Figure 7.1. Various factors affecting design of preclinical drug disposition research protocol.
Drivers of Preclinical Drug Disposition Studies
Generally, the main driver of preclinical drug disposition studies is a drug project or demand for innovative scientific tool (e.g. model development for novel metabolic pathway).
The scope of drug project-driven preclinical studies largely depends on the type of compound (e.g. small chemical molecule versus large biotech-derived macromolecule) and the disease area targeted (chronic disease condition versus life-threatening disease). Preclinical studies are performed with or without use of animals and are aimed to limit the risk of the NCE when used as medicinal product in humans. They should be designed in order to achieve fast, risk-free, unproblematic transition of the NME from preclinical stage to clinical trials during the drug development programme. For this purpose, the legitimate interest of subjects, patients, manufacturers (sponsors) and the scientific community, as well as regulatory and competent authorities, must be harmonised to achieve a course of action that is medically, ethically (also with respect to animal testing) and legally justified. As every NME is unique, investigations and testing strategies must be designed individually. Important considerations for determining the nature of such studies and their timing with respect to clinical trials include the following1:
1. Targeted disease area (chronic disease, immunocompromised, cancer, etc.)
2. Duration of drug exposure and total drug exposure
3. Characteristics of drug (t½, bioavailability, biodistribution, protein binding, etc.)
4. Special population targeted (older patients, children, pregnant women, etc.)
5. Route of administration (intravenous, intramuscular, oral, etc.)
Today rational drug design, integrated knowledge and new technologies from all scientific disciplines (combinatorial chemistry, target discovery and functional analysis, genomics and proteomics and so forth) produce a multitude of new and highly optimised compounds.2 Unfortunately, these compounds are often developed in an old regulatory paradigm where chemical development is hampered by patient scarcity. This necessitates development of innovative preclinical approaches for better models to give scientifically sound results with minimal expense of animals (three R concept, vide infra), money and man hours. More commonly, these types of basic research models are developed in academia and validated by industry to suit regulatory standards.
Preclinical Research Environment
The preclinical research has a highly dynamic environment of early stage development. The focus shifts from selecting a drug candidate (lead selection) to maximising the potential of selected drug candidate for approval for clinical studies (lead optimisation). The following specific considerations are important in writing protocols for preclinical drug disposition studies.
Preclinical Drug Disposition Science
The concepts of drug ADME are central to preclinical drug disposition science. These phenomena, taken together, comprise a vast number of subjects that can be selected to answer intriguing questions, and add to the valuable knowledge base of current pharmacokinetics. Thorough and deep understanding of these interdependent concepts is fundamental to posing any question that can be drafted as a preclinical research protocol. For example, existence of P-glycoprotein transporter in the intestine, liver, blood–brain barrier, placenta and kidney cells makes this transporter significant in drug ADME pathways.
Animal Ethics
Preclinical disposition studies are conducted in animal species to adjudge if the proposed drug candidate is safe and effective for use in humans. This comprises various types of studies like interspecies metabolism characterisation to help identify most relevant toxicological species. Young researchers must make themselves aware of local institutional animal ethical standards and follow relevant certification courses, if required. For example, the American Association for Laboratory Animal Science based in the United States, Animal Federation of European Laboratory Animal Science Associations in Europe and the Committee for the Purpose of Control and Supervision on Experiments on Animals (CPCSEA) in India3 are useful sources of information in this regard. The preclinical protocols with animal experimentation are carefully designed to select appropriate study design and determine the required number of animals based on statistical considerations. The biology and husbandry of laboratory animals, behaviour, stress and well-being, standardisation of experimentation, nutritional aspects, genetic and microbial standardisation and diseases in laboratory animals should be described, as and where appropriate, in the protocols.4 The ‘Three R concept’ (Replacement, Reduction and Refinement) proposed by Russell and Burch should be the deciding rule to select appropriate experimentation methodology to achieve scientific goals.5 Replacement refers to the substitution of living animals by in vitro techniques, computerised models or surrogate procedures using microchip or nanotechnology. Reduction refers to a decrease in the number of animals required for a given experiment. Refinement refers to any decrease in incidence or severity of painful or distressing procedures applied to animals.
