Complete vital signs, including height (Visit 1 only), weight, respiratory rate, temperature, and orthostatic (sitting, immediate standing and 2-min standing) BP and pulse rate measurements. Subjects administered one ECG at Visits 1 and Day 2; subjects instructed to remain in a supine position 2 min prior to each ECG evaluation. Echocardiogram scheduled 2 weeks prior to Visit 3 to insure results are available at this visit.
First 24-h urine collection to begin 24 h prior to dose and second 24-h urine collection to begin at the time of dose. Urine samples are collected and labelled at 2-, 4- and 24-h increments.
Visit 1 clinical laboratory tests include chemistry, haematology, endocrinology, urinalysis, and HIV, hepatitis B & C antibody screen. Day 1 and Day 2 clinical laboratory tests include chemistry, haematology, endocrinology, and urinalysis as well as plasma rennin, aldosterone, PK and cGMP (baseline/predose, 0.25 h, 0.5 h, 1 h, 2 h, 4 h, 6 h, 8 h, 12 h and 24 h), BNP, endothelin, catecholamines, baseline sample for anti-drug A antibody formation and 24 h urine (including measurement of electrolytes). Eight week follow-up clinical laboratory test includes chemistry, urinalysis, as well as an analysis for potential anti-drug A antibody formation. Samples for immunology analysis (i.e., anti-drug A antibodies) are batched and processed at the end of the study.
Dosing begins with two of the four subjects in the first cohort. Dosing for the next two subjects begins 2 h after the first two subjects are dosed. The monitor is set to take a measurement at baseline, 5, 10, 15, 20, 30 min and then every 15 min. Should a clinically significant change in BP is detected, it is confirmed with a manual cuff measurement within 5 min.
Pre-Study Screening
During Visit 1 (screening), informed consent is obtained, a complete medical history (including medications) is recorded and the following screening safety assessments made: physical examination, ECG, echocardiogram, clinical laboratory tests, sitting, standing and orthostatic BP and pulse measurements.
Study Procedures
Please refer to the schedule of study procedures and assessments (Table 14.1).
Blinding
All study personnel and the sponsor remain blinded to treatment allocation until the study is completed, and the database is locked. If a serious adverse effect (SAE), which is unexpected and attributable to the study drug, is reported, the sponsor may ask the investigator to unblind the treatment assignment for that individual subject in order to fulfill expedited regulatory reporting requirements.
In the event that subjects experience the stopping criterion specified for hypotension (a decrease in >15% systolic BP from baseline), the treatment allocation for the affected subjects are unblinded to determine whether to proceed to the next dose level.
In an emergency, when the knowledge of the study drug is essential for the clinical management or welfare of the subject, the investigator will contact the project medical monitor/medical director to unblind an individual subject’s treatment assignment.
If the blinding is broken for any reason, the investigator must record the date and reason for breaking the blinding on the appropriate page of the CRF.
Concomitant Medications
A list of medications which are prohibited during the study should be provided here. For example, α-adrenoceptor antagonists, antidepressants, antihistamines, antipsychotics, β-adrenoceptor antagonists, calcium channel antagonists, catecholamines/related compounds, diuretics, monoamine oxidase inhibitors, stimulants (including caffeine and nicotine), sympathomimetics, vasodilators, antihypertensive agents, dopamine agonists.
Concomitant treatment with an investigational drug or device is not permitted 30 days prior to signing consent, at any time during the study and until after Visit 3.
Pharmacokinetic Procedures
No human PK information is currently available. This is the first-in-human study of a new chemical entity. The study determines the PK of drug A in healthy male subjects after single SC administration.
Pharmacodynamic Procedures
This is the first-in-human study of a new chemical entity.
Endpoints
The following safety endpoints are defined for this protocol: incidence, severity and seriousness of AEs, changes in BP and heart rate, physical exam, ECGs, echocardiograms, clinical laboratory tests, diuresis and natriuresis, immunogenicity of drug A and findings in clinical registry.
Sample Size
The sample size estimation is based on the minimum number of subjects necessary to draw a clinical conclusion regarding the drug’s safety profile within this dosage range.
The planned sample size is 29–37 healthy males: 0.01 µg/kg (3 active and 1 placebo), 0.03 µg/kg (3 active and 1 placebo), 0.1 µg/kg (6 active and 1 placebo), 0.3 µg/kg (6 active and 1 placebo) and 1.0 µg/kg (6 active and 1 placebo).
In the event that a subject in Group I or II satisfies the protocol-defined criterion of hypotension, a second group of 4 subjects (3 active and 1 placebo) is randomised so that six subjects may receive the study medication in these groups.
