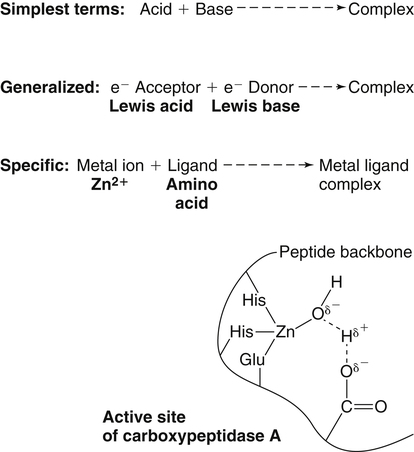
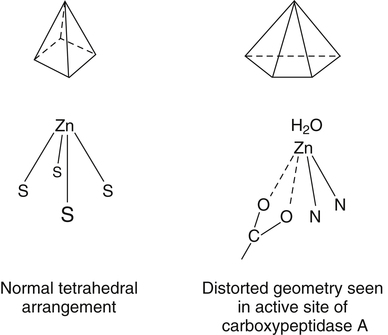
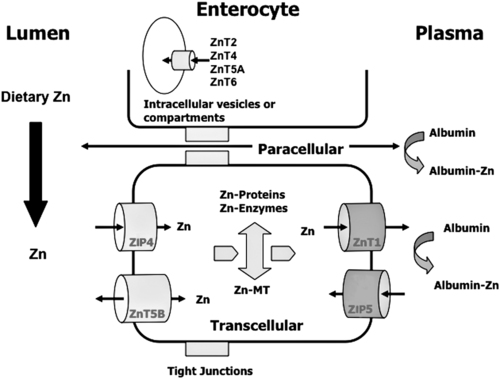
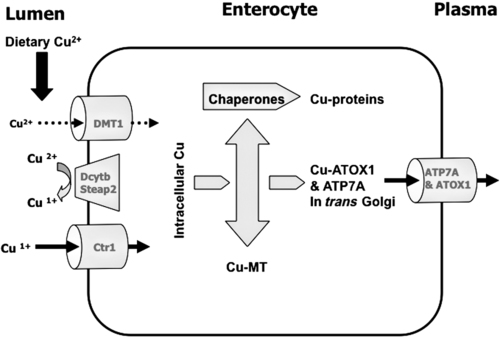
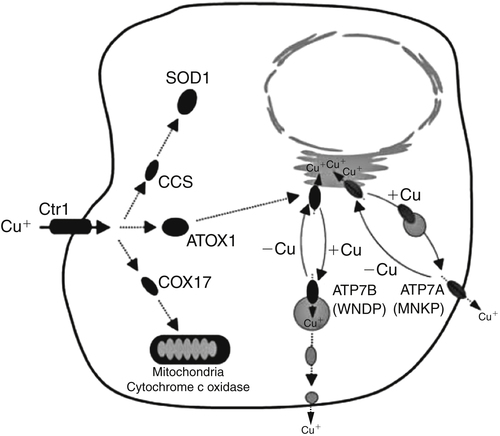
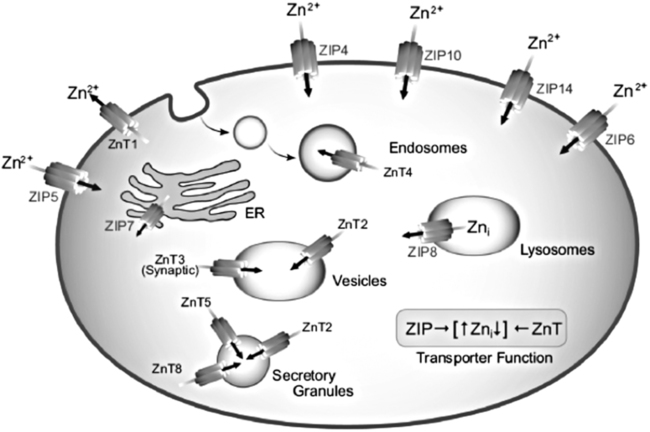
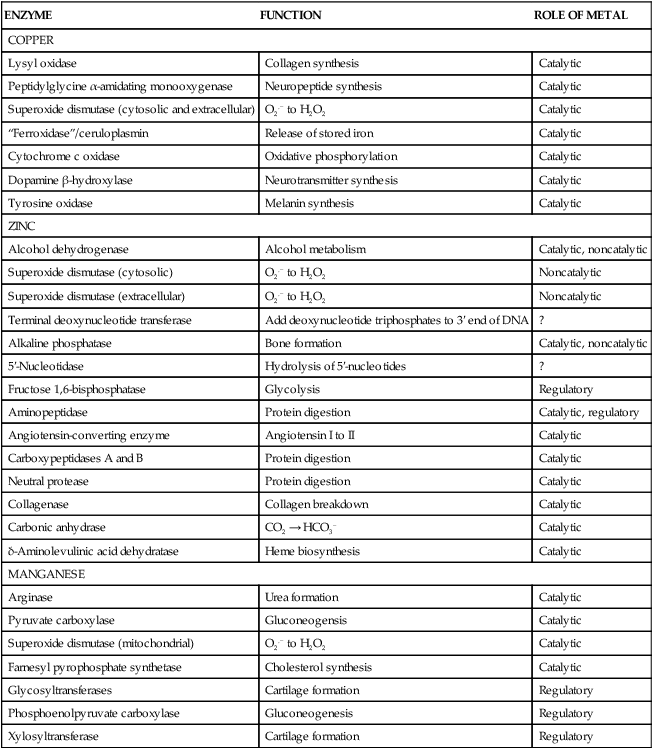
Trace elements function in the body as components of enzymes and proteins involved in various biochemical pathways. Manganese is essential for certain enzymes involved in urea formation, carbohydrate metabolism, cartilage formation, and protection from reactive oxygen species. Copper is associated with numerous enzyme systems, such as those involved in collagen formation, neuropeptide and neurotransmitter synthesis, oxidative phosphorylation, iron metabolism, and protection from reactive oxygen species. Zinc is necessary for the activity of a number of enzymes, including those involved in alcohol metabolism, DNA metabolism, protein metabolism, glycolysis, bone formation, protection from reactive oxygen species, and signal transduction. Clearly, these trace elements, though present in relatively small concentrations, play a major role in maintaining our health and well-being. Zinc (Zn), copper (Cu), and manganese (Mn) are found within the periodic table as transition metals, defined as those elements containing d or f orbitals that are progressively filled with electrons. Manganese and copper contain partially filled d orbitals, whereas the d orbitals of zinc are completely filled. Zinc, copper, and manganese function as electron-pair acceptors (Lewis acids). In biological systems, the electron-pair donors (Lewis bases) are amino acids or water (Figure 37-1). A partial list of enzymes dependent on these minerals is contained in Table 37-1. Because more than 200 zinc-containing metalloenzymes with at least 20 distinct biological functions have been identified in various species, the metalloenzyme function is particularly associated with zinc. However, the metalloenzyme function is central to our understanding of the biology of copper, manganese, and zinc, and the loss of specific metalloenzyme function may account for the symptoms associated with deficiencies of these three metals. TABLE 37-1 Vertebrate Enzymes Containing or Activated by Copper, Zinc, or Manganese There are four biological roles in which metals function: signaling, structural, catalytic, and regulatory. The last three roles relate directly to the functions of metals in proteins and enzyme systems. What makes zinc, copper, and manganese so useful in enzyme systems? Some general guidelines that can be followed to assess the likelihood that a mineral will fit a particular biological role include (1) the charge of the ion (determines the stability and reactivity of the metal in an enzyme), (2) the size of the atom (limits the sites a metal can fit), and (3) the natural abundance of a metal and its location within a cell (e.g., cytosolic versus extracellular will define the likelihood of incorporation into specific enzymes) (Glusker, 1991). When the chemical features of zinc are examined, it becomes apparent why this metal is so prevalent in proteins and enzyme systems. First, with the exception of potassium (K+) and magnesium (Mg2+), zinc is the most common intracellular metal ion. It is found in the cytosol, in vesicles and organelles, and in the nucleus. Therefore it is in the correct proximity to be incorporated into many cellular enzymes. Next, zinc’s flexible coordination geometry makes it ideal for the active site of enzymes. One hypothesis regarding metalloenzymes is that the active site of metalloenzymes is “poised for catalysis,” a condition called the entatic state (Vallee and Galdes, 1984). Researchers have defined the entatic state as the condition in which the geometry of the metal binding site in an enzyme is distorted and asymmetrical (Figure 37-2). When this strain is released by allowing the metal binding site to return to a less distorted form, the