue to lend support to its use in brain abscess. In some developing countries, chloramphenicol is used more extensively because of its low cost and proven efficacy in diseases such as typhoid fever and bacterial meningitis.
Use is sharply restricted
A related class of drugs, the pleuromutilins, also blocks peptidyl transferase at the 50S ribosomal subunit. Retapamulin is used topically for relatively superficial streptococcal and staphylococcal skin infections.
Retapamulin Ribosomal binding blocks translocation
Macrolides
The macrolides, erythromycin, azithromycin, and clarithromycin, differ in their composition of a large 14- or 15-member ring structure. They affect protein synthesis at the ribosomal level by binding to the 50S subunit and blocking the translocation reaction. Their effect is primarily bacteriostatic. Macrolides, which are concentrated in phagocytes and other cells, are effective against some intracellular pathogens.
Erythromycin, the first macrolide, has a spectrum of activity that includes many pathogenic Gram-positive bacteria and some Gram-negative organisms. Its Gram-negative spectrum includes Neisseria, Bordetella, Campylobacter, and Legionella, but not the Enterobacteriaceae. Erythromycin and related drugs are also effective against Chlamydia and Mycoplasma.
Erythromycin is active against Gram-positives and Legionella
Bacteria that have developed resistance to erythromycin are usually resistant to the newer macrolides azithromycin and clarithromycin as well. These newer agents have the same spectrum as erythromycin, with some significant additions. Azithromycin has quantitatively greater activity (lower MICs) against most of the same Gram-negative bacteria. Clarithromycin is the most active of the three against both Gram-positive and Gramnegative pathogens, and it is also active against mycobacteria. In addition, both azithro-mycin and clarithromycin have demonstrated efficacy against Borrelia burgdorferi, the causal agent of Lyme disease and the protozoan parasite Toxoplasma gondii, which causes toxoplasmosis. Both azithromycin and clarithromycin may have undesirable side effects, including GI upset and cardiac arrhythmias. A related drug, telithromycin, belongs to the ketolide class; it is less susceptible to bacterial resistance mechanisms, but has been associated with liver toxicity.
Azithromycin and clarithromycin have enhanced Gram-negative spectrum
Clindamycin
Clindamycin is a lincosamide, chemically unrelated to the macrolides but with a similar mode of action and spectrum. It has greater activity than the macrolides against Gram-negative anaerobes, including the important Bacteroides fragilis group. Although clindamycin is a perfectly adequate substitute for a macrolide in many situations, its primary use is in instances where anaerobes are or may be involved. In addition, there is experimental evidence that clindamycin may mitigate toxin production by highly virulent Staphylococcus aureus and Streptococcus pyogenes strains. For this reason, many clinicians add it to a bactericidal agent such as nafcillin or vancomycin for treatment of serious deep-tissue infections caused by these organisms.
Spectrum is similar to macrolides with addition of anaerobes
May mitigate toxin production
Oxazolidinones
Linezolid is the most widely used of a new class of antibiotics that act by binding to the bacterial 50S ribosome of Gram-positive organisms, and many mycobacteria and anaerobes. It does not cover Gram-negatives. Oxazolidinones are clinically useful in pneumonia and other soft tissue infections, particularly those caused by resistant strains of staphylococci, pneumococci, and enterococci. Risk of bone marrow suppression is notorious for linezolid, especially when dosed for more than 2 weeks. Optic and peripheral neuropathy have been reported. It also acts as a monoamine oxidase inhibitor, and thus may precipitate a systemic reaction called the serotonin syndrome when given to patients simultaneously taking antidepressants.
Activity against Gram-positive bacteria resistant to other agents
Streptogramins
Quinupristin and dalfopristin are used in a synergistic combination known as synercid. They inhibit protein synthesis by binding to different sites on the 50S bacterial ribosome of certain Gram positives, including MRSA and vancomycin-resistant enterococci (VRE); quinupristin inhibits peptide chain elongation, and dalfopristin interferes with peptidyl transferase. Muscle pain is a common and often-limiting side effect. Their clinical use thus far has been limited generally to the treatment of VRE.
Useful against vancomycin-resistant enterococci
 Inhibitors of Nucleic Acid Synthesis (Figure 23–4)
Inhibitors of Nucleic Acid Synthesis (Figure 23–4)
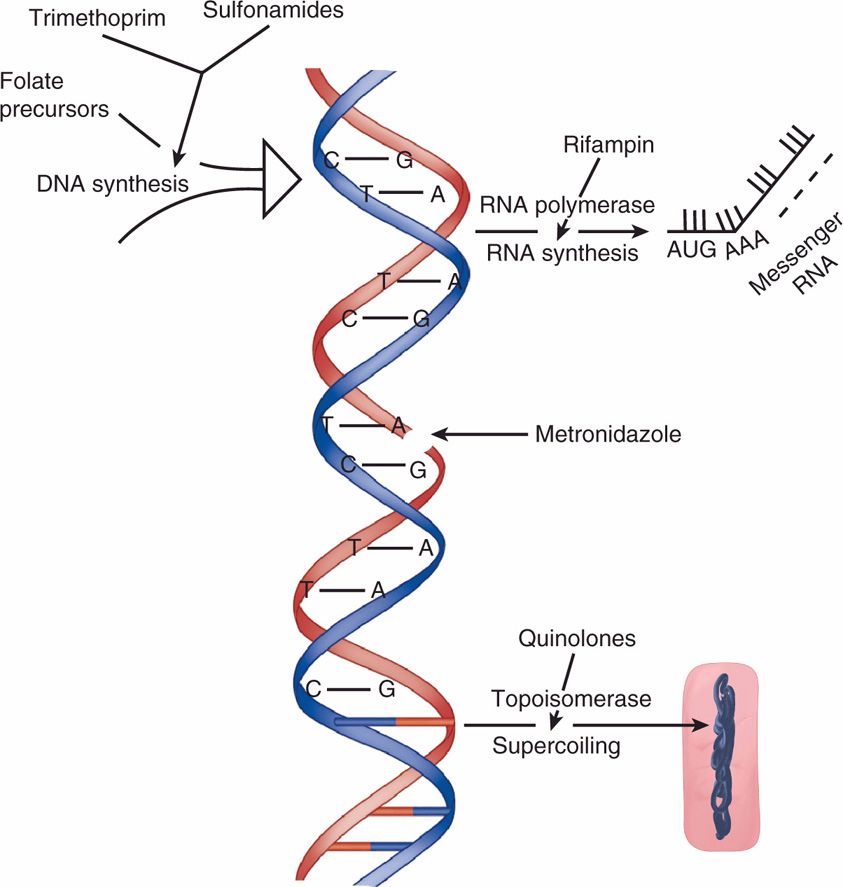
FIGURE 23–4. Antimicrobials acting on nucleic acids. Sulfonamides block the folate precursors of DNA synthesis, metronidazole inflicts breaks in the DNA itself, rifampin inhibits the synthesis of RNA from DNA by inhibiting RNA polymerase, and quinolones inhibit DNA topoisomerase and thus prevent the supercoiling required for the DNA to “fit” inside the bacterial cell.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