Chemically Hazardous Substances and Radioactivity
Large numbers of chemical compounds with unknown toxicities or known potential toxicities are handled in preclinical disposition research studies. Many of these compounds (e.g. trypan blue stain for cell culture work) are commercially available from various suppliers and prevalently used in common procedures. To ensure safety of personnel performing these experiments, the dose, frequency and method of exposure of investigative or control compounds in the study should be carefully evaluated before incorporating them into a protocol. For example, compounds classified by the supplier into category of risk phrase ‘R45’ (may cause cancer) require a full risk assessment of exposure and may require approval from biosafety or environmental safety and health (ESH) committees before they can be included in the study design or implemented. The protocol should refer to appropriate procedures to prepare, store, sample or handle the solutions of these substances using personal protective equipment (PPE) where needed.
Radioactive analysis is one of the most widely used methods to perform various kinds of pre-clinical drug disposition studies. The type of isotope required and final radioactivity in working solutions/samples/specimen should be anticipated well in advance and reflected in the protocol and approved by radiation safety committees. This is important in managing radioactive waste disposal, isotope compound inventory management and budget forecasting. The use of radioactivity in final procedures should be sufficient to warrant detectable limits well above lower limits of quantitation of analysing equipment.
Biohazards
Use of recombinant biological systems is becoming very popular in preclinical disposition studies. This comprises a wide range of genetically modified systems and includes whole animals (e.g. P-glycoprotein knock-out mice), cell lines (e.g. Caco-2) and human protein expressing membrane fractions (e.g. BD supersomes™ expressing CYP enzymes). These systems need to be thoroughly validated using known science (e.g. literature-quoted substrates or inhibitors of enzymes) before implementing them to define observations of unknown science (new proposed enzyme inhibitor). The protocol should comply with all biosafety measures required for safe working practices involving various biohazards and should be approved by recombinant-DNA-use committee.
Allied Emerging Sciences
Developments in instrumental methods of analysis, software packages for in silico predictions and simulations or experimental techniques are a few examples of knowledge-based areas that are always in a dynamic state of improvement. A proactive approach to incorporate the most up-to-date technology in the protocol will ensure that the outcome of the study exhibits the highest standards of innovation and most relevant results, at par with contemporary science.
Good Laboratory Practices
The principles of good laboratory practices (GLP) are promulgated by regulatory authorities to assure that the high quality standards are practiced in research to generate the information that supports the approval of medicinal products for human use and thus minimise or prevent any possible potential health hazards and risks. It is a statutory requirement to follow GLP principles for contract research organisations (CROs) and companies in order to get drug candidates approved by regulatory authorities. The preclinical research conducted by researchers in academic or industry-sponsored academic projects is generally exploratory in nature and might not be comprehensively GLP compliant. However, it is emphasised that researchers should work in the spirit of GLP and write methodologies in protocols that reflect application of GLP in their work practices. Further information on GLP can be obtained from the websites of regulatory agencies (e.g. US FDA and OECD).
Preclinical Drug Disposition Protocol
The structure of protocols is described comprehensively in Chapter 5. The preclinical drug disposition protocol essentially constitutes the following: title, introduction to the study, literature survey, objective(s), hypothesis, description of test article(s), justification on test system(s), experimental details, methods of data analysis and acceptance criteria (if any) for results and references. The basic philosophy behind the protocol structure and content is the same for all biomedical studies. However, they are framed according to the specific terminology of each scientific discipline. In addition, based on institutional requirements, different protocols may vary in the pattern of formatting and/or final document style. The following are a few key points that should be kept in mind when preparing preclinical drug disposition protocols. Specific preclinical drug disposition studies might involve additional headings.
Preamble
The title of the protocol should be a brief and precise statement of the research work and proposed study plan including species or the cell line used. For example, ‘In vitro inter-species metabolic clearance of drug candidate X in hepatocytes’ or ‘In vitro evaluation of drug candidate X as cytochrome P450 inducer in cultured human hepatocytes’. The title should be followed by introduction to the preclinical research problem, significance of the problem and the scientific justification to perform the proposed work. Previous studies on the preclinical drug candidate done at the discovery stage are a relevant source of information for NCE/NMEs, whereas a general literature review might be helpful for marketed drugs. The protocol should clearly underline key facts, terms and information to delineate the research issue and then lead up to the objectives.