energy released may lower the energy of activation of the enzymatic reaction. Theoretically this permits a faster, more efficient enzymatic reaction. Zinc can sit in this entatic state because it has several possible coordination geometries, and because the coordination geometry is easily distorted. In addition, zinc is a strong Lewis acid (only copper is better), and its presence at an active site can supply a hydroxyl group (OH–), which is important for many enzymatic reactions (see Figure 37-1). In this instance zinc uses water as a fourth ligand (the other three being amino acid residues in the enzyme). The hydroxyl group results when the water molecule forms a partial dipole that is loosely associated with zinc and with a negatively charged group in the enzyme (e.g., a carboxyl group from an aspartate residue). Zinc, copper, and manganese are each absorbed throughout the length of the small intestine but mainly in the jejunum. Copper may also be absorbed in the stomach. Absorption is regulated at the intestinal level for copper and zinc; despite limited evidence, this is also likely to be true for manganese. Absorption can be separated into a saturable, regulated portion and a nonregulated, diffusional component. Because of the existence of both carrier-mediated and nonregulated diffusional absorption of these minerals, the efficiency of absorption falls (i.e., lower fractional absorption), although the total amount of mineral entering the body increases as the dietary level of the mineral increases. Specific zinc and copper transporters have been identified as subsequently described. Intestinal manganese absorption may occur through the divalent metal transporter 1 (DMT1/SLC11A2), which transports iron, manganese, nickle, zinc, and the toxic metals cadmium and lead. Rats expressing a functionally impaired DMT1 exhibit reduced intestinal uptake of manganese (Bressler et al., 2007). (See Chapter 36 for a discussion of iron absorption.) Transcellular and paracellular transport are the two mechanisms for the intestinal transport of minerals from the lumen of the intestine to the portal circulation. Transcellular transport, the movement of zinc across the apical membrane through the cell and exiting at the basolateral membrane, is a carrier-mediated process. Paracellular transport occurs by simple diffusion as the concentration of zinc in the lumen exceeds the ability of the transcellular mechanism to transport zinc into the intestinal cell at its apical surface; zinc diffuses through the tight junctions between intestinal cells. The model for intestinal zinc absorption is shown in Figure 37-3. Two zinc transporters that facilitate carrier-mediated zinc uptake into the intestinal cell have been identified, ZIP (Zrt/Irt-like protein 4; SLC39A4) and ZnT5 (zinc transporter 5; SLC30A5). Molecular studies of patients with the genetic disease acrodermatitis enteropathica (AE) have revealed that the ZIP4/SLC394A4 gene is mutated in these individuals (Dufner-Beattie et al., 2003). This transporter protein is located at the apical surface of intestinal cells, and its presence is responsive to dietary zinc, increased with zinc deficiency, and decreased during zinc sufficiency (Kim et al., 2004). The B splice variant of ZnT5 was found to be present at the apical surface of the Caco-2 intestinal cell line and the brush border membrane of human intestinal biopsies. Its messenger RNA (mRNA) expression was increased in these cells with 100 μM zinc supplementation, and it was shown to function as a zinc uptake transporter (Cragg et al., 2002). However, ZnT5 mRNA levels decreased when the cultured cells were grown in 200 μM zinc compared to 100 μM zinc (Cragg et al., 2005). Similarly, subjects consuming 25 mg zinc for 14 days exhibited reduced ZnT5 mRNA and ZnT5 protein in their intestinal biopsies (Cragg et al., 2005). Although ZnT5 is a member of the cation diffusion facilitator family, it may function as both an influx