Clinical and Laboratory Studies
The clinical and laboratory evaluation schedule is shown in the study procedures (Table 14.1).
Indications for Discontinuation
A generic list of reasons for discontinuation is given below.
Subjects who discontinue the study may be replaced at the sponsor’s discretion.
Efficacy Considerations
Efficacy variable—not applicable.
Substudy Evaluations
Not applicable.
Proposed Observation Period
Each subject enrolled into the study is kept under observation for 24 h post-dose, in the clinical research unit. Subjects return to the clinic 30 days later for a laboratory sample to be taken for potential anti-drug A antibody formation and for an echocardiogram (for potential atrio-ventricular cysts which have been reported in preclinical toxicology studies). Subjects are followed for 2 years by a clinical registry.
Sample Protocol 2
Phase 1 (first-in-man clinical study) protocol for drug B (anticoagulant) for the treatment of atrial fibrillation (AF), deep vein thrombosis (DVT) and pulmonary embolism (PE).
Title: Placebo-controlled, ascending single-dose study, in healthy subjects, to evaluate the safety, PK, and PD of drug B, a reversible inhibitor of factor Xa
Objectives of the Study
The primary objective is to assess the safety and tolerability of drug B when administered as a single oral dose to healthy subjects. The secondary objectives are to assess: (i) the single-dose PK of drug B, (ii) the oral bioavailability of drug B as a suspension formulation compared to a solution formulation and (iii) the single-dose PD of drug B.
Research Hypothesis
The research hypothesis is that drug B, a selective, reversible inhibitor of coagulation factor Xa (and subsequently decreases the conversion of prothrombin to active thrombin and diminishes fibrin formation and platelet activation), administered orally once and/or twice daily, reduces thromboembolic events in patients with AF, and reduces DVT and PE in surgical and non-surgical settings.
Rationale for the Study
In the United States in 2001, there were 416,000 cases of AF, 931,000 cases of stroke and 250,000 cases of VTE. Of the 250,000 cases of VTE, about one-third were due to either DVT or PE.6 The thrombotic or thromboembolic events appeared to influence the prognosis. The anticoagulants currently available are limited by a narrow therapeutic index or the need for parenteral administration.
To overcome these limitations, oral agents are being sought for, particularly to inhibit factor Xa activity because factor Xa plays a pivotal role in being activated by both intrinsic and extrinsic pathways of the coagulation system. The proposed study drug is an orally active, selective, reversible inhibitor of factor Xa. Inhibition of factor Xa decreases the conversion of prothrombin to active thrombin, and thus diminishes thrombin-mediated activation of the coagulation process, including fibrin formation and platelet activation.
Monitors and Their Qualifications
The sponsor’s medical monitor is __________ (name and address).
Principal Investigator
The clinical investigator is __________, MD. The study will be conducted at __________ (provide address).
Institutional Review Board
The IRB is identified as __________ (provide name and address).
Experimental Design
This is a randomised, double-blind, placebo-controlled, sequential, ascending single oral dose study. Subjects will be assigned to one of seven sequential panels as shown in Figure 14.1 below.
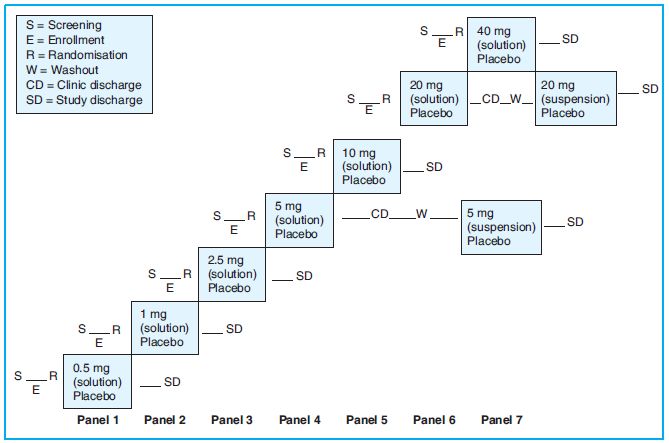
Procedure
On the morning of Day 1, eight subjects are randomly assigned in a ratio of 3:1 to receive either a single oral dose of drug B (N = 6) or placebo (N = 2). Subjects stay at the clinical facility for at least 96 h after study drug administration. Physical examinations, vital signs, 12-lead ECGs and clinical labs are done at specified times. Blood samples for PK analyses are collected for up to 96 h.
