Use of Vasopressors and Inotropes in the Pharmacotherapy of Shock
KEY CONCEPTS
![]() Continuous hemodynamic monitoring with an arterial catheter or a central venous catheter capable of measuring mixed venous oxygen saturation (SVO2) or central venous oxygen saturation (SCVO2) should be used early and throughout the course of septic shock to assess intravascular fluid status and ventricular filling pressures, determine cardiac output (CO), and monitor arterial and venous oxygenation. They can be used for monitoring the response to drug therapy and guiding dosage titration.
Continuous hemodynamic monitoring with an arterial catheter or a central venous catheter capable of measuring mixed venous oxygen saturation (SVO2) or central venous oxygen saturation (SCVO2) should be used early and throughout the course of septic shock to assess intravascular fluid status and ventricular filling pressures, determine cardiac output (CO), and monitor arterial and venous oxygenation. They can be used for monitoring the response to drug therapy and guiding dosage titration.
![]() Early goal-directed therapy with aggressive fluid resuscitation in the emergency department within the first 6 hours of presentation improves survival of patients with sepsis and septic shock.
Early goal-directed therapy with aggressive fluid resuscitation in the emergency department within the first 6 hours of presentation improves survival of patients with sepsis and septic shock.
![]() Lactate production is increased under anaerobic conditions. Elevated serum lactate concentrations represent global perfusion abnormalities. Lactate clearance may be used to assess repayment of oxygen to the tissues. GI tonometry and sublingual capnometry represent methods of assessing regional perfusion but are used infrequently.
Lactate production is increased under anaerobic conditions. Elevated serum lactate concentrations represent global perfusion abnormalities. Lactate clearance may be used to assess repayment of oxygen to the tissues. GI tonometry and sublingual capnometry represent methods of assessing regional perfusion but are used infrequently.
![]() Derangements in adrenergic receptor sensitivity or activity frequently result in resistance to catecholamine vasopressor and inotropic therapy in critically ill patients. These changes may be a function of endogenous catecholamine concentrations, dosage/duration of exposure to and type of exogenously administered vasopressors, stage of septic shock, preexisting illness, and other factors.
Derangements in adrenergic receptor sensitivity or activity frequently result in resistance to catecholamine vasopressor and inotropic therapy in critically ill patients. These changes may be a function of endogenous catecholamine concentrations, dosage/duration of exposure to and type of exogenously administered vasopressors, stage of septic shock, preexisting illness, and other factors.
![]() In refractory septic shock, rational use of vasopressor or inotropic agents should be guided by receptor activity, pharmacologic and pharmacokinetic characteristics, and regional and systemic hemodynamic effects of the drug and should be tailored to the patient’s physiologic needs. Pharmacologically sound combinations of vasopressor and/or inotrope agents should be initiated early to optimize and facilitate rapid response.
In refractory septic shock, rational use of vasopressor or inotropic agents should be guided by receptor activity, pharmacologic and pharmacokinetic characteristics, and regional and systemic hemodynamic effects of the drug and should be tailored to the patient’s physiologic needs. Pharmacologically sound combinations of vasopressor and/or inotrope agents should be initiated early to optimize and facilitate rapid response.
![]() Goals of therapy with vasopressors and inotropes should be predetermined and should optimize global and regional perfusion parameters (e.g., cardiac, renal, mesenteric, and periphery) to normalize cellular metabolism. This can be accomplished by continuous or intermittent measurements. Targeted goals should be central venous pressure (CVP) of 8 to 12 mm Hg (up to 15 mm Hg in mechanically ventilated patients, patients with preexisting left ventricular dysfunction, or patients with abdominal distension), mean arterial pressure (MAP) ≥65 mm Hg, SVO2 ≥65% or SCVO2 ≥70%, and lactate clearance of ≥20%.
Goals of therapy with vasopressors and inotropes should be predetermined and should optimize global and regional perfusion parameters (e.g., cardiac, renal, mesenteric, and periphery) to normalize cellular metabolism. This can be accomplished by continuous or intermittent measurements. Targeted goals should be central venous pressure (CVP) of 8 to 12 mm Hg (up to 15 mm Hg in mechanically ventilated patients, patients with preexisting left ventricular dysfunction, or patients with abdominal distension), mean arterial pressure (MAP) ≥65 mm Hg, SVO2 ≥65% or SCVO2 ≥70%, and lactate clearance of ≥20%.
![]() Dose titration and monitoring of vasopressor and inotropic therapy should be guided by the “best clinical response” while observing for and minimizing evidence of myocardial ischemia (e.g., tachydysrhythmias, electrocardiographic changes, troponin elevation), renal (decreased glomerular filtration rate and/or urine production), splanchnic/gastric (low intramucosal pH, bowel ischemia), or peripheral (cold extremities) hypoperfusion, and worsening of partial pressure of arterial oxygen (PaO2), pulmonary artery occlusive pressure (PAOP), and other hemodynamic variables.
Dose titration and monitoring of vasopressor and inotropic therapy should be guided by the “best clinical response” while observing for and minimizing evidence of myocardial ischemia (e.g., tachydysrhythmias, electrocardiographic changes, troponin elevation), renal (decreased glomerular filtration rate and/or urine production), splanchnic/gastric (low intramucosal pH, bowel ischemia), or peripheral (cold extremities) hypoperfusion, and worsening of partial pressure of arterial oxygen (PaO2), pulmonary artery occlusive pressure (PAOP), and other hemodynamic variables.
![]() Much higher dosages of all vasopressors and inotropes than traditionally recommended are required to improve hemodynamic and oxygen-transport variables in patients with septic shock. Arbitrarily targeting vasopressor and inotrope therapy to supranormal values of global oxygen-transport variables cannot be recommended because of the lack of clear benefit and possible increased morbidity.
Much higher dosages of all vasopressors and inotropes than traditionally recommended are required to improve hemodynamic and oxygen-transport variables in patients with septic shock. Arbitrarily targeting vasopressor and inotrope therapy to supranormal values of global oxygen-transport variables cannot be recommended because of the lack of clear benefit and possible increased morbidity.
![]() First-line therapy of septic shock is aggressive volume resuscitation with crystalloid or colloid types of fluids. Norepinephrine is the preferred initial vasopressor agent for hemodynamic support. Norepinephrine achieves greater hemodynamic response than dopamine and is less likely to cause tachydysrhythmias and a decrease in splanchnic oxygen utilization. Dopamine is also limited by its inability to adequately increase CO and complications of increased PAOP and decreased splanchnic oxygen use. Low-dose dopamine should not be used to prevent renal failure.
First-line therapy of septic shock is aggressive volume resuscitation with crystalloid or colloid types of fluids. Norepinephrine is the preferred initial vasopressor agent for hemodynamic support. Norepinephrine achieves greater hemodynamic response than dopamine and is less likely to cause tachydysrhythmias and a decrease in splanchnic oxygen utilization. Dopamine is also limited by its inability to adequately increase CO and complications of increased PAOP and decreased splanchnic oxygen use. Low-dose dopamine should not be used to prevent renal failure.
![]() Phenylephrine may be a particularly useful alternative in patients who cannot tolerate tachycardia or tachydysrhythmia associated with the use of other agents. Its effects on cardiac performance and splanchnic oxygen utilization are variable.