and an efflux zinc transporter in the intestinal cell (Valentine et al., 2007). The control for intestinal zinc transport is not fully understood. The mechanism is likely to be complex, as recent cellular studies suggest. Elegant cell culture studies using cells transfected with the mouse SLC39A4 gene showed that, under normal zinc levels, the ZIP4 transporter recycled rapidly via endocytosis between the plasma membrane and an endosomal vesicular compartment (Kim et al., 2004). Under zinc-deficient culture conditions, more of the ZIP4 transporter remained at the plasma membrane, whereas less was observed within the intracellular vesicular compartment. Conversely, when the cultured cells were exposed to an increasing concentration of zinc in the culture medium, less of the transporter was located at the plasma membrane, with a corresponding increase in the transporter within intracellular endosomal vesicles. Furthermore, the increased presence of the transporter at the plasma membrane corresponded with increased zinc uptake, with the converse also being true. Therefore these current studies with ZIP4 agree with whole animal and small intestine cell culture studies performed earlier, showing that zinc deficiency increases intestinal zinc transport and absorption. However, the zinc-dependent trigger for the translocation of the ZIP4 zinc transporter between the intracellular vesicles and the plasma membrane is not known at this time. The mechanism of the intracellular transport of zinc from the apical to the basolateral intracellular surface for transport to the portal circulation is not known at this time. The transport of zinc from the enterocyte cytosol to the serosa across the basolateral membrane occurs via ZnT1 (SLC30A1), of the cation diffusion family of zinc transporters (McMahon and Cousins, 1998; Palmiter and Findley, 1995; Lichten and Cousins, 2009). AE (acrodermatitis enteropathica) is caused by an autosomal recessive mutation and is characterized by symptoms normally associated with severe zinc deficiency. Symptoms include dermatitis, alopecia, poor growth, immune deficiencies, hypogonadism, night blindness, impaired taste, and diarrhea. Studies have shown that a primary defect in AE patients is reduced intestinal zinc absorption. Other studies have shown that cellular zinc uptake is reduced in intestinal biopsies and in cultured fibroblasts of patients with AE (Grider and Young, 1996). The AE mutation has been mapped to chromosomal region 8q24.3. Several mutations within the AE gene (SLC39A4, which encodes ZIP4) have been identified. The mutations, found in genomic DNA from patients with AE, include missense mutations and a premature termination codon. Several of the missense mutations have been studied thus far using transfected cell culture models; the mutations affect either the translocation of ZIP4 to the plasma membrane or its endocytosis from the plasma membrane (Wang et al., 2004). The posttranslational regulation of trafficking between membrane compartments has also been shown for the copper transporting P-type ATPases ATP7A and ATP7B. 1. Uptake from the intestinal lumen across the brush border membrane 2. Intracellular transport to the basolateral membrane 3. Transport across the plasma membrane to the portal circulation Copper versus Zinc Transporters: Lethal versus Nonlethal Mutations 1. Where do these transporters reside within the intestinal cell and how does their position within the cell affect the transport of their respective metal? 2. Why would an accumulation of intracellular copper be observed in the intestinal cells of MD patients but an accumulation of zinc not be observed in AE patients? 