Chapter 13 Water, electrolytes and acid–base balance
Distribution and composition of body water
 Intracellular fluid (28 L, about 35% of lean bodyweight)
Intracellular fluid (28 L, about 35% of lean bodyweight)
 Extracellular – the interstitial fluid that bathes the cells (9.4 L, about 12%)
Extracellular – the interstitial fluid that bathes the cells (9.4 L, about 12%)

The intracellular and interstitial fluids are separated by the cell membrane; the interstitial fluid and plasma are separated by the capillary wall (Fig. 13.1). In the absence of solute, water molecules move randomly and in equal numbers in either direction across a semi-permeable membrane. However, if solutes are added to one side of the membrane, the intermolecular cohesive forces reduce the activity of the water molecules. As a result, water tends to stay in the solute-containing compartment because there is less free diffusion across the membrane. This ability to hold water in the compartment can be measured as the osmotic pressure.
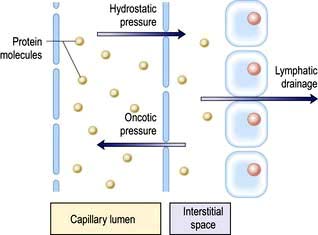
Osmotic pressure
 The intracellular fluid contains mainly potassium (K+) (most of the cell Mg2+ is bound and osmotically inactive)
The intracellular fluid contains mainly potassium (K+) (most of the cell Mg2+ is bound and osmotically inactive)

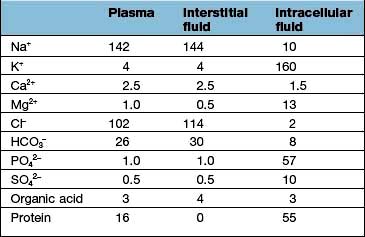
Regulation of the plasma volume is somewhat more complicated because of the tendency of the plasma proteins to hold water in the vascular space by an oncotic effect which is, in part, counterbalanced by the hydrostatic pressure in the capillaries that is generated by cardiac contraction (Fig. 13.1). The composition of intracellular and extracellular fluids is shown in Table 13.1.
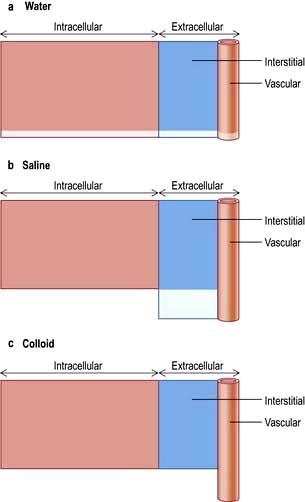
Distribution of different types of replacement fluids
Figure 13.2 shows the relative effects on the compartments of the addition of identical volumes of water, saline and colloid solutions. Thus, 1 L of water given intravenously as 5% glucose is distributed equally into all compartments, whereas the same amount of 0.9% saline remains in the extracellular compartment. The latter is thus the correct treatment for extracellular water depletion – sodium keeping the water in this compartment. The addition of 1 L of colloid with its high oncotic pressure stays in the vascular compartment and is a treatment for hypovolaemia.
Regulation of extracellular volume (Fig. 13.3)
 Plasma fluid constitutes one-third of extracellular volume (4.6 L), and of this,
Plasma fluid constitutes one-third of extracellular volume (4.6 L), and of this,
 85% (3.9 L) lies in the venous side and only 15% (0.7 L) resides in the arterial circulation.
85% (3.9 L) lies in the venous side and only 15% (0.7 L) resides in the arterial circulation.
These changes in Na+ excretion can result from alterations both in the filtered load, determined primarily by the glomerular filtration rate (GFR), and in tubular reabsorption, which is affected by multiple factors. In general, it is changes in tubular reabsorption that constitute the main adaptive response to fluctuations in the effective circulating volume. How this occurs can be appreciated from Table 13.2 and Figure 13.4 and Figure 12.2 (see p. 563), which depicts the sites and determinants of segmental Na+ reabsorption. Although the loop of Henle and distal tubules make a major overall contribution to net Na+ handling, transport in these segments primarily varies with the amount of Na+ delivered; that is, reabsorption is flow-dependent. In comparison, the neurohumoral regulation of Na+ reabsorption according to body needs occurs primarily in the proximal tubules and collecting ducts.