Phenylephrine may be a particularly useful alternative in patients who cannot tolerate tachycardia or tachydysrhythmia associated with the use of other agents. Its effects on cardiac performance and splanchnic oxygen utilization are variable.
![]() Epinephrine is an effective initial agent and as an add-on agent. It is particularly useful in the young, in patients with otherwise healthy myocardium, and potentially in patients when used early in the course of treatment. However, because epinephrine causes a significant increase in lactate and worsening of splanchnic oxygen utilization, it is not the agent of first choice in patients with septic shock and is reserved as adjunctive therapy when other vasopressors do not adequately increase MAP. It should be used cautiously in patients with a history of coronary artery disease or underlying cardiac disturbances.
Epinephrine is an effective initial agent and as an add-on agent. It is particularly useful in the young, in patients with otherwise healthy myocardium, and potentially in patients when used early in the course of treatment. However, because epinephrine causes a significant increase in lactate and worsening of splanchnic oxygen utilization, it is not the agent of first choice in patients with septic shock and is reserved as adjunctive therapy when other vasopressors do not adequately increase MAP. It should be used cautiously in patients with a history of coronary artery disease or underlying cardiac disturbances.
![]() Dobutamine may be used as adjunctive therapy for its inotropic effect. It enhances CO and may increase SVO2/SCVO2. Concurrent vasopressor therapy is needed because dobutamine causes vasodilation. Dobutamine therapy may be limited by tachycardia and dysrhythmias.
Dobutamine may be used as adjunctive therapy for its inotropic effect. It enhances CO and may increase SVO2/SCVO2. Concurrent vasopressor therapy is needed because dobutamine causes vasodilation. Dobutamine therapy may be limited by tachycardia and dysrhythmias.
![]() Therapy with vasopressors and inotropes is continued until the myocardial depression and vascular hyporesponsiveness of septic shock improve, usually measured in hours to days. Discontinuation of vasopressor or inotropic therapy should be executed slowly; therapy should be “weaned” to avoid a precipitous worsening in regional and systemic hemodynamics.
Therapy with vasopressors and inotropes is continued until the myocardial depression and vascular hyporesponsiveness of septic shock improve, usually measured in hours to days. Discontinuation of vasopressor or inotropic therapy should be executed slowly; therapy should be “weaned” to avoid a precipitous worsening in regional and systemic hemodynamics.
![]() Vasopressin produces vasoconstriction independent of adrenergic receptors and reduces the dosages of catecholamine vasopressors. Replacement dosages of vasopressin (0.01 to 0.04 units/min) can be considered in patients with septic shock refractory to catecholamine vasopressors despite adequate fluid resuscitation. Dosage rates should not be titrated upward. Vasopressin may enhance urine production but it may worsen splanchnic and peripheral perfusion. Given the current data, corticosteroids can be administered to patients with septic shock refractory to vasopressors or when adrenal insufficiency is suspected. Side effects of short-term corticosteroids are minimal.
Vasopressin produces vasoconstriction independent of adrenergic receptors and reduces the dosages of catecholamine vasopressors. Replacement dosages of vasopressin (0.01 to 0.04 units/min) can be considered in patients with septic shock refractory to catecholamine vasopressors despite adequate fluid resuscitation. Dosage rates should not be titrated upward. Vasopressin may enhance urine production but it may worsen splanchnic and peripheral perfusion. Given the current data, corticosteroids can be administered to patients with septic shock refractory to vasopressors or when adrenal insufficiency is suspected. Side effects of short-term corticosteroids are minimal.
Shock is an acute, generalized state of inadequate perfusion of critical organs that can produce serious pathophysiologic consequences, including death, when therapy is not optimal. Shock is defined as systolic blood pressure <90 mm Hg or reduction of at least 40 mm Hg from baseline with perfusion abnormalities despite adequate fluid resuscitation.1 Previously, mortality from septic or cardiogenic shock exceeded 70% but now ranges between 20% and 40%.2–5 This chapter reviews the theory and current status of hemodynamic monitoring and presents an update on the optimal use of inotropes and vasopressor drugs in shock states, specifically septic shock.6–15
The general goal of therapy during resuscitation from shock is to achieve and maintain mean arterial pressure (MAP) above 65 mm Hg while ensuring adequate perfusion to the critical organs.6–15 Hemodynamic and perfusion monitoring can be categorized into two broad areas: global versus regional monitoring. Global parameters, such as systemic blood pressure, oxygen tension, and lactate, assess perfusion and oxygen utilization of the entire body. Regional monitoring techniques, such as tonometry or cardiac function, focus on oxygen delivery and subsequent changes in metabolism of individual organs and tissues. Patients in shock generally have several modes of monitoring so therapies are based on all gathered information and correlating with the patient’s clinical response. Normal values for commonly monitored parameters are listed in Table 13-1. Evidence-based goals of therapy are listed in Table 13-2.6–21 The adequacy of regional perfusion can be assessed by indices of specific organ perfusion.6,15–21 These measurements include coagulation abnormalities (disseminated intravascular coagulation), altered