If a dose level is found to be safe and tolerated, then the succeeding panel of eight subjects receives the next higher dose of drug B or placebo. There is no intra-subject dose escalation. Subjects who do not complete the study (except those discontinued for AEs) may be replaced. Subjects are not enrolled at the next dose level until the safety data through Day 5 evaluations for at least six subjects from the current dose level are reviewed by the sponsor and the investigator.
Subjects in the 5- and 20-mg dose panels return to the clinic after a 7-day washout for an additional dose of drug B (or placebo) administered as a suspension formulation (Period 2).
Patient Population
Healthy males and females aged 18–45 years.
Inclusion Criteria
List criteria. The following is a generic list for such a clinical trial: These include healthy subjects as determined by medical history, physical examination, 12-lead ECG and clinical evaluations and a BMI of 18–30 kg/m2. Female subjects must not be nursing, pregnant, nor of childbearing potential (i.e., postmenopausal (amenorrhea ≥12 consecutive months or, if on hormone replacement therapy, serum FSH level >35 mIU/mL) or surgically sterile (hysterectomy, bilateral tubal ligation or bilateral oophorectomy)).
Exclusion Criteria
List criteria. The following is a generic list for such a clinical trial: These include women of childbearing potential (including those using oral, implanted or injectable contraceptive hormones or mechanical products such as intrauterine device or barrier methods (diaphragm, condoms, spermicides) or practicing abstinence, or where the partner is sterile (e.g. vasectomy)), women who are pregnant or breastfeeding and women with a positive pregnancy test at screening.
Formulation
Study drug is administered as an oral solution formulation (Period 1) and, for subjects in the 5-mg and 20-mg panels, as a suspension formulation (Period 2).
Controls
This is a double-blind, placebo-controlled, parallel group, single-dose trial.
Dose Administration
Study drug is administered at 9:00 am after an overnight fast of at least 10 h. The total volume per dose panel remains constant (20 mL). After dosing, all subjects are given a total of 240 mL water (including the volume used to rinse the syringe at least twice for Period 1 and at least 3 times for Period 2). No food is allowed for at least 2 h post-dose.
Informed Consent
Informed consent procedure should be discussed in detail in the protocol.
Compliance
After dosing (followed by 240 mL of water), a mouth check is performed to verify that the subject had swallowed the dose.
Pre-Study Screening
This is a Phase I study. Subjects undergo screening evaluations to determine eligibility within 21 days prior to study enrollment.
Pharmacokinetic Procedures
The single-dose PK of drug B is derived from plasma concentration versus time data, including Cmax, Tmax, AUC(inf), AUC(0-t), T1/2, C24, C24/Cmax, and FREL (relative bioavailability of suspension versus solution).
Pharmacodynamic Procedures
The single-dose PD of drug B is derived from determination of international normalised ratio (INR) and modified prothrombin time (mPT) in platelet-poor plasma samples collected at specified times.
Endpoints
The endpoints are safety:
Sample Size
Administration of drug B to six subjects in each panel provides an 80% probability of observing at least one occurrence of any AE in a panel that would occur with a 24% incidence in the population from which the sample is drawn.
Safety Considerations
Similar to that described in endpoints.
Clinical and Laboratory Studies
These include ECGs (PR, QRS, QTc intervals), haematology, serum chemistry, urinalysis and coagulation tests (INR and aPTT), evidence of bleeding from bleeding time, occult faecal blood and haematuria.
Indications for Discontinuation
Subjects must be discontinued for (provide list, example of a generic list of indications for discontinuation is given below):
Efficacy Considerations
Not applicable.
Substudy Evaluations
Not applicable.
Proposed Observation Period
Subjects enrolled into 0.5, 1, 2.5, 10 and 40 mg dose panels are observed for a duration of 7 days, and those enrolled in the 5 and 20 mg panels are observed for a duration of ≥21 days.
Sample Protocol 3
Phase 2 protocol for drug C (anticoagulant) for the prevention of DVT and PE in patients undergoing knee replacement surgery.
Title: A Phase 2 randomised, double-blinded (test drug C and antithrombin drug X), active-controlled (antithrombin drug X and reference anticoagulant drug Y), parallel-arm, dose–response study of the oral factor Xa inhibitor drug C in subjects undergoing elective total knee replacement surgery
Objectives of the Study
The primary objective is to determine the dose–response relationship among the three QD and three BD doses of oral test drug C on the composite endpoint of adjudicated venous thromboembolic (VTEs) events (asymptomatic and symptomatic DVT and non-fatal PE) and all-cause death in subjects treated for 12 ± 2 days following elective unilateral total knee replacement surgery.