3. MD is lethal despite dietary copper supplementation because these patients die from copper deficiency but AE is treatable with dietary zinc supplementation. Why do the MD patients die while the AE patients thrive with the dietary supplementation of their respective metals? The model for intestinal copper absorption is shown in Figure 37-4. Two potential apical membrane copper transporters have been identified in intestinal absorptive cells: (1) the Ctr1 (SLC31A1) copper transporter, and (2) the divalent metal transporter 1 (DMT1/SLC11A2; also called Nramp2). Ctr1 transports monovalent copper (Cu1+) (Lee et al., 2002a, 2002b). DMT1 transports divalent iron but can also transport divalent copper (Cu2+) (Gunshin et al., 2009). Most of the dietary copper is in its cupric state (Cu2+). Luminal reduction of Cu2+ to Cu1+ may occur at the brush border membrane via duodenal cytochrome b (Dcytb/CYBRD1) or six-transmembrane epithelial antigen of the prostate 2 (Steap2), both serving as Fe3+ reductases that may also reduce Cu2+ (Prohaska, 2008). Once inside the cell, copper is bound by chaperones that carry copper to various copper-binding proteins, cuproenzymes, or ATP7A, a membrane-associated copper transporting ATPase that is defective in Menkes syndrome (Figure 37-5). ATP7A is also called Menkes protein or MNKP. ATOX1 is a cytosolic copper chaperone that delivers copper from Ctr1 to ATP7A located at the trans-Golgi network. The copper is then exported from the intestinal cell at its basolateral side by this copper transporting ATPase. Menkes syndrome is an X-linked recessive genetic disorder caused by a mutation in the gene encoding for ATP7A that results in a lethal reduction in copper absorption (Tumer et al., 2003) (see subsequent text). At low zinc status, transcellular zinc absorption is at its most efficient; at high zinc status, zinc absorption is inhibited. Research suggests that this downregulation of zinc absorption is due to the production of metallothionein, a zinc-binding protein. Metallothionein is a low-molecular-weight protein (6.1 kDa) found in the cell cytosol, and it is produced in response to high levels of dietary zinc or copper, as well as toxic heavy metals such as cadmium (Cd2+) and mercury (Hg2+) (Cousins, 1985). It has been proposed that high levels of metallothionein in enterocytes act as a mucosal block by binding zinc and preventing its movement through the cell, thereby limiting absorption. The “blocked” metal is later lost from the body as the enterocyte is sloughed off into the intestine. Similarly, the induced production of metallothionein by copper may downregulate copper absorption by this same mechanism. Functions for metallothionein in tissues other than the intestine have also been proposed (discussed in subsequent text). Some studies have shown that high levels of zinc can inhibit intestinal copper absorption; this observation has been used to reduce copper absorption and help minimize the copper toxicity that is characteristic of Wilson disease. This rare autosomal recessive genetic disorder is due to mutations in ATP7B (referred to as Wilson Disease Protein, WNDP), which encodes an ATPase similar to ATP7A in its copper transporting functions (see subsequent text). The high-zinc diet stimulates the production of metallothionein, which then reduces copper absorption by blocking transcellular transport. In contrast, high levels of dietary copper do not reduce zinc absorption. One explanation for this latter observation is that a high copper level is handled differently than a high zinc level. Zinc is a much stronger inducer of intestinal metallothionein than copper (Leone et al., 1985). In addition, copper may not reach the metal-regulatory element of the metallothionein gene as rapidly as zinc, when copper intake is excessive, because of its shuttling to various copper-binding protein compartments via chaperones. Just as the biological roles of copper, zinc, and manganese are defined by their chemical interactions with ligands, so are the interactions of these metals with dietary components. These interactions can influence how well copper, zinc, or manganese is absorbed from the diet, but the luminal interactions leading to increased or decreased absorption are