renal function with reduced urine production or increased serum concentrations of blood urea nitrogen and creatinine, altered hepatic parenchymal function with increased serum concentrations of transaminases and bilirubin, altered GI perfusion manifested by ileus and diminished bowel sounds, cool extremities, cardiac ischemia with elevated troponin concentrations and electrocardiographic changes, and altered sensorium. Although none of these indices alone is a reliable indicator of adequate resuscitation, they offer immediate detection and may be prognostic of recovery when combined and defined at the level of organ function. As a result, these indices are frequently used as surrogate endpoints for the goals of resuscitation.22 While it is assumed that normalization of these parameters infers benefit, the clinician must first treat the patient clinically rather than relying solely on data from continuous monitoring to guide therapy.22
TABLE 13-1 Hemodynamic and Oxygen-Transport Monitoring Parameters
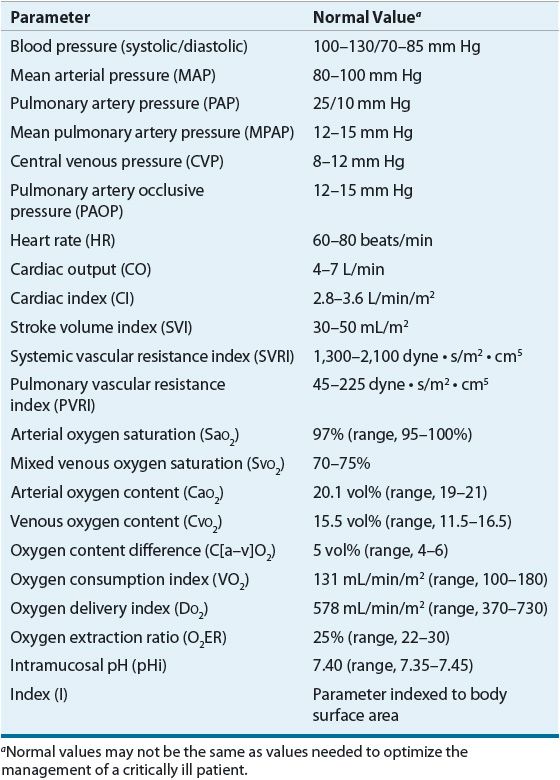
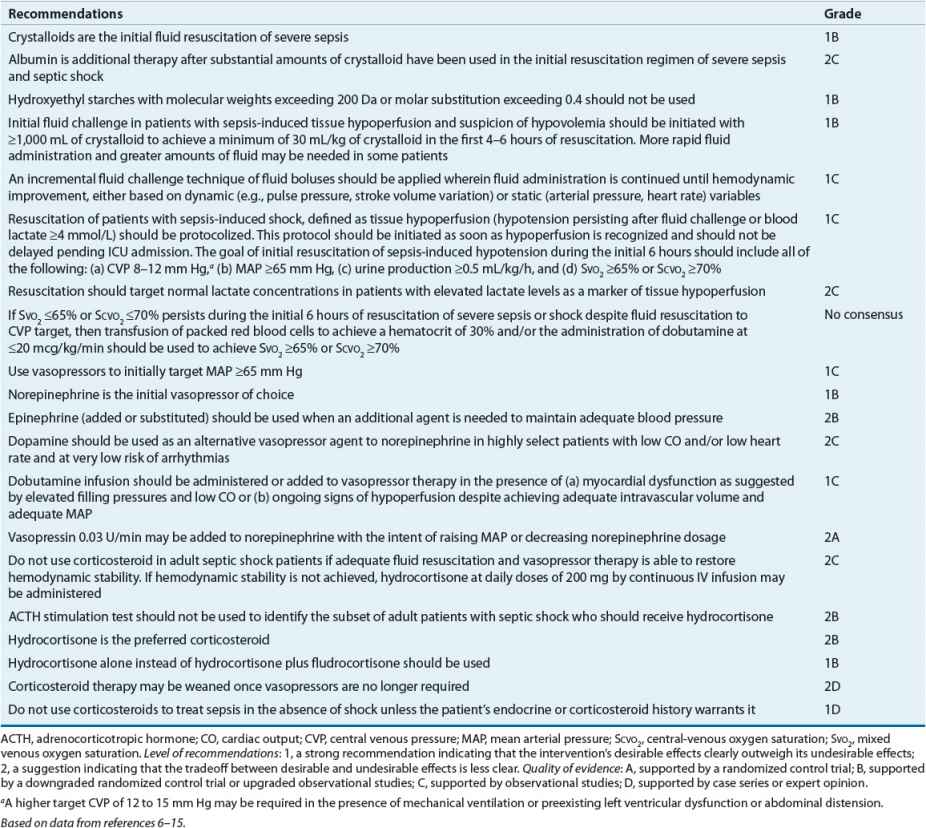
TABLE 13-2 Evidence-Based Treatment Recommendations for Management of Severe Sepsis or Septic Shock
GLOBAL PERFUSION MONITORING
Arterial Blood Pressure Measurement
MAP is the product of cardiac output (CO) and systemic vascular resistance (SVR). Conditions that may lower blood pressure through diminished CO in critically ill patients include cardiac failure (etiology may be myocardial infarction, arrhythmia, acute heart failure, or valvular disease) and hypovolemia (etiology may be hemorrhage, intractable diarrhea, or heat stroke). Vasodilatory conditions such as sepsis, anaphylaxis, pancreatitis, acute hepatic failure, or neurotrauma, lower blood pressure by reducing SVR. Arterial blood pressure is the commonly used end point of therapy; however, restoration of adequate perfusion pressure is the primary criterion of effectiveness.6–21 Profound hypotension (MAP <60 mm Hg) is associated with pressure-dependent decreases in coronary, cerebral, and renal blood flow and may rapidly produce myocardial, cerebral, and renal ischemia. Therefore, a goal MAP of 65 mm Hg is often targeted for shock to maintain perfusion.
Arterial blood pressure can be determined by noninvasive and invasive methods. All noninvasive blood pressure monitoring techniques depend on the use of an occluding cuff. Systolic and diastolic blood pressures are further determined by oscillometry, auscultation, palpation (systolic pressure only), or Doppler technique (systolic pressures are most reliable). Oscillometry is the only noninvasive method used in the intensive care unit (ICU) to measure MAP because the data are valid during low-flow states and the method provides automatic cycling and serial measurements (every 1 to 3 minutes) that do not require operator intervention, a key component in ICU monitoring. The oscillometry method operates by sensing arterial blood pressure changes, or oscillations, against an inflated cuff. Rapid changes in oscillation amplitude correspond to systolic and diastolic pressure. The use of narrow cuffs or cuffs applied too loosely can result in falsely high readings, whereas wide cuffs may produce falsely low readings. Fingertip devices offer another avenue for continuous indirect blood pressure measurement, but their accuracy in ICU patients may be significantly diminished by concurrent administration of vasoactive drugs.
![]() The use of invasive arterial catheters makes possible the continuous measurement of MAP as well as procurement of blood samples for laboratory testing. The radial artery is the most commonly used vessel, but the dorsalis pedis, femoral, brachial, and axillary arteries and the umbilical artery in the newborn also can be accessed. This method of blood pressure monitoring is the standard technique used in the ICU against which all other methods are compared. Major complications of peripheral artery catheterization include infection and distal ischemia. Acute distal ischemia and catheter-related bacteremia occur in <1% of catheter insertions. This translates to 2.9% of bloodstream infections per 1,000 catheter-days.23 Ischemia is most common in patients with multiple or prolonged arterial cannulations, hypertension, or vasopressor therapy.8 Invasive techniques are labor intensive, require aseptic techniques, and offer potential sources of equipment errors, such as length and quality of tubing, air bubbles, stopcocks, thrombus formation, tube kinking, and transducer placement. Hypertension, advanced age, and atherosclerosis also may affect the accuracy of invasive blood pressure readings.
The use of invasive arterial catheters makes possible the continuous measurement of MAP as well as procurement of blood samples for laboratory testing. The radial artery is the most commonly used vessel, but the dorsalis pedis, femoral, brachial, and axillary arteries and the umbilical artery in the newborn also can be accessed. This method of blood pressure monitoring is the standard technique used in the ICU against which all other methods are compared. Major complications of peripheral artery catheterization include infection and distal ischemia. Acute distal ischemia and catheter-related bacteremia occur in <1% of catheter insertions. This translates to 2.9% of bloodstream infections per 1,000 catheter-days.23 Ischemia is most common in patients with multiple or prolonged arterial cannulations, hypertension, or vasopressor therapy.8 Invasive techniques are labor intensive, require aseptic techniques, and offer potential sources of equipment errors, such as length and quality of tubing, air bubbles, stopcocks, thrombus formation, tube kinking, and transducer placement. Hypertension, advanced age, and atherosclerosis also may affect the accuracy of invasive blood pressure readings.
