Copper chelators and zinc are mainstay of medical therapy
– Without treatment, progresses to cirrhosis and death
Increased serum free copper and urinary copper are characteristic
Increased hepatic copper concentration is most reliable test and is diagnostic in most cases
• Laboratory tests
Low serum ceruloplasmin level is characteristic but is neither sensitive nor specific
Increased serum free copper and urinary copper
Increased hepatic copper concentration is most reliable test
Microscopic
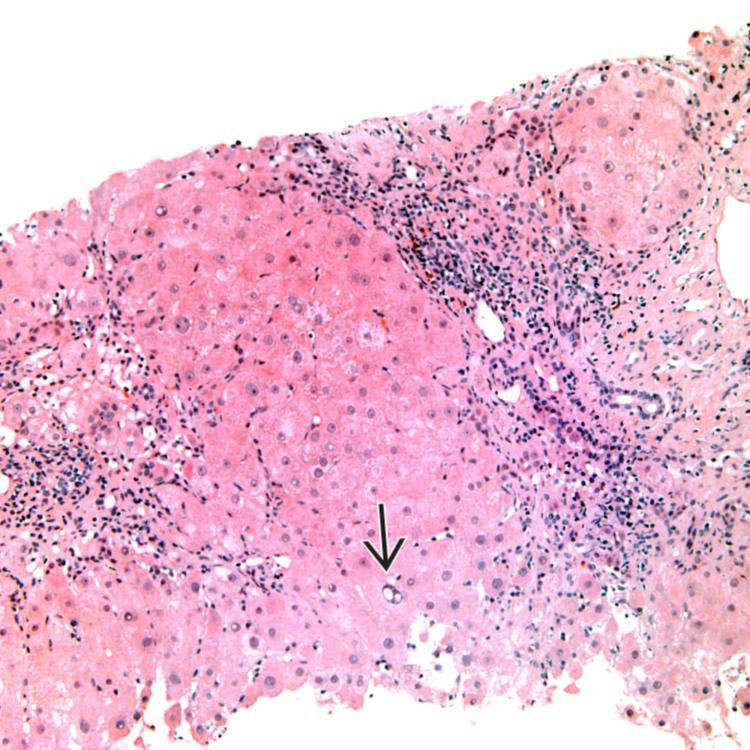
• Early disease is characterized by steatosis, variably present Mallory hyaline, and glycogenated nuclei
• Intermediate stage is characterized by chronic hepatitis with fibrosis or cirrhosis
• Histologic features on routine staining are very nonspecific, so diagnosis easily missed
• Rhodanine or rubeanic acid stains for copper and orcein or aldehyde fuchsin stains for copper-associated protein may be helpful
Staining may be very variable within liver; thus negative stain does not exclude disease
Kayser-Fleischer Ring Clinical photograph of Kayser-Fleischer ring shows brown deposits of copper at the periphery of the iris . (Courtesy S. Uwaydat, MD.) Almost half of patients with Wilson disease lack this finding, however.
Rhodanine Stain Red-brown granular staining is seen within hepatocytes on rhodanine stain, characteristic of Wilson disease. However, staining may be focal within the liver, and thus a negative stain does not exclude disease.
Features of Steatohepatitis Liver biopsies in Wilson disease frequently show features of steatohepatitis, with steatosis and glycogenated nuclei .
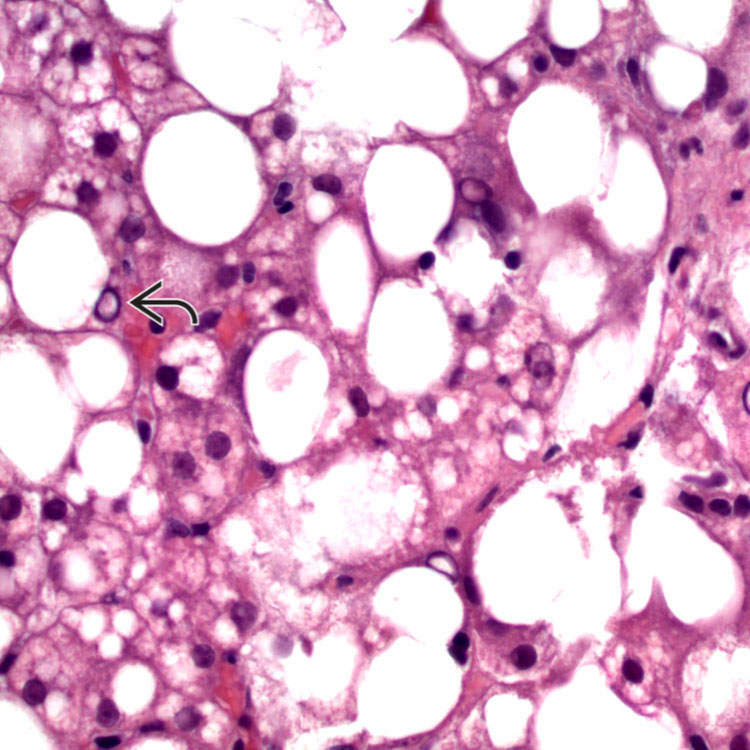
Inflammation Many biopsies from Wilson disease patients show only nonspecific portal-based chronic inflammation, as seen here. Note the rare, large glycogenated nuclei .