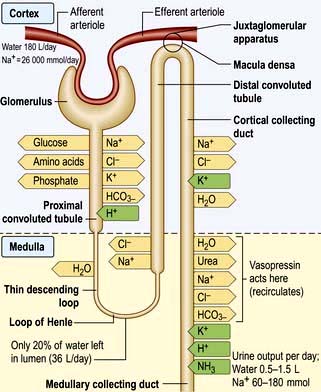
Neurohumoral regulation of extracellular volume
 Intrarenal receptors. Receptors in the walls of the afferent glomerular arterioles respond, via the juxtaglomerular apparatus, to changes in renal perfusion, and control the activity of the renin-angiotensin-aldosterone system (see p. 1006). In addition, sodium concentration in the distal tubule and sympathetic nerve activity alter renin release from the juxtaglomerular cells. Prostaglandins I2 and E2 are also generated within the kidney in response to angiotensin II, acting to maintain glomerular filtration rate and sodium and water excretion, modulating the sodium-retaining effect of this hormone.
Intrarenal receptors. Receptors in the walls of the afferent glomerular arterioles respond, via the juxtaglomerular apparatus, to changes in renal perfusion, and control the activity of the renin-angiotensin-aldosterone system (see p. 1006). In addition, sodium concentration in the distal tubule and sympathetic nerve activity alter renin release from the juxtaglomerular cells. Prostaglandins I2 and E2 are also generated within the kidney in response to angiotensin II, acting to maintain glomerular filtration rate and sodium and water excretion, modulating the sodium-retaining effect of this hormone.
 Extrarenal receptors. These are located in the vascular tree in the left atrium and major thoracic veins, and in the carotid sinus body and aortic arch. These volume receptors respond to a slight reduction in effective circulating volume and result in increased sympathetic nerve activity and a rise in catecholamines. In addition, volume receptors in the cardiac atria control the release of a powerful natriuretic hormone – atrial natriuretic peptide (ANP) – from granules located in the atrial walls (see p. 943).
Extrarenal receptors. These are located in the vascular tree in the left atrium and major thoracic veins, and in the carotid sinus body and aortic arch. These volume receptors respond to a slight reduction in effective circulating volume and result in increased sympathetic nerve activity and a rise in catecholamines. In addition, volume receptors in the cardiac atria control the release of a powerful natriuretic hormone – atrial natriuretic peptide (ANP) – from granules located in the atrial walls (see p. 943).
A salt load, for example, leads to an increase in the effective circulatory and extracellular volume, raising both renal perfusion pressure, and atrial and arterial filling pressure. The increase in the renal perfusion pressure reduces the secretion of renin, and subsequently that of angiotensin II and aldosterone (see Fig. 12.5), whereas the rise in atrial and arterial filling pressure increases the release of ANP. These factors combine to reduce Na+ reabsorption in the collecting duct, thereby promoting excretion of excess Na+.