Central Venous Catheter
![]() The central venous catheter is used to measure the central venous pressure (CVP), to obtain venous samples for laboratory testing, and to administer drugs or fluids directly to the central circulation. A triple-lumen catheter frequently is used, whereby drugs with known incompatibility can be administered. Blood volume, venous wall compliance, right-sided cardiac function, intraabdominal and intrathoracic pressures, and vasopressor therapy affect CVP. The CVP is not a reliable estimate of blood volume but can be used to qualitatively assess blood volume changes in patients during the early phases of fluid resuscitation.24,25 The goal of fluid administration is to maintain the CVP at 8 to 12 mm Hg, but values of 15 mm Hg may be targeted in mechanically ventilated patients or patients with abdominal distension or preexisting ventricular dysfunction.24,25 Sustained elevated pressures may be indicative of fluid overloading. Few data support the use of continuous CVP monitoring in the ICU. However, CVP monitoring of fluid therapy during resuscitation of septic shock is associated with reduced mortality.6–8,24,25
The central venous catheter is used to measure the central venous pressure (CVP), to obtain venous samples for laboratory testing, and to administer drugs or fluids directly to the central circulation. A triple-lumen catheter frequently is used, whereby drugs with known incompatibility can be administered. Blood volume, venous wall compliance, right-sided cardiac function, intraabdominal and intrathoracic pressures, and vasopressor therapy affect CVP. The CVP is not a reliable estimate of blood volume but can be used to qualitatively assess blood volume changes in patients during the early phases of fluid resuscitation.24,25 The goal of fluid administration is to maintain the CVP at 8 to 12 mm Hg, but values of 15 mm Hg may be targeted in mechanically ventilated patients or patients with abdominal distension or preexisting ventricular dysfunction.24,25 Sustained elevated pressures may be indicative of fluid overloading. Few data support the use of continuous CVP monitoring in the ICU. However, CVP monitoring of fluid therapy during resuscitation of septic shock is associated with reduced mortality.6–8,24,25
Pulmonary Artery Catheter
![]() Pulmonary artery catheterization provides multiple cardiovascular parameters, including CVP, pulmonary artery pressure, pulmonary artery occlusive pressures (PAOP, commonly called the “wedge pressure”), CO, SVR, and the mixed-venous oxygen saturation (SVO2).26 Ideally, the pulmonary artery catheter should be positioned fluoroscopically; however, satisfactory placement also may be obtained by observing pulmonary artery pressure readings and electrocardiographic waveforms during catheter advancement. Proper positioning, or wedging, in the lower lung (zone 3) is essential to measure PAOP and to prevent distal pulmonary artery collapse. Inflation of the balloon at the catheter tip occludes the pulmonary artery, isolates the distal catheter tip from the right side of the heart, and allows the user to measure the PAOP, an approximate measure of the left ventricular end-diastolic volume and a major determinant of left ventricular preload. Poor wedging may be caused by catheter migration, patient movement, mechanical ventilation, or eccentric balloon inflation. Pulmonary artery catheters equipped with a distal thermistor also allow measurement of CO by thermodilution. Rapid injection of cold saline or dextrose solutions via the right atrial port allows complete mixing of blood with the injectate, and the resulting change in blood temperature is measured in the pulmonary artery. From the temperature change, the patient’s CO can be calculated. Newer pulmonary artery catheters contain a temperature coil or filament that intermittently warms the blood in the right ventricle for near-continuous CO measurement. Significant tricuspid regurgitation, an intracardiac shunt, the respiratory phase, and significant positive end-expiratory pressure decrease the validity of CO measurements. The most common complications of pulmonary artery catheterization include mural thrombus formation (14% to 91%), transient ventricular tachydysrhythmias (11% to 63%), pulmonary infarction (1% to 7%), pulmonary artery rupture (0.06% to 2.0%), and sepsis (0.3% to 0.5%).27,28 Most pulmonary artery catheters are heparin bonded which requires consideration in patients with unexplained thrombocytopenia. The relative risk (RR) of infection is 2.6 per 1,000 patient-days, similar to the risk with central venous catheters of 2.3 per 1,000 patient-days.27,28 Controversy surrounds the utility and safety of the pulmonary artery catheter, including issues surrounding correct placement and impact of the device on patient outcome. Recent studies failed to demonstrate beneficial outcomes with the use of the pulmonary artery catheter.29 The most recent guidelines suggest careful evaluation of the indications and the risk of placing a pulmonary artery catheter for resuscitation of critically ill patients.30
Pulmonary artery catheterization provides multiple cardiovascular parameters, including CVP, pulmonary artery pressure, pulmonary artery occlusive pressures (PAOP, commonly called the “wedge pressure”), CO, SVR, and the mixed-venous oxygen saturation (SVO2).26 Ideally, the pulmonary artery catheter should be positioned fluoroscopically; however, satisfactory placement also may be obtained by observing pulmonary artery pressure readings and electrocardiographic waveforms during catheter advancement. Proper positioning, or wedging, in the lower lung (zone 3) is essential to measure PAOP and to prevent distal pulmonary artery collapse. Inflation of the balloon at the catheter tip occludes the pulmonary artery, isolates the distal catheter tip from the right side of the heart, and allows the user to measure the PAOP, an approximate measure of the left ventricular end-diastolic volume and a major determinant of left ventricular preload. Poor wedging may be caused by catheter migration, patient movement, mechanical ventilation, or eccentric balloon inflation. Pulmonary artery catheters equipped with a distal thermistor also allow measurement of CO by thermodilution. Rapid injection of cold saline or dextrose solutions via the right atrial port allows complete mixing of blood with the injectate, and the resulting change in blood temperature is measured in the pulmonary artery. From the temperature change, the patient’s CO can be calculated. Newer pulmonary artery catheters contain a temperature coil or filament that intermittently warms the blood in the right ventricle for near-continuous CO measurement. Significant tricuspid regurgitation, an intracardiac shunt, the respiratory phase, and significant positive end-expiratory pressure decrease the validity of CO measurements. The most common complications of pulmonary artery catheterization include mural thrombus formation (14% to 91%), transient ventricular tachydysrhythmias (11% to 63%), pulmonary infarction (1% to 7%), pulmonary artery rupture (0.06% to 2.0%), and sepsis (0.3% to 0.5%).27,28 Most pulmonary artery catheters are heparin bonded which requires consideration in patients with unexplained thrombocytopenia. The relative risk (RR) of infection is 2.6 per 1,000 patient-days, similar to the risk with central venous catheters of 2.3 per 1,000 patient-days.27,28 Controversy surrounds the utility and safety of the pulmonary artery catheter, including issues surrounding correct placement and impact of the device on patient outcome. Recent studies failed to demonstrate beneficial outcomes with the use of the pulmonary artery catheter.29 The most recent guidelines suggest careful evaluation of the indications and the risk of placing a pulmonary artery catheter for resuscitation of critically ill patients.30
![]() The optimal PAOP needs to be individualized for each patient. Administering a fluid bolus followed by simultaneous PAOP and CO measurements with the goal of increasing the PAOP until CO does not change can be accomplished and is based on Starling’s law of the heart. However, clinical experience suggests that most patients have an optimal response to PAOP values in the range from 12 to 15 mm Hg. Limited data are available comparing the use of CVP and PAOP for guiding therapy in patients with shock. The results of recent studies of critically ill patients suggest CVP and PAOP are equivalent in terms of clinical outcomes, including mortality.31 Therefore, a pulmonary artery catheter should be inserted when hemodynamic data are needed that cannot be obtained from a central venous catheter or when the validity of measurements from the central venous catheter is questionable.
The optimal PAOP needs to be individualized for each patient. Administering a fluid bolus followed by simultaneous PAOP and CO measurements with the goal of increasing the PAOP until CO does not change can be accomplished and is based on Starling’s law of the heart. However, clinical experience suggests that most patients have an optimal response to PAOP values in the range from 12 to 15 mm Hg. Limited data are available comparing the use of CVP and PAOP for guiding therapy in patients with shock. The results of recent studies of critically ill patients suggest CVP and PAOP are equivalent in terms of clinical outcomes, including mortality.31 Therefore, a pulmonary artery catheter should be inserted when hemodynamic data are needed that cannot be obtained from a central venous catheter or when the validity of measurements from the central venous catheter is questionable.