TERMINOLOGY
Synonyms
• Hepatolenticular degeneration
Definitions
• Inherited autosomal recessive inherited mutation of copper transport protein
ETIOLOGY/PATHOGENESIS
Genetic Defect
• Mutations of ATP7B gene, which codes for copper-dependent P-type ATPase, copper transport protein found on Golgi apparatus and on canalicular membrane
Inability to excrete copper in bile leads to its accumulation in liver and various tissues
Inability to transport copper into Golgi apparatus makes it unavailable for synthesis of ceruloplasmin, leading to release of apoceruloplasmin into serum and its rapid degradation
– Ceruloplasmin functions as plasma ferroxidase, oxidizing ferrous iron for subsequent transfer to plasma apotransferrin, making it available for hemoglobin biosynthesis
CLINICAL ISSUES
Epidemiology
• Incidence
1 in 30,000
Only gold members can continue reading. Log In or Register to continue








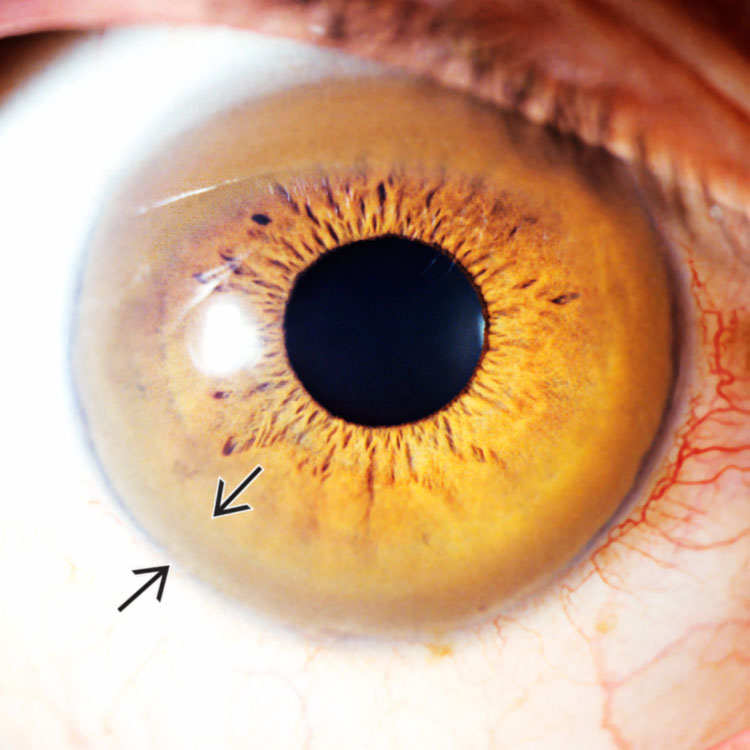
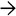 . (Courtesy S. Uwaydat, MD.) Almost half of patients with Wilson disease lack this finding, however.
. (Courtesy S. Uwaydat, MD.) Almost half of patients with Wilson disease lack this finding, however.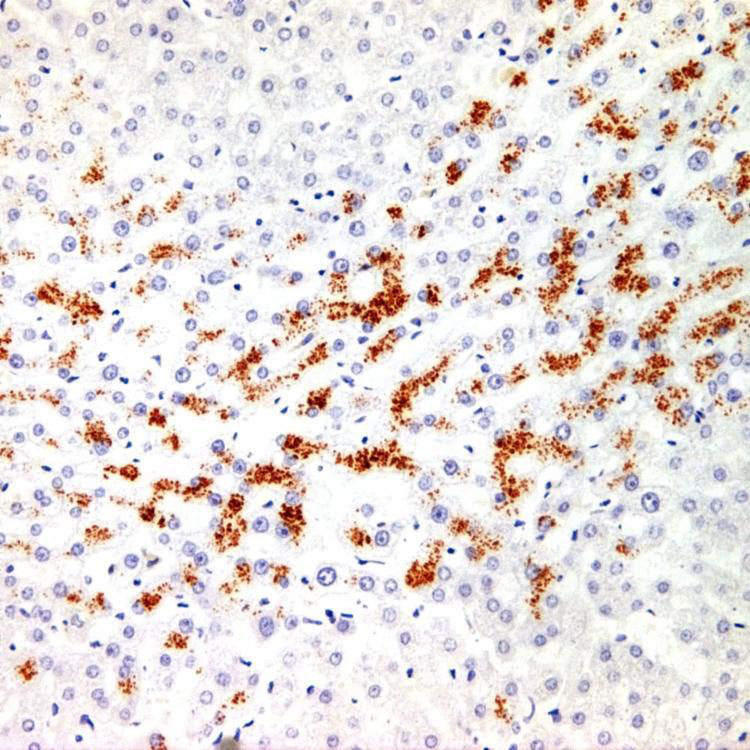
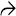 .
.
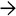 .
. Inability to transport copper into Golgi apparatus makes it unavailable for synthesis of ceruloplasmin, leading to release of apoceruloplasmin into serum and its rapid degradation
Inability to transport copper into Golgi apparatus makes it unavailable for synthesis of ceruloplasmin, leading to release of apoceruloplasmin into serum and its rapid degradation