With more marked hypovolaemia, a decrease in GFR leads to an increase in proximal and thin ascending limb Na+ reabsorption which contributes to Na+ retention. This is brought about by enhanced sympathetic activity acting directly on the kidneys and indirectly by stimulating the secretion of renin/angiotensin II (see Fig. 13.3b) and non-osmotic release of antidiuretic hormone (ADH), also called vasopressin. The pressure natriuresis phenomenon may be the final defence against changes in the effective circulating volume. Marked persistent hypovolaemia leads to systemic hypotension and increased salt and water absorption in the proximal tubules and ascending limb of Henle. This process is partly mediated by changes in renal interstitial hydrostatic pressure and local prostaglandin and nitric oxide production.
Volume regulation in oedematous conditions
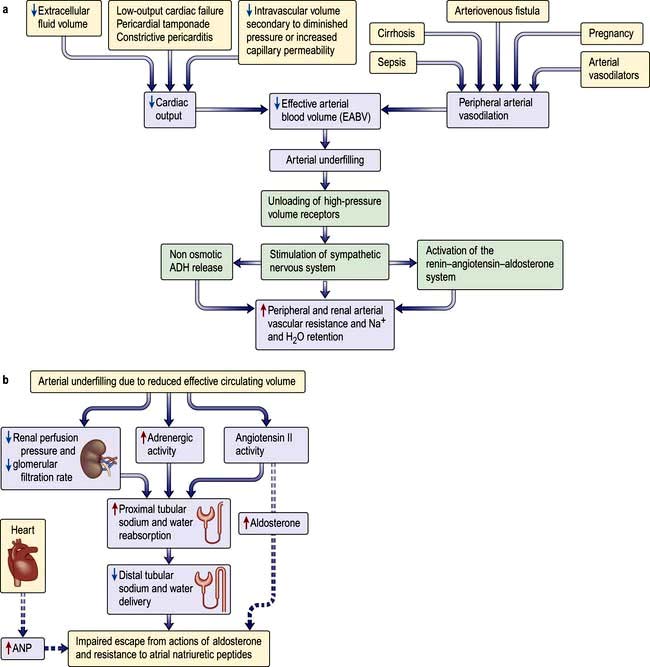
Sodium and water are retained despite increased extracellular volume in oedematous conditions such as cardiac failure, hepatic cirrhosis and hypoalbuminaemia. Here the principal mediator of salt and water retention is the concept of arterial underfilling due either to reduced cardiac output or diminished peripheral arterial resistance. Arterial underfilling in these settings leads to reduction of pressure or stretch (i.e. ‘unloading’ of arterial volume receptors), which results in activation of the sympathetic nervous system, activation of the renin-angiotensin-aldosterone system and non-osmotic release of ADH. These neurohumoral mediators promote salt and water retention in the face of increased extracellular volume. The common nature of the degree of arterial fullness and neurohumoral pathway in the regulation of extracellular volume in health and disease states forms the basis of Schrier’s unifying hypothesis of volume homeostasis (Fig. 13.3a).
Mechanism of impaired escape from actions of aldosterone and resistance to ANP
Not only is the activity of the renin-angiotensin-aldosterone system increased in oedematous conditions such as cardiac failure, hepatic cirrhosis and hypoalbuminaemia, but also the action of aldosterone is more persistent than in normal subjects and patients with Conn’s syndrome, who have increased aldosterone secretion (see p. 989).
In patients with the above oedematous conditions, e.g. heart failure, escape from the sodium-retaining actions of aldosterone does not occur and therefore they continue to retain sodium in response to aldosterone. Accordingly they have substantial natriuresis when given spironolactone, which blocks mineralocorticoid receptors. Alpha-adrenergic stimulation and elevated angiotensin II increase sodium transport in the proximal tubule, and reduced renal perfusion and GFR further increases sodium absorption from the proximal tubules by presenting less sodium and water in the tubular fluid. Sodium delivery to the distal portion of the nephron, and thus the collecting duct, is reduced. Similarly, increased cardiac ANP release in these conditions requires optimum sodium concentration at the site of its action in the collecting duct for its desired natriuretic effects. Decreased sodium delivery to the collecting duct is therefore the most likely explanation for the persistent aldosterone-mediated sodium retention, absence of escape phenomenon and resistance to natriuretic peptides in these patients (Fig. 13.3b).
Regulation of water excretion