Research Hypothesis
The research hypothesis is that drug C, a direct and selective inhibitor of coagulation factor Xa (and also inhibits other coagulation proteases and structurally related enzymes involved in fibrinolysis), administered orally once and/or twice daily, elicits a dose-dependent reduction in VTEs, asymptomatic and symptomatic DVT, non-fatal PE, and all-cause death in the treated subjects following elective unilateral total knee replacement surgery.
Rationale for the Study
Currently available anticoagulants (warfarin and other Vitamin K antagonists, unfractionated heparin (UFH) and low-molecular-weight heparins (LMWHs), e.g. Lovenox®/Clexane®, enoxaparin) are limited by a narrow therapeutic index or multiple interactions with food and drugs or the need for frequent monitoring and dose adjustment or the need for parenteral administration.
The proposed study drug, drug C, is an orally active, selective, direct (i.e. does not require antithrombin III for efficacy) inhibitor of factor Xa. Safety and tolerability of drug C have been demonstrated in Phase 1 healthy volunteer studies up to 7 days duration and these studies have provided data to establish the PK and PD profile of drug C.
Monitors and Their Qualifications
The sponsor’s medical monitor is __________ (provide name and address).
Principal Investigator
The clinical investigator is __________, MD. The study is conducted at approximately __________ centres in the United States and 6–8 (example) countries. An average of 8–10 subjects are expected to be randomised per site (total = 1,200 subjects).
Institutional Review Board
The IRB is identified as __________ (provide name and address).
Experimental Design
This is a randomised, eight-arm, double-blind (drug C and drug X), active-controlled (drug X and reference drug Y), multicentre study.
This eight-arm study evaluates 3 BD dose regimens (2.5, 5 and 10 mg BD) of drug C, 3 QD dose regimens (5, 10 and 20 mg QD) of drug C, BD drug X and QD drug Y. Subjects are assigned to one of eight treatment arms.
Patient Population
Demographics: Males and females (18–90 years old) who are not of childbearing potential, of any race, and scheduled to undergo elective unilateral total knee replacement are eligible.
Inclusion Criteria
List criteria. The following is a generic list for such a clinical trial: Subjects 18–90 years of age of non-childbearing potential (postmenopausal or surgically sterile women) who are undergoing elective unilateral total knee replacement, willing and able to provide written consent, able to undergo bilateral ascending contrast venography and able to inject (self or caregiver) drug X or placebo SC.
Exclusion Criteria
List criteria. The following is a generic list for such a clinical trial: These include women of childbearing potential (including those using oral, implanted or injectable contraceptive hormones or mechanical products such as intrauterine device or barrier methods—diaphragm, condoms, spermicides—or practicing abstinence, or where the partner is sterile, e.g. vasectomy), women who are pregnant or breastfeeding and women with a positive pregnancy test on enrollment or prior to study drug administration.
There are also exclusion criteria based on medical history and concurrent diseases, physical and laboratory test findings (e.g. Hb <10 g/dL, platelet count <100,000/mm3, alanine aminotransferase (ALT) or aspartate aminotransferase (AST) or bilirubin >1.5-fold higher than the upper limit of normal (ULN) or INR ≥1.4 or aPTT ≥1.4-fold higher than control value), hypersensitivity to heparin and its products, warfarin and vitamin K analogues, porcine products or iodinated contrast medium (for venogram), and the need for medications that affect coagulation or platelet function including aspirin, nonsteroidal anti-inflammatory drugs, fondaparinux and ximelegatran, within 7 days prior to surgery, and cholestyramine, phenytoin, barbiturates or rifampin within 3 months prior to surgery (due to interaction with drug Y).
Exclusion criteria in the postoperative screening period include use of epidural or intrathecal catheter, traumatic or difficult (≥2 attempts) lumbar puncture, current active bleeding (gastrointestinal, genito-urinary, wound), uncontrolled hypertension requiring medical supervision or supine systolic BP >180 mm Hg or supine diastolic BP >105 mm Hg, simultaneous arthroplasty of an additional joint, use of pneumatic compression devices or tourniquets after skin wound closure, and unwilling or unable to comply with medication or procedures such as bilateral ascending contrast venography.
Procedure: Formulation
Drug C tablets (2.5–10 mg) or matching placebo.
Controls
Drug X 30 mg or matching placebo for SC injection. Drug Y (scored 1.0 and 2.5 mg tablets) will be open-label.
Dose Administration
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