less well characterized than those for ligand interactions of copper, zinc, and manganese with enzymes (Lonnerdal and Sandstrom, 1995). Chemical similarities with other nutrients may result in competition for common binding sites between related minerals. This could explain the inhibition of manganese absorption caused by high dietary iron or the zinc–copper antagonism discussed previously. Binding of minerals to organic components of the diet in the lumen of the gastrointestinal tract can alter absorption. The classic example of this is the inhibitory effect that the phosphate-rich plant compound phytate (myo-inositol pentaphosphates and hexaphosphates) has on the absorption of zinc and many other minerals (e.g., copper and iron) from the diet (Torre et al., 1992). The presence of calcium and phytate together in the same meal enhances the inhibitory effect on mineral absorption because calcium stabilizes phytate. Others have suggested that binding of zinc or copper to low-molecular-weight ligands such as amino acids (e.g., histidine) may enhance intestinal absorption, but it is not clear how this enhancement would occur. Although ascorbate enhances iron absorption, it inhibits copper absorption. The chemical effects of ascorbate on iron and copper are the same; ascorbate promotes the reduction of both metals (to Fe2+ and Cu1+). Dietary components can also influence other points of zinc or copper utilization. For example, high dietary molybdenum has been shown to increase urinary copper losses, and high dietary calcium increases exogenous zinc losses in the intestine. After reaching the liver, zinc is repackaged and released into the circulation bound to α2-macroglobulin. At any given time, the relative distribution of zinc in the circulation is approximately 57% bound to albumin, 40% to α2-macroglobulin, and 3% to low-molecular-weight ligands such as amino acids. There is evidence that the uptake of zinc into cells is regulated (note the discussion in the preceding text concerning the ZIP4 zinc transporter). Close to 200 zinc transporters, belonging either to the SLC30 or SLC39 gene families, have been identified, and are found in fungi, plants, insects, nematodes, and mammals (Liuzzi and Cousins, 2004; Lichten and Cousins, 2009). A total of 24 mammalian zinc transporters belonging to the SLC30 (10 members; cation diffusion family, or CDF/ZnT family) and SLC39 (14 members; zinc-responsive transport/iron responsive transport protein family, or ZIP family) gene families have been identified to date (reviewed in Kambe et al., 2004 and Lichten and Cousins, 2009). Figure 37-6 shows the membrane compartments containing these transporters and the direction of zinc transport. None of these transporters appears to require energy for its function. In endothelial cells, albumin is taken up by endocytosis as a part of transcytosis (Frank et al., 2009). This mechanism may provide for the majority of the internalization of zinc by cells because most zinc in the circulation is bound to albumin. A portion of this endocytosis occurs through non–clathrin-coated pits and may involve caveolae or lipid rafts, or both. These caveolae are specialized plasma membrane compartments that contain an unusually high amount of cholesterol and sphingolipids. They are involved in numerous signal transduction pathways and may be involved in some aspect of zinc uptake (Doherty and McMahon, 2009). After endocytosis the albumin likely releases the zinc, allowing it to be transported across the endosomal membrane and into the cytosol. Once in the cytosol it binds to proteins within the zinc-binding protein pool, including metallothionein. The two types of transporters differ in several ways. The ZIP transporters that have been characterized thus far transport zinc into the cytosol, either into the cell from the extracellular milieu or into the cytosol from within intracellular membrane compartments. The ZnT transporters exhibit the opposite