The cortical collecting duct has two cell types (see also p. 597) with very different functions:
 Principal cells (about 65%) have sodium and potassium channels in the apical membrane and, as in all sodium-reabsorbing cells, Na+/K+-ATPase pumps in the basolateral membrane.
Principal cells (about 65%) have sodium and potassium channels in the apical membrane and, as in all sodium-reabsorbing cells, Na+/K+-ATPase pumps in the basolateral membrane.

The ADH-induced increase in collecting duct water permeability occurs primarily in the principal cells. ADH acts on V2 (vasopressin) receptors located on the basolateral surface of principal cells, resulting in the activation of adenyl cyclase. This leads to protein kinase activation and to preformed cytoplasmic vesicles that contain unique water channels (called aquaporins) moving to and then being inserted into the luminal membrane. Four renal aquaporins have been well characterized and are localized in different areas of the cells of the collecting duct. The water channels span the luminal membrane and permit water movement into the cells down a favourable osmotic gradient (Fig. 13.5). This water is then rapidly returned to the systemic circulation across the basolateral membrane. When the ADH effect has worn off, the water channels aggregate within clathrin-coated pits, from which they are removed from the luminal membrane by endocytosis and returned to the cytoplasm. A defect in any step in this pathway, such as in attachment of ADH to its receptor or the function of the water channel, can cause resistance to the action of ADH and an increase in urine output. This disorder is called nephrogenic diabetes insipidus.
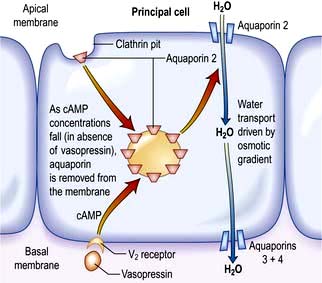
Plasma osmolality
1. Ingestion of a water load leads to an initial reduction in the plasma osmolality, thereby diminishing the release of ADH. The ensuing reduction in water reabsorption in the collecting ducts allows the excess water to be excreted in a dilute urine.
2. Water loss resulting from sweating is followed by, in sequence, a rise in both plasma osmolality and ADH secretion, enhanced water reabsorption, and the appropriate excretion of a small volume of concentrated urine. This renal effect of ADH minimizes further water loss but does not replace the existing water deficit. Thus, optimal osmoregulation requires an increase in water intake, which is mediated by a concurrent stimulation of thirst. The importance of thirst can also be illustrated by studies in patients with central diabetes insipidus, who are deficient in ADH. These patients often complain of marked polyuria, which is caused by the decline in water reabsorption in the collecting ducts. However, they do not typically become hypernatraemic, because urinary water loss is offset by the thirst mechanism.
Osmoregulation versus volume regulation
The roles of these two pathways should be considered separately when evaluating patients.
 A water load is rapidly excreted (in 4–6 h) by inhibition of ADH release so that there is little or no water reabsorption in the collecting ducts. This process is normally so efficient that volume regulation is not affected and there is no change in ANP release or in the activity of the renin-angiotensin-aldosterone system. Thus, a dilute urine is excreted, and there is little alteration in the excretion of Na+.
A water load is rapidly excreted (in 4–6 h) by inhibition of ADH release so that there is little or no water reabsorption in the collecting ducts. This process is normally so efficient that volume regulation is not affected and there is no change in ANP release or in the activity of the renin-angiotensin-aldosterone system. Thus, a dilute urine is excreted, and there is little alteration in the excretion of Na+.

This principle of separate volume and osmoregulatory pathways is also evident in the syndrome of inappropriate ADH secretion (SIADH). Patients with SIADH (see p. 993) have impaired water excretion and hyponatraemia (dilutional) caused by the persistent presence of ADH. However, the release of ANP and aldosterone is not impaired and, thus, Na+ handling remains intact. These findings have implications for the correction of the hyponatraemia in this setting which initially requires restriction of water intake.
Increased extracellular volume
 Venous tone, which determines the capacitance of the blood compartment and thus hydrostatic pressure
Venous tone, which determines the capacitance of the blood compartment and thus hydrostatic pressure

 Oncotic pressure – mainly dependent on serum albumin
Oncotic pressure – mainly dependent on serum albumin