![]() Other methods used to assess CO include carbon dioxide (CO2) partial rebreathing, esophageal Doppler, and transpulmonary (ultrasound) indicator dilution.15–22 The CO2 partial rebreathing technique compares end-tidal CO2 partial pressure obtained during a nonrebreathing period with that obtained during a subsequent rebreathing period. The ratio of change in end-tidal CO2 and CO2 elimination estimates CO but must be corrected for blood shunting. Poor to acceptable agreement exists between this method and the thermodilution method of assessing CO in critically ill patients. In addition, low minute ventilation, a high shunt fraction, or a high CO produce inaccurate results. The esophageal Doppler technique measures flow velocity in the descending aorta by means of a Doppler transducer. CO is calculated based on the diameter of the aorta, the distribution of CO to the aorta, and the flow velocity of blood in the aorta. The CO reported by this method correlates with therapeutic interventions and demonstrates excellent agreement with the pulmonary artery catheter. Unfortunately, this method is technologically difficult and may not produce reliable measurements over time. The transpulmonary (ultrasound) indicator dilution method is functionally similar to the pulmonary artery catheter in that it employs thermodilution to calculate CO but it uses a central venous catheter rather than introducing a catheter into the pulmonary artery. This method of measuring CO correlates well with the values obtained from the pulmonary artery catheter and may display less respiratory phase variations. It also may estimate global end diastolic volume but other hemodynamic variables are not readily obtained.
Other methods used to assess CO include carbon dioxide (CO2) partial rebreathing, esophageal Doppler, and transpulmonary (ultrasound) indicator dilution.15–22 The CO2 partial rebreathing technique compares end-tidal CO2 partial pressure obtained during a nonrebreathing period with that obtained during a subsequent rebreathing period. The ratio of change in end-tidal CO2 and CO2 elimination estimates CO but must be corrected for blood shunting. Poor to acceptable agreement exists between this method and the thermodilution method of assessing CO in critically ill patients. In addition, low minute ventilation, a high shunt fraction, or a high CO produce inaccurate results. The esophageal Doppler technique measures flow velocity in the descending aorta by means of a Doppler transducer. CO is calculated based on the diameter of the aorta, the distribution of CO to the aorta, and the flow velocity of blood in the aorta. The CO reported by this method correlates with therapeutic interventions and demonstrates excellent agreement with the pulmonary artery catheter. Unfortunately, this method is technologically difficult and may not produce reliable measurements over time. The transpulmonary (ultrasound) indicator dilution method is functionally similar to the pulmonary artery catheter in that it employs thermodilution to calculate CO but it uses a central venous catheter rather than introducing a catheter into the pulmonary artery. This method of measuring CO correlates well with the values obtained from the pulmonary artery catheter and may display less respiratory phase variations. It also may estimate global end diastolic volume but other hemodynamic variables are not readily obtained.
Oxygen Tension and Saturation Monitoring
Partial pressure of arterial oxygen (PaO2) and arterial oxygen saturation (SaO2) can be assessed subjectively by assessing capillary refill or invasively by obtaining an arterial blood sample. Arterial blood gases measured by conventional arterial sampling are considered standard, but their accuracy and usefulness are affected by poor sampling techniques, transportation and analysis delays, analyzer accuracy, sample cellular metabolism, and inability to trend results. Indwelling fiberoptic and electrochemical systems that allow continuous monitoring and trend analyses of blood pH, PaO2, and partial pressure of arterial carbon dioxide (PaCO2) while decreasing patient blood loss from less frequent sampling are in development. Mixed-venous oxygen saturation (SVO2) and central-venous oxygen saturation (SCVO2) reflect oxygen delivery (DO2, or DO2I, indexed to body surface area) with low values indicative of inadequate tissue perfusion that may occur during the early stages of septic shock, cardiogenic shock, or hypovolemic shock. Both measurements depend on CO, oxygen demand, hemoglobin, and SaO2.
![]() SVO2 is measured in patients using a pulmonary artery catheter. Initially, critically ill septic patients may present with a low SVO2 value (<65%), indicating high extraction of oxygen by tissues or lack of adequate DO2 to tissues. In patients with sepsis and other conditions who present with a low SVO2 value, rapid intervention should be undertaken to increase DO2 to tissues, with the goal of obtaining SVO2 ≥65%.24,32 The length of time SVO2 is <65% is associated with mortality.24 As sepsis worsens, however, SVO2 may be ≥65%. This occurs because extraction of oxygen in the arteriolar beds is hampered and is indicative of poor outcome.
SVO2 is measured in patients using a pulmonary artery catheter. Initially, critically ill septic patients may present with a low SVO2 value (<65%), indicating high extraction of oxygen by tissues or lack of adequate DO2 to tissues. In patients with sepsis and other conditions who present with a low SVO2 value, rapid intervention should be undertaken to increase DO2 to tissues, with the goal of obtaining SVO2 ≥65%.24,32 The length of time SVO2 is <65% is associated with mortality.24 As sepsis worsens, however, SVO2 may be ≥65%. This occurs because extraction of oxygen in the arteriolar beds is hampered and is indicative of poor outcome.
![]() SCVO2 is a less invasive measure of venous oxygen saturation because the catheter is placed at the junction of the inferior and superior venae cavae rather than at the pulmonary artery.18–21 It is as accurate as SVO2 but provides slightly higher normal values. Concentrations of SCVO2 <70% reliably indicate inadequate oxygenation in shock states and detect subclinical (“cryptic”) shock much earlier than hypotension. Targeting fluid and hemodynamic resuscitation to achieve SCVO2 ≥70% is a sensitive indicator and measure of the extent of global tissue hypoxia, a determinant of the adequacy of hemodynamic resuscitation, and is associated with improved survival in patients with sepsis and septic shock.15–21,24,32
SCVO2 is a less invasive measure of venous oxygen saturation because the catheter is placed at the junction of the inferior and superior venae cavae rather than at the pulmonary artery.18–21 It is as accurate as SVO2 but provides slightly higher normal values. Concentrations of SCVO2 <70% reliably indicate inadequate oxygenation in shock states and detect subclinical (“cryptic”) shock much earlier than hypotension. Targeting fluid and hemodynamic resuscitation to achieve SCVO2 ≥70% is a sensitive indicator and measure of the extent of global tissue hypoxia, a determinant of the adequacy of hemodynamic resuscitation, and is associated with improved survival in patients with sepsis and septic shock.15–21,24,32
Oxygen Delivery and Consumption
Tissue oxygen debt is indicative of organ damage in critical illness. In normal individuals, oxygen consumption (VO2 or VO2I [oxygen consumption index], indexed to body surface area) depends on DO2 (or DO2I) up to a certain critical level (VO2 flow dependency). At this point, tissue oxygen requirements apparently are satisfied and further increases in DO2 will not alter VO2 (VO2 flow independency). The point that VO2 becomes dependent on DO2 represents a pathologic transition from aerobic to anaerobic cellular metabolism and lactate production.21,24,33,34 Although animal models of sepsis substantiate this relationship, studies in critically ill humans show a continuous, pathologic dependence relationship of VO2 with DO2.21,24,33,34 The VO2/DO2 ratio, or oxygen extraction ratio (O2ER), can be used to assess adequacy of perfusion and metabolic response.21,24,33,34 Maintaining the O2ER at <25% without a changing VO2 may be helpful in maintaining or improving the body’s reserve in meeting the oxygen demands. However, low VO2 and O2ER values are indicative of poor oxygen utilization and lead to greater mortality. Patients who are able to increase VO2 when DO2 is increased show improved survival. This finding became the basis for targeting supranormal DO2 and VO2 values in the treatment of ICU patients in the 1970s and 1980s. However, a meta-analysis of randomized clinical trials involving 1,016 adult ICU patients failed to show that achievement of this goal improved mortality.35 This may have been due in part to the heterogeneous nature of the ICU patients studied and therapies provided, lack of study blinding, crossover patients (control patients who achieve supranormal DO2 and VO2 values by themselves), or lack of adequate control of cointerventions. The debate continues in more homogeneous patient populations. For example, in high-risk surgical patients, supranormal DO2 values decrease mortality except in the subgroup of patients exceeding 75 years of age in whom achieving DO2I >600 mL/m2/min shows mixed mortality results.35 Of note, the systematic assessment of DO2 and VO2 dependence is rarely done in practice but the concepts are frequently applied indirectly.