function, facilitating zinc efflux from the cytosol to the outside of the cell or into intracellular membrane compartments. Both transporter families are transmembrane proteins. The ZIP family contains proteins exhibiting seven or eight transmembrane domains, with extracellular amino and carboxyl ends. The ZnT family is made up mostly of proteins containing six transmembrane regions. Their amino and carboxyl ends are located intracellularly. The zinc transport/binding site for both families is an intracellular histidine-rich loop. However, this loop is located between transmembrane regions three and four in the ZIP family and between transmembrane regions four and five in the ZnT family. One of the members of the ZnT family, ZnT5, does not fit the general description of the other members of the ZnT family. It has 12 transmembrane regions and has been implicated in intestinal cell zinc influx and efflux at the brush border membrane (Cragg et al., 2002; Valentine et al., 2007). These transporter families are intimately involved in zinc acquisition from the environment as well as control of intracellular zinc influx and efflux within organelles (see Figure 37-6). The proliferation of these transporter families is indicative of the importance of zinc in cellular metabolism. Once copper reaches the liver, it is incorporated into ceruloplasmin within the trans-Golgi network facilitated by ATP7B, a membrane-associated copper-transporting ATPase that is defective in Wilson disease. Then the copper–ceruloplasmin complex is released into the circulation for delivery to peripheral tissues. Scientists have traditionally thought that ceruloplasmin is a critical protein in the metabolism and function of copper (Vulpe and Packman, 1995). It is a large glycoprotein of 132 kDa, which contains 6% to 7% carbohydrate and which can bind six atoms of copper; 90% to 95% of serum copper is bound to ceruloplasmin. Ceruloplasmin is produced in the liver, and its synthesis is regulated by copper as well as inflammatory mediators such as interleukin 1 (IL1) and glucocorticoids. These and other factors associated with the acute-phase response are presumably responsible for the increase in serum copper and ceruloplasmin that occurs following acute inflammation or infection. In cell culture studies, the mechanism of copper uptake into cells occurred through the binding of ceruloplasmin–copper to a cell-surface receptor (Percival and Harris, 1990). Whereas the circulating transferrin–iron complex is internalized after it binds to the transferrin receptor on the cell surface, copper is reduced and released from ceruloplasmin, at which point the copper can be taken up by the cell as the free metal through the Ctr1 copper transporter. Paradoxically, individuals who have a genetic mutation in the ceruloplasmin gene, leading to a total lack of ceruloplasmin in the serum (i.e., aceruloplasminemia), do not have overt symptoms of copper deficiency as one might predict from the cell culture studies (Harris et al., 1995). Instead, these individuals have altered iron metabolism. This raises questions regarding the biological role of ceruloplasmin in serum copper transport. Other proteins are also involved in copper movement. Most if not all copper ions in cells are bound to SOD or other proteins. Studies in animals and cells have shown that a copper chaperone for SOD (CCS) is necessary for the insertion of copper into SOD. Rats fed copper-deficient diets displayed a dose-dependent increase in CCS expression in liver and erythrocytes (Bertinato et al., 2003). Role of Ceruloplasmin in Iron Metabolism As patients were referred for molecular diagnosis, it became clear that some of the patients with low or absent ceruloplasmin did not have Wilson disease. Further molecular genetic analysis of patients with non–Wilson disease aceruloplasminemia revealed mutations in the ceruloplasmin gene that resulted in truncation of the open reading frame. Harris and