Causes
Heart failure
Reduction in cardiac output and the consequent fall in effective circulatory volume and arterial filling lead to activation of the renin-angiotensin-aldosterone system, non-osmotic release of ADH, and increased activity of the renal sympathetic nerves via volume receptors and baroreceptors (Fig. 13.3a). Sympathetic overdrive also indirectly augments ADH and renin-angiotensin-aldosterone response in these conditions. The cumulative effect of these mediators results in increased peripheral and renal arteriolar resistance and water and sodium retention. These factors result in extracellular volume expansion and increased venous pressure, causing oedema formation.
Hepatic cirrhosis
The mechanism is complex, but involves peripheral vasodilatation (possibly owing to increased nitric oxide generation) resulting in reduced effective arterial blood volume (EABV) and arterial filling. This leads to an activation of a chain of events common to cardiac failure and other conditions with marked peripheral vasodilatation (Fig. 13.3). The cumulative effect results in increased peripheral and renal resistance, water and sodium retention, and oedema formation.
Nephrotic syndrome
Interstitial oedema is a common clinical finding with hypoalbuminaemia, particularly in the nephrotic syndrome. Expansion of the interstitial compartment is secondary to the accumulation of sodium in the extracellular compartment. This is due to an imbalance between oral (or parenteral) sodium intake and urinary sodium output, as well as alterations of fluid transfer across capillary walls. The intrarenal site of sodium retention is the cortical collecting duct (CCD) where Na+/K+-ATPase expression and activity are increased threefold along the basolateral surface (Fig. 13.4). In addition, amiloride-sensitive epithelial sodium channel activity is also increased in the CCD. The renal sodium retention should normally be counterbalanced by increased secretion of sodium in the inner medullary collecting duct, brought about by the release of ANP. This regulatory pathway is altered in patients with nephrotic syndrome by enhanced kidney specific catabolism of cyclic GMP (the second messenger for ANP) following phosphodiesterase activation.
Oedema generation was classically attributed to the decrease in the plasma oncotic pressure and the subsequent increase in the transcapillary oncotic gradient. However, the oncotic pressure and transcapillary oncotic gradient remain unchanged and the transcapillary hydrostatic pressure gradient is not altered. Conversely, capillary hydraulic conductivity (a measure of permeability) is increased. This is determined by intercellular macromolecular complexes between the endothelial cells consisting of tight junctions (made of occludins, claudins and ZO proteins) and adherens junctions (made of cadherin, catenins and actin cytoskeleton). Elevated TNF-α levels in nephrotic syndrome activate protein kinase C, which changes phosphorylation of occludin and capillary permeability. In addition, increased circulating ANP can increase capillary hydraulic conductivity by altering the permeability of intercellular junctional complexes. Furthermore, reduction in effective circulatory volume and the consequent fall in cardiac output and arterial filling can lead to a chain of events as in cardiac failure and cirrhosis (see above and Fig. 13.3). These factors result in extracellular volume expansion and oedema formation.
Sodium retention
A decreased GFR decreases the renal capacity to excrete sodium. This may be acute, as in the acute nephritic syndrome (see p. 582), or may occur as part of the presentation of chronic kidney disease. In end-stage renal failure, extracellular volume is controlled by the balance between salt intake and its removal by dialysis.
 Oestrogens cause mild sodium retention, due to a weak aldosterone-like effect. This is the cause of weight gain in the premenstrual phase.
Oestrogens cause mild sodium retention, due to a weak aldosterone-like effect. This is the cause of weight gain in the premenstrual phase.


 Thiazolidinediones (TZD) (see p. 1011) are widely used to treat type 2 diabetes. Their mechanism of action is attributed to binding and activation of the PPAR-γ system. PPARs are nuclear transcription factors essential to the control of energy metabolism that are modulated via binding with tissue-specific fatty acid metabolites. Of the three PPAR isoforms, γ has been extensively studied and is expressed at high levels in adipose and liver tissues, macrophages, pancreatic-β cells and principal cells of the collecting duct. These drugs have been asociated with salt and water retention and are contraindicated in patients with heart failure. Recent evidence suggests that TZD-induced oedema (like insulin) is also due to upregulation of epithelial Na transporter channel (ENaC) but by different pathways. Diuretics of choice for TZD-induced oedema are amiloride and triamterene.
Thiazolidinediones (TZD) (see p. 1011) are widely used to treat type 2 diabetes. Their mechanism of action is attributed to binding and activation of the PPAR-γ system. PPARs are nuclear transcription factors essential to the control of energy metabolism that are modulated via binding with tissue-specific fatty acid metabolites. Of the three PPAR isoforms, γ has been extensively studied and is expressed at high levels in adipose and liver tissues, macrophages, pancreatic-β cells and principal cells of the collecting duct. These drugs have been asociated with salt and water retention and are contraindicated in patients with heart failure. Recent evidence suggests that TZD-induced oedema (like insulin) is also due to upregulation of epithelial Na transporter channel (ENaC) but by different pathways. Diuretics of choice for TZD-induced oedema are amiloride and triamterene.
Other causes of oedema
 Initiation of insulin treatment for type 1 diabetes and refeeding after malnutrition are both associated with the development of transient oedema. The mechanism is complex but involves upregulation of ENaC in the principal cell of the collecting duct. This transporter is amiloride sensitive which makes amiloride or triamterene the diuretic of choice in insulin-induced oedema.
Initiation of insulin treatment for type 1 diabetes and refeeding after malnutrition are both associated with the development of transient oedema. The mechanism is complex but involves upregulation of ENaC in the principal cell of the collecting duct. This transporter is amiloride sensitive which makes amiloride or triamterene the diuretic of choice in insulin-induced oedema.