Objective(s)
The study is directed to achieve well-defined and specific objectives. Generally, these objectives are inspired from specific questions raised in drug projects with regard to their ADME. For example, if the new compound proposed as a drug candidate is administered as co-medication with marketed drugs, which are metabolised primarily by CYP3A4, then a question under evaluation will be whether the new drug candidate causes induction and/or inhibition of CYP3A4 in liver. Irrespective of direct requirements of drug project, investigative study that might be relevant in the aforementioned example will be to answer a question, if the sandwich cultures of hepatocytes can better predict induction of CYP enzymes in liver compared with simple plated primary cultures of hepatocytes. Some of the selected possible topics in preclinical drug disposition research are listed below.
(a) Structure ADME relationships
(b) Structure CYP activity relationships
(c) Structure transporter activity relationships
(d) In vitro–in silico or in vivo–in silico correlations in pharmacokinetic properties
(a) High throughput and automated sampling from in vitro incubations
(b) Online data processing and report generation
(a) Metabolite profiling of drug candidates
(b) Hepatic clearances and/or uptake mechanism
(c) Induction of CYPs/transporters
(a) Novel experimental approaches to predict CYP inhibition
(b) Novel experimental approaches to predict transporter-based interactions
(c) Modelling of enzyme-transporter interplay in hepatocytes
(d) In vivo and/or in vitro models for clinically relevant drug transporters
(a) Perpetual cell lines (transfected with single or multiple transporters)
(b) Primary co-cultures in BBB cells (endothelial, glioma and neuronal cells)
(a) Novel methods for unstable compounds and/or lipophilic compounds with short experiment run times
Hypothesis
As described in Chapter 5, the hypothesis is an integral part of systematic research. It, however, might not be pertinent in certain studies where the primary question is focused on characterisation of pharmacokinetic properties of drug candidates rather than looking into exploratory mechanistic aspects of the observed pharmacokinetic properties. For example, a study to determine the absorption rate constant of a new compound, using single-pass intestinal perfusion in rats, to help define its place in the biopharmaceutics classification system (BCS) might not have a hypothesis. It forms inherent basis of investigative studies where the objectives of the study need a hypothesis to prove or disprove a certain belief. For example, yellow pigmentation in liver observed in toxicology studies might be hypothesised to be due to interaction of drug candidate with bilirubin secreting hepatobiliary transporters (OATP1B1 and/or MRP2).
Choice of Test Systems
Preclinical drug disposition studies are performed using different biological systems such as cellular membranes, subcellular fractions, primary cultures, transgenic cell lines and different animal species. Each of the test systems has its own characteristics and that should be mentioned in the protocol. For example, acclimatisation period, housing, day-light schedule and diet are different for each species. In case of transgenic cell lines, it is pertinent to know the most suitable incubation medium, selection of antibiotics in the medium, doubling time, etc. It might be possible to have a choice of various test systems to perform a particular research objective. For example, an extremely large number of in vitro models are reported in the literature for transporter studies (a few of which are listed in Table 7.1) and might appear confounding prima facie. Finally, it is left to the discretion of the researcher to select a model that best solves the problem under investigation.
Table 7.1. In vitro models reported in the literature for transporter studies
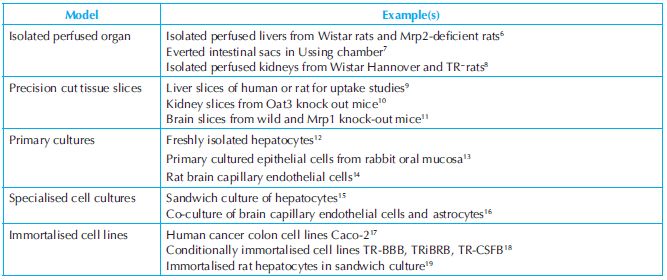

Transporters referred to in the above table are abbreviations of protein names from classical convention. Full protein names and more recent nomenclature recommendations based on gene names can be found at http://www.genenames.org/cgi-bin/hgnc_search.pl.
One must have a good reason to justify the appropriateness of the test system selected for the investigation and this must be specified in the protocol citing previously conducted studies (e.g. validation report or published article). Hepatocytes and microsomes are commonly used in drug metabolism studies (Table 7.2).
Table 7.2. Test systems and the justification for their use

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