A review of alternative potential mechanisms of beneficial effect of supranormal DO2 suggests that catecholamines exert antiinflammatory actions by modulating cytokine response.6–15 In general, catecholamines inhibit the production of inflammatory cytokines (e.g., interleukin [IL]-6, tumor necrosis factor [TNF]-α) and may enhance synthesis of antiinflammatory cytokines (e.g., IL-4 and IL-10). The actions of epinephrine on these cytokines are blocked by propranolol and thus are mediated by adrenergic β-receptors.
Another issue with therapy directed to achieve supranormal oxygen transport values is that the apparent linear relationship between DO2 and VO2 has been questioned because both share variables, and this mathematical coupling can produce artifactual relationships between variables.21,24,33,34 The DO2 and VO2 indexed parameters are calculated as follows:
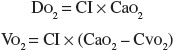
where CI is the cardiac index, CaO2 is the arterial oxygen content determined by hemoglobin concentration and SaO2, and CVO2 is the mixed venous oxygen content determined by hemoglobin concentration and SVO2.
However, variable relationships between DO2 and VO2 are observed when VO2 is measured independently by indirect calorimetry. Therefore, a linear relationship between DO2 and VO2 may be the result of mathematical coupling or flow-dependent VO2. Currently available data do not support the concept that patient outcome or survival is improved by treatment measures directed toward achieving supranormal DO2 and VO2 values.35 In fact, a consensus conference concluded that although pulmonary artery catheterization is useful for guiding therapy, routinely increasing cardiac index to predetermined supranormal values does not improve outcome.7 Furthermore, achievement of a supranormal DO2 does not ensure parallel improvements in regional organ blood flow and oxygenation. One approach that may decrease the effect of mathematical coupling and provide individualized therapy may lie in titrated therapy, with sequential measurements of DO2 and VO2 to achieve VO2 flow independency along with normalization of blood lactate and hemodynamic parameters.
![]() The most recent data regarding goal-directed therapy in the hemodynamic support of sepsis relates to the importance of achieving predetermined parameters early in the management of sepsis.36 In a meta-analysis of early (defined as 8 to 12 hours postoperatively or before the development of organ failure) versus late (defined as after the onset of organ failure) resuscitative efforts targeting supranormal oxygen-transport variables, early goal-directed therapy reduced mortality and the development of organ failure in patients who were more severely ill when therapeutic interventions produced differences in DO2.36 Survival outcome was not improved significantly in less severely ill patients (control group mortality <15% and normal DO2 values as goals) or when therapy did not improve DO2.
The most recent data regarding goal-directed therapy in the hemodynamic support of sepsis relates to the importance of achieving predetermined parameters early in the management of sepsis.36 In a meta-analysis of early (defined as 8 to 12 hours postoperatively or before the development of organ failure) versus late (defined as after the onset of organ failure) resuscitative efforts targeting supranormal oxygen-transport variables, early goal-directed therapy reduced mortality and the development of organ failure in patients who were more severely ill when therapeutic interventions produced differences in DO2.36 Survival outcome was not improved significantly in less severely ill patients (control group mortality <15% and normal DO2 values as goals) or when therapy did not improve DO2.
![]()
![]() The rapid initiation of therapy to optimize the components of DO2 (CO, hemoglobin, and SaO2) improves survival. In a prospective, randomized controlled trial, Rivers et al. demonstrated a significant reduction in mortality (30.5% vs. 46.5%; P < 0.001) in patients with severe sepsis and septic shock randomized to receive therapy based on goal-directed hemodynamic end points that were achieved within 6 hours of hospital presentation.32 They used a strategy of serial administering (a) fluids rapidly to achieve CVP 8 to 12 mm Hg, (b) vasopressor agents to achieve MAP at least 65 mm Hg, (c) red blood cell transfusion to maintain hematocrit ≥30%, and (d) dobutamine to achieve SCVO2 ≥70%. During the 6-hour window, the goal-directed therapy group received substantially more fluid, blood transfusions, and dobutamine administration but required less vasopressor and ventilator support later. This approach demonstrates the benefits of initiating therapy early in the course of sepsis and directs therapy toward clearly defined goals of optimizing DO2 in a consistent manner. The results of several evaluations of protocols or order sets designed to achieve the hemodynamic end points of early goal-directed therapy show that implementation is easily accomplished, cost is reduced, and patient outcomes, including survival, are improved.37–43 A meta-analysis of nine studies showed reduced mortality when quantitative resuscitation goals are used to guide therapy (odds ratio [OR] 0.64; 95% confidence interval [CI]: 0.43 to 0.96; P = 0.03).43 Therefore, healthcare facilities should implement strategies to achieve early goal-directed therapy using the predefined hemodynamic variables of the study by Rivers et al.32
The rapid initiation of therapy to optimize the components of DO2 (CO, hemoglobin, and SaO2) improves survival. In a prospective, randomized controlled trial, Rivers et al. demonstrated a significant reduction in mortality (30.5% vs. 46.5%; P < 0.001) in patients with severe sepsis and septic shock randomized to receive therapy based on goal-directed hemodynamic end points that were achieved within 6 hours of hospital presentation.32 They used a strategy of serial administering (a) fluids rapidly to achieve CVP 8 to 12 mm Hg, (b) vasopressor agents to achieve MAP at least 65 mm Hg, (c) red blood cell transfusion to maintain hematocrit ≥30%, and (d) dobutamine to achieve SCVO2 ≥70%. During the 6-hour window, the goal-directed therapy group received substantially more fluid, blood transfusions, and dobutamine administration but required less vasopressor and ventilator support later. This approach demonstrates the benefits of initiating therapy early in the course of sepsis and directs therapy toward clearly defined goals of optimizing DO2 in a consistent manner. The results of several evaluations of protocols or order sets designed to achieve the hemodynamic end points of early goal-directed therapy show that implementation is easily accomplished, cost is reduced, and patient outcomes, including survival, are improved.37–43 A meta-analysis of nine studies showed reduced mortality when quantitative resuscitation goals are used to guide therapy (odds ratio [OR] 0.64; 95% confidence interval [CI]: 0.43 to 0.96; P = 0.03).43 Therefore, healthcare facilities should implement strategies to achieve early goal-directed therapy using the predefined hemodynamic variables of the study by Rivers et al.32
Clinical Controversy…
Blood Lactate