 Oedema is also caused by increased interstitial oncotic pressure as a result of increased capillary permeability to proteins. This can occur as part of a rare complement-deficiency syndrome; with therapeutic use of interleukin 2 in cancer chemotherapy; or in ovarian hyperstimulation syndrome (see p. 981).
Oedema is also caused by increased interstitial oncotic pressure as a result of increased capillary permeability to proteins. This can occur as part of a rare complement-deficiency syndrome; with therapeutic use of interleukin 2 in cancer chemotherapy; or in ovarian hyperstimulation syndrome (see p. 981).
Treatment
The mainstay of treatment is the use of diuretic agents, which increase sodium, chloride and water excretion in the kidney (Table 13.3). These agents act by interfering with membrane ion pumps which are present on numerous cell types; they mostly achieve specificity for the kidney by being secreted into the proximal tubule, resulting in much higher concentrations in the tubular fluid than in other parts of the body.
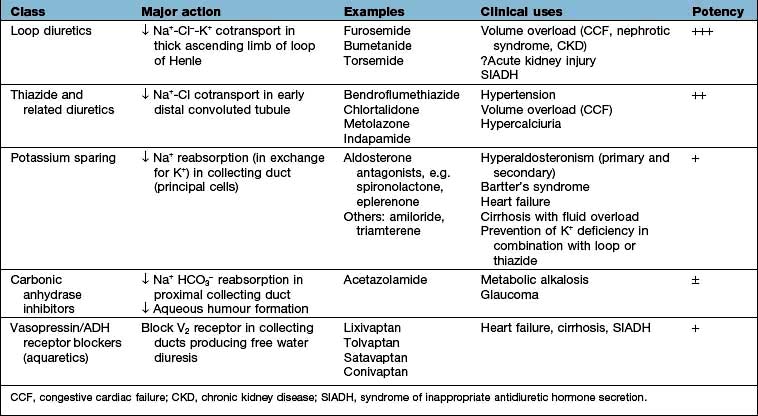
Clinical use of diuretics
Loop diuretics
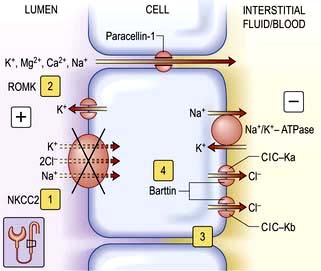
These potent diuretics are useful in the treatment of any cause of systemic extracellular volume overload. They stimulate excretion of both sodium chloride and water by blocking the sodium-potassium-2-chloride (NKCC2) channel in the thick ascending limb of Henle (Fig. 13.6) and are useful in stimulating water excretion in states of relative water overload. They also act by causing increased venous capacitance, resulting in rapid clinical improvement in patients with left ventricular failure, preceding the diuresis. Unwanted effects include:




 Allergic tubulointerstitial nephritis and other allergic reactions
Allergic tubulointerstitial nephritis and other allergic reactions
 Myalgia – especially with high-dose bumetanide
Myalgia – especially with high-dose bumetanide