colleagues (1995) described one of two sisters with undetectable ceruloplasmin. This patient was homozygous for a mutation in exon 7 of the ceruloplasmin gene. She had developed degeneration of the retina and basal ganglia during the fifth decade of life. Liver biopsy revealed a normal hepatic copper concentration but elevated iron stores. Indirect evidence for iron deposition in the basal ganglia of the brain was also obtained. The association of defects in ceruloplasmin synthesis with abnormalities in iron metabolism in this and other patients confirms an essential role of ceruloplasmin in human iron metabolism and supports a role of ceruloplasmin as a ferroxidase (Harris et al., 1995). However, the major problem in patients with aceruloplasminemia seems to be excessive iron deposition or storage, with much milder problems related to iron delivery or utilization. How a plasma protein can be involved in release of iron from tissue ferritin is not yet clear. 1. Explain how a lack of ceruloplasmin could result in iron overload in liver and other tissues. 2. A low serum iron concentration and an elevated serum ferritin concentration are found in patients with defects in the ceruloplasmin gene. Anemia is seen in some but not all patients with defects in the ceruloplasmin gene. (a) Explain how each of these clinical/biochemical manifestations could result from a defect in ferroxidase activity. (b) Are these changes in measures of iron status similar to or different from the changes that would occur in iron deficiency anemia? (See Chapter 36 for more information about iron metabolism.) 3. Copper metabolism and transport seem to be relatively normal in patients with defects in ceruloplasmin synthesis. What might this tell us about the role of ceruloplasmin in copper transport in the body?
Zinc, Copper, and Manganese
Zinc, Copper, and Manganese in Enzyme Systems
ENZYME
FUNCTION
ROLE OF METAL
COPPER
Lysyl oxidase
Collagen synthesis
Catalytic
Peptidylglycine α-amidating monooxygenase
Neuropeptide synthesis
Catalytic
Superoxide dismutase (cytosolic and extracellular)
O2.− to H2O2
Catalytic
“Ferroxidase”/ceruloplasmin
Release of stored iron
Catalytic
Cytochrome c oxidase
Oxidative phosphorylation
Catalytic
Dopamine β-hydroxylase
Neurotransmitter synthesis
Catalytic
Tyrosine oxidase
Melanin synthesis
Catalytic
ZINC
Alcohol dehydrogenase
Alcohol metabolism
Catalytic, noncatalytic
Superoxide dismutase (cytosolic)
O2.− to H2O2
Noncatalytic
Superoxide dismutase (extracellular)
O2.− to H2O2
Noncatalytic
Terminal deoxynucleotide transferase
Add deoxynucleotide triphosphates to 3′ end of DNA
?
Alkaline phosphatase
Bone formation
Catalytic, noncatalytic
5′-Nucleotidase
Hydrolysis of 5′-nucleotides
?
Fructose 1,6-bisphosphatase
Glycolysis
Regulatory
Aminopeptidase
Protein digestion
Catalytic, regulatory
Angiotensin-converting enzyme
Angiotensin I to II
Catalytic
Carboxypeptidases A and B
Protein digestion
Catalytic
Neutral protease
Protein digestion
Catalytic
Collagenase
Collagen breakdown
Catalytic
Carbonic anhydrase
CO2 → HCO3–
Catalytic
δ-Aminolevulinic acid dehydratase
Heme biosynthesis
Catalytic
MANGANESE
Arginase
Urea formation
Catalytic
Pyruvate carboxylase
Gluconeogensis
Catalytic
Superoxide dismutase (mitochondrial)
O2.− to H2O2
Catalytic
Farnesyl pyrophosphate synthetase
Cholesterol synthesis
Catalytic
Glycosyltransferases
Cartilage formation
Regulatory
Phosphoenolpyruvate carboxylase
Gluconeogenesis
Regulatory
Xylosyltransferase
Cartilage formation
Regulatory
Absorption, Transport, Storage, and Excretion of Zinc, Copper, and Manganese
Absorption
Zinc Absorption
Copper Absorption
 CLINICAL CORRELATION
CLINICAL CORRELATION
Inhibition of Intestinal Zinc and Copper Absorption by Metallothionein
Bioavailability
Transport in Plasma and Tissue Uptake
Cellular Zinc Transporters
Copper and Ceruloplasmin
Storage
 CLINICAL CORRELATION
CLINICAL CORRELATION
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Zinc, Copper, and Manganese