![]() Lactate is a metabolic product of pyruvate. Its production is increased under anaerobic conditions when VO2 exceeds DO2, such as may occur during shock.21,24,33,34 Blood lactate concentrations are used as a diagnostic and prognostic tool in sepsis; they also are used to measure the repayment of oxygen debt to tissues.15–21 Several studies have demonstrated risk stratification of mortality rates based on initial lactate concentrations.44,45 Serial lactate concentrations may show better correlation with outcome than oxygen transport parameters and may be superior to hemodynamic markers in determining adequacy of restoration of systemic oxygenation. Continuously elevated concentrations are predictive of morbidity and mortality. Maintaining lactate elimination (commonly termed “clearance”) of 10% for 6 hours during initial resuscitation produces similar survival outcomes as achieving SCVO2 ≥70%.46 The utility of blood lactate measurements in guiding therapy was recently demonstrated in a study of 348 septic patients that showed targeting a 20% lactate reduction during the first 2 hours of resuscitation reduced hospital mortality compared to conventional assessment methods (hazard ratio [HR], 0.61; 95% CI: 0.43 to 0.87; P = 0.006).47
Lactate is a metabolic product of pyruvate. Its production is increased under anaerobic conditions when VO2 exceeds DO2, such as may occur during shock.21,24,33,34 Blood lactate concentrations are used as a diagnostic and prognostic tool in sepsis; they also are used to measure the repayment of oxygen debt to tissues.15–21 Several studies have demonstrated risk stratification of mortality rates based on initial lactate concentrations.44,45 Serial lactate concentrations may show better correlation with outcome than oxygen transport parameters and may be superior to hemodynamic markers in determining adequacy of restoration of systemic oxygenation. Continuously elevated concentrations are predictive of morbidity and mortality. Maintaining lactate elimination (commonly termed “clearance”) of 10% for 6 hours during initial resuscitation produces similar survival outcomes as achieving SCVO2 ≥70%.46 The utility of blood lactate measurements in guiding therapy was recently demonstrated in a study of 348 septic patients that showed targeting a 20% lactate reduction during the first 2 hours of resuscitation reduced hospital mortality compared to conventional assessment methods (hazard ratio [HR], 0.61; 95% CI: 0.43 to 0.87; P = 0.006).47
Several caveats guide the use of lactate concentrations in septic patients. First, lactate may accumulate in patients with other conditions, such as significant hepatic dysfunction or acute respiratory distress syndrome, who are not in shock. Second, both well-perfused and poorly perfused tissues contribute to arterial and mixed venous lactate concentrations and therefore are not reflective of regional perfusion. Third, elevated lactate concentrations may result from cellular metabolic failure rather than global hypoperfusion in shock.
Clinical Controversy…
REGIONAL PERFUSION MONITORING
GI Tonometry
Blood pressures, CO, blood lactate, and global oxygen homeostasis parameters do not offer information about the function of individual organs. Organ-specific hypoxia may be evident by coagulopathy as indicated by thrombocytopenia (platelet count <100,000/L and/or prolonged clotting times [international normalized ratio >1.5 or activated partial thromboplastin time at least 1.5-fold the upper limit of normal]), impaired renal function with urine production <0.5 mL/kg/h and/or increased serum concentrations of blood urea nitrogen and creatinine, altered hepatic function with substantially increased serum concentrations of transaminases and bilirubin, altered GI perfusion manifested by ileus and diminished bowel sounds, cardiac ischemia with elevated troponin levels and electrocardiogram changes, and altered sensorium.6,22 Objective measurement of regional perfusion to detect inadequate tissue oxygenation has focused on the mesenteric/splanchnic circulation, which is sensitive to changes in blood flow and oxygenation for several reasons.15–20,34,48 Normally, most blood flow to the gut mucosa is redistributed toward the serosa and muscularis. Second, the gut may have a higher critical DO2 threshold than other organs. Third, the tip of the villus has a countercurrent oxygen-exchange mechanism, rendering it highly sensitive to alterations in regional blood flow and oxygenation.
![]() Gastric tonometry measures gut luminal partial pressure of carbon dioxide (PCO2) at equilibrium by placing a saline- or air-filled gas-permeable balloon in the gastric lumen. Assuming that carbon dioxide (CO2) permeates freely among tissues and that the arterial bicarbonate
Gastric tonometry measures gut luminal partial pressure of carbon dioxide (PCO2) at equilibrium by placing a saline- or air-filled gas-permeable balloon in the gastric lumen. Assuming that carbon dioxide (CO2) permeates freely among tissues and that the arterial bicarbonate ![]() concentration is equal to that of the gut mucosa, the intramucosal pH (pHi) may be calculated using the Henderson–Hasselbalch equation:
concentration is equal to that of the gut mucosa, the intramucosal pH (pHi) may be calculated using the Henderson–Hasselbalch equation:
![]()
Increases in mucosal PCO2 and calculated decreases in pHi are associated with mucosal hypoperfusion and perhaps increased mortality.15–20,34,48 Calculation of pHi can be confounded by increases in luminal PCO2, such as may occur when buffering antacids are used. Histamine2-receptor antagonists or proton pump inhibitors can be used instead. The presence of respiratory acid–base disorders; systemic bicarbonate administration; arterial blood gas measurement errors; or enteral feeding products, blood, or stool in the gut may confound pHi determinations. As a result, the change in gastric mucosal PCO2 may be more accurate than pHi. Furthermore, because mucosal PCO2 is influenced by arterial PCO2, the mucosal–arterial PCO2 difference (PCO2 gap) likely is the optimal measurement.15–20,34,48 Gastric tonometry can be performed using either a saline- or air-filled balloon. The time delay (30 minutes) associated with equilibration of saline inside the balloon makes this method impractical for monitoring of resuscitation and inconvenient for routine bedside monitoring. An air-filled balloon requires a shorter equilibrium time, is simpler to use, and is equally accurate. However, the clinical utility of gastric tonometry remains uncertain. Clinical trials of pHi-directed therapy do not show that it aids resuscitation when other goals are concomitantly targeted. Gastric tonometry, in general, inconsistently predicts mortality.
![]() Evidence suggests that the most proximal part of the GI tract, the sublingual mucosa, may be an acceptable location for monitoring regional perfusion and PCO2.15–20,34,48 Unlike GI circulation, limited intra- and interpatient variability exists in the microvasculature and only few arterioles are available for assessment. Sublingual capnometry is noninvasive, is not technically complex, and provides results within minutes. The device consists of a disposable sublingual carbon dioxide pressure (PslCO2) sensor, a fiberoptic cable that connects the disposable sensor to a blood gas analyzer, and a blood gas monitoring instrument. Small studies of critically ill patients with and without sepsis and septic shock show that the PslCO2 and the sublingual-to-arterial PCO2 gap correlate better with the enhancement of DO2 with dobutamine than the mucosal PCO2 and the mucosal-to-arterial PCO2 gap.15–20,34,48 The initial sublingual-to-arterial PCO2 gap is a better predictor of mortality. These pilot studies must be expanded before this technology becomes part of routine practice, but it offers the possibility of noninvasive measurement of regional perfusion. Of note, some institutions have incorporated the PslCO2 into their algorithms of early goal-directed therapy in an effort to provide additional information about the effectiveness of therapies during resuscitation.