 Interference with excretion of lithium, resulting in toxicity.
Interference with excretion of lithium, resulting in toxicity.
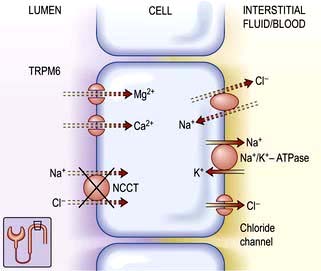
Thiazide diuretics (see p. 719)
These are less potent than loop diuretics. They act by blocking a sodium chloride channel in the distal convoluted tubule (Fig. 13.7). They cause relatively more urate retention, glucose intolerance and hypokalaemia than loop diuretics. They interfere with water excretion and may cause hyponatraemia, particularly if combined with amiloride or triamterene. This effect is clinically useful in diabetes insipidus. Thiazides reduce peripheral vascular resistance by mechanisms that are not completely understood but do not appear to depend on their diuretic action, and are widely used in the treatment of essential hypertension. They are also used extensively in mild to moderate cardiac failure. Thiazides reduce calcium excretion. This effect is useful in patients with idiopathic hypercalciuria, but may cause hypercalcaemia. Numerous agents are available, with varying half-lives but little else to choose between them. Metolazone is not dependent for its action on glomerular filtration, and therefore retains its potency in renal impairment.
Potassium-sparing diuretics (see Fig. 13.8)
 Aldosterone antagonists, which compete with aldosterone in the collecting ducts and reduce sodium absorption, e.g. spironolactone and eplerenone (which has a shorter half-life). Spironolactone is used in patients with heart failure because it significantly reduces the mortality in these patients by antagonizing the fibrotic effect of aldosterone on the heart. Eplerenone is devoid of antiandrogenic or antiprogesterone properties.
Aldosterone antagonists, which compete with aldosterone in the collecting ducts and reduce sodium absorption, e.g. spironolactone and eplerenone (which has a shorter half-life). Spironolactone is used in patients with heart failure because it significantly reduces the mortality in these patients by antagonizing the fibrotic effect of aldosterone on the heart. Eplerenone is devoid of antiandrogenic or antiprogesterone properties.

Aquaretics (vasopressin or antidiuretic hormone antagonists)
Vasopressin V2 receptor antagonists are very useful agents in the treatment of conditions associated with elevated levels of vasopressin, such as heart failure, cirrhosis and SIADH (see p. 993). Non-peptide vasopressin V2 receptor antagonists are efficacious in producing free water diuresis in humans. Studies in patients with heart failure and cirrhosis suggest that such agents will allow normalization of serum osmolality with less water restriction (see p. 650).
Resistance to diuretics
Resistance may occur as a result of:






Decreased extracellular volume
Clinical features
 Loss of interstitial fluid leads to loss of skin elasticity (’turgor’) – the rapidity with which the skin recoils to normal after being pinched. Skin turgor decreases with age, particularly at the peripheries. The turgor over the anterior triangle of the neck or on the forehead is a very useful sign in all ages.
Loss of interstitial fluid leads to loss of skin elasticity (’turgor’) – the rapidity with which the skin recoils to normal after being pinched. Skin turgor decreases with age, particularly at the peripheries. The turgor over the anterior triangle of the neck or on the forehead is a very useful sign in all ages.

Loss of more than this causes the following:
Postural hypotension
Normally the blood pressure rises if a subject stands up, as a result of increased venous return due to venoconstriction (this maintains cerebral perfusion). Loss of extracellular fluid (underfill) prevents this and causes a fall in blood pressure. This is one of the earliest and most reliable signs of volume depletion, as long as the other causes of postural hypotension are excluded (Table 13.4).
Table 13.4 Postural hypotension: some causes of a fall in blood pressure from lying to standing
Causes
Salt and water may be lost from the kidneys, from the gastrointestinal tract, or from the skin. Examples are given in Table 13.5.
Table 13.5 Causes of extracellular volume depletion



Investigations
Assessment of volume status is shown in Box 13.1.
 Box 13.1
Box 13.1