Evidence suggests that the most proximal part of the GI tract, the sublingual mucosa, may be an acceptable location for monitoring regional perfusion and PCO2.15–20,34,48 Unlike GI circulation, limited intra- and interpatient variability exists in the microvasculature and only few arterioles are available for assessment. Sublingual capnometry is noninvasive, is not technically complex, and provides results within minutes. The device consists of a disposable sublingual carbon dioxide pressure (PslCO2) sensor, a fiberoptic cable that connects the disposable sensor to a blood gas analyzer, and a blood gas monitoring instrument. Small studies of critically ill patients with and without sepsis and septic shock show that the PslCO2 and the sublingual-to-arterial PCO2 gap correlate better with the enhancement of DO2 with dobutamine than the mucosal PCO2 and the mucosal-to-arterial PCO2 gap.15–20,34,48 The initial sublingual-to-arterial PCO2 gap is a better predictor of mortality. These pilot studies must be expanded before this technology becomes part of routine practice, but it offers the possibility of noninvasive measurement of regional perfusion. Of note, some institutions have incorporated the PslCO2 into their algorithms of early goal-directed therapy in an effort to provide additional information about the effectiveness of therapies during resuscitation.
Myocardial Dysfunction
![]() Although loss of vascular tone is the hallmark of septic shock, myocardial dysfunction characterized by transient impairment of contractility is a recognized complication.5,49 The range of left ventricular ejection fraction (LVEF) upon presentation is wide, but approximately 35% of patients with septic shock have left ventricular hypokinesis (mean ejection fraction 38% ± 17%) and low CO.5,49 Because LVEF also is affected by preload and afterload, the low SVR of septic shock may mask depressed myocardial contractility that may be revealed upon restoration of MAP by administration of fluid and vasopressors. Therefore, CO may not reflect the extent of myocardial dysfunction. While it requires technical and interpretive training, echocardiography is a relatively simple method of assessing cardiac function and ventricular response to therapies.50 It can assess chamber size, ventricular contractility, valve function, blood flow, and CO. Patients with tissue hypoxia or a hypercontractile left ventricle may benefit from fluid administration or vasopressor therapy; whereas, patients with poor left ventricular function may require inotropic intervention. Like sublingual capnometry, some institutions use echocardiography to direct resuscitation therapies.
Although loss of vascular tone is the hallmark of septic shock, myocardial dysfunction characterized by transient impairment of contractility is a recognized complication.5,49 The range of left ventricular ejection fraction (LVEF) upon presentation is wide, but approximately 35% of patients with septic shock have left ventricular hypokinesis (mean ejection fraction 38% ± 17%) and low CO.5,49 Because LVEF also is affected by preload and afterload, the low SVR of septic shock may mask depressed myocardial contractility that may be revealed upon restoration of MAP by administration of fluid and vasopressors. Therefore, CO may not reflect the extent of myocardial dysfunction. While it requires technical and interpretive training, echocardiography is a relatively simple method of assessing cardiac function and ventricular response to therapies.50 It can assess chamber size, ventricular contractility, valve function, blood flow, and CO. Patients with tissue hypoxia or a hypercontractile left ventricle may benefit from fluid administration or vasopressor therapy; whereas, patients with poor left ventricular function may require inotropic intervention. Like sublingual capnometry, some institutions use echocardiography to direct resuscitation therapies.
Cardiac troponin release in septic patients occurs in the absence of flow-limiting disease, likely due to a loss in membrane integrity with subsequent leakage or microvascular thrombosis. Elevation of cardiac troponin concentrations in patients with sepsis indicates left ventricular dysfunction and portends a poor prognosis.49–51 Troponin concentrations also correlate with the duration of hypotension and the intensity of vasopressor therapy. Early recognition of myocardial dysfunction is crucial for administration of appropriate therapy. In the absence of other mechanisms for assessing cardiac function, echocardiographic findings and troponin concentrations may help guide and monitor therapy. Whereas cardiac troponins may be integrated into the monitoring of myocardial dysfunction to identify patients requiring aggressive therapy, natriuretic peptides show variable correlation with LVEF and should not be routinely monitored.49–51
VASOPRESSORS AND INOTROPES
![]() Vasopressors and inotropes in patients with septic shock are required when volume resuscitation fails to maintain adequate blood pressure (MAP ≥65 mm Hg) and organs and tissues remain hypoperfused despite optimizing CVP to 8 to 12 mm Hg or PAOP to 12 to 15 mm Hg.6 However, vasopressors may be needed temporarily to treat life-threatening hypotension when filling pressures are inadequate despite aggressive fluid resuscitation. Inotropes are frequently used to optimize cardiac function in cases of cardiogenic shock. The clinician must decide on the choice of agent, therapeutic end points, and safe and effective doses of vasopressors and inotropes to be used. This section reviews adrenergic receptor pharmacology, exogenous catecholamine use, and alterations in receptor function in critically ill patients. It also provides guidelines for the clinical use of adrenergic agents, optimization of pharmacotherapeutic outcomes, and minimization of adverse effects in critically ill patients with septic shock. Therapies of hypovolemic shock and cardiogenic shock are discussed in other chapters.
Vasopressors and inotropes in patients with septic shock are required when volume resuscitation fails to maintain adequate blood pressure (MAP ≥65 mm Hg) and organs and tissues remain hypoperfused despite optimizing CVP to 8 to 12 mm Hg or PAOP to 12 to 15 mm Hg.6 However, vasopressors may be needed temporarily to treat life-threatening hypotension when filling pressures are inadequate despite aggressive fluid resuscitation. Inotropes are frequently used to optimize cardiac function in cases of cardiogenic shock. The clinician must decide on the choice of agent, therapeutic end points, and safe and effective doses of vasopressors and inotropes to be used. This section reviews adrenergic receptor pharmacology, exogenous catecholamine use, and alterations in receptor function in critically ill patients. It also provides guidelines for the clinical use of adrenergic agents, optimization of pharmacotherapeutic outcomes, and minimization of adverse effects in critically ill patients with septic shock. Therapies of hypovolemic shock and cardiogenic shock are discussed in other chapters.
Of note, agents other than catecholamines have been used as inotropes and vasopressors in shock states. They include phosphodiesterase III inhibitors, naloxone, nitric oxide (NO) synthase (NOS) inhibitors, and calcium sensitizers. This chapter focuses on catecholamines. Vasopressin and corticosteroids, as they relate to septic shock, also are emphasized because they have pharmacologic interactions with catecholamines, possess hemodynamic effects, and are frequently used.
Catecholamine Receptor Pharmacology
![]() Comparative receptor activities of endogenous and exogenously administered catecholamines are summarized in Table 13-3.6–15,52–54 Endogenous catecholamines are responsible for regulation of vascular and bronchiolar smooth muscle tone and myocardial contractility. These