Treatment
The overriding principle is to replace what is missing.
Loss of plasma
Loss of plasma, as occurs in burns or severe peritonitis, should be treated with human plasma or a plasma substitute (see p. 390).
Loss of water and electrolytes
Sodium bicarbonate (500 mg, 6 mmol each of Na+ and HCO3− per tablet) is used in doses of 6–12 tablets/day with 2–3 L of water. This is used in milder chronic sodium depletion with acidosis (e.g. chronic kidney disease, post-obstructive renal failure, renal tubular acidosis). Sodium bicarbonate is less effective than sodium chloride in causing positive sodium balance. Oral rehydration solutions are described in Box 4.10.
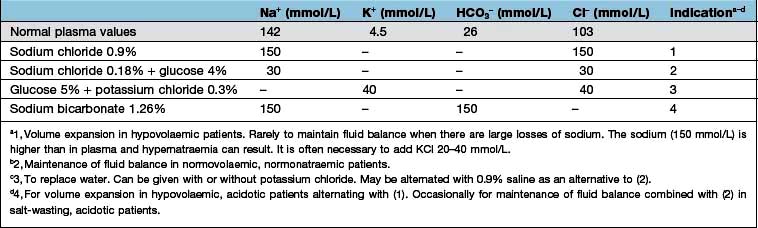
Intravenous fluids are sometimes required (Table 13.6). Rapid infusion (e.g. 1000 mL per hour or even faster) is necessary if there is hypotension and evidence of impaired organ perfusion (e.g. oliguria, confusion); in these situations, plasma expanders (colloids) are often used in the first instance to restore an adequate circulating volume (see p. 887). Repeated clinical assessments are vital in this situation, usually complemented by frequent measurements of central venous pressure (see p. 872, for the management of shock). Severe hypovolaemia induces venoconstriction, which maintains venous return; over-rapid correction does not give time for this to reverse, resulting in signs of circulatory overload (e.g. pulmonary oedema) even if a total body extracellular fluid (ECF) deficit remains. In less severe ECF depletion (such as in a patient with postural hypotension complicating acute tubular necrosis), the fluid should be replaced at a rate of 1000 mL every 4–6 h, again with repeated clinical assessment. If all that is required is avoidance of fluid depletion during surgery, 1–2 L can be given over 24 h, remembering that surgery is a stimulus to sodium and water retention and that over-replacement may be as dangerous as under-replacement. Regular monitoring by fluid balance charts, bodyweight and plasma biochemistry is mandatory.
Loss of water alone
FURTHER READING
Ahmed MS, Wong CF, Pai P. Cardiorenal syndrome – a new classification and current evidence on its management. Clin Nephrol 2010; 74(4):245–257.
Bie P. Blood volume, blood pressure and total body sodium: internal signaling and output control. Acta Physiol (Oxford) 2009; 195(1):187–196.
Bie P, Damkjaer M. Renin secretion and total body sodium: pathways of integrative control. Clin Exp Pharmacol Physiol 2010; 37(2):e34–42.
Schrier RW. Molecular mechanisms of clinical concentrating and diluting disorders. Prog Brain Res 2008; 170:539–550.
Wakil A, Atkin SL. Serum sodium disorders: safe management. Clin Med 2010; 10:79–82.
Disorders of sodium concentration
Hyponatraemia



Table 13.7 Causes of hyponatraemia with decreased extracellular volume (hypovolaemia)
| Extrarenal (urinary sodium <20 mmol/L) | Kidney (urinary sodium >20 mmol/L) |
|---|---|
Table 13.8 Causes of hyponatraemia with normal extracellular volume (euvolaemia)
Table 13.9 Causes of hyponatraemia with increased extracellular volume (hypervolaemia)
Heart failure | Oliguric kidney injury |
Liver failure | Hypoalbuminaemia |
Hyponatraemia with hypovolaemia
This is due to salt loss in excess of water loss; the causes are listed in Table 13.7. In this situation, ADH secretion is initially suppressed (via the hypothalamic osmoreceptors); but as fluid volume is lost, volume receptors override the osmoreceptors and stimulate both thirst and the release of ADH. This is an attempt by the body to defend circulating volume at the expense of osmolality.
Clinical features
With sodium depletion the clinical picture is usually dominated by features of volume depletion (see p. 638). The diagnosis is usually obvious where there is a history of gut losses, diabetes mellitus or diuretic abuse. Examination of the patient is often more helpful than the biochemical investigations, which include plasma and urine electrolytes and osmolality.
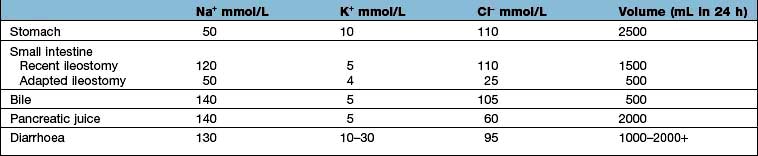
Table 13.10 shows the potential daily losses of water and electrolytes from the gut. Losses due to renal or adrenocortical disease may be less easily identified but a urinary sodium concentration of >20 mmol/L, in the presence of clinically evident volume depletion, suggests a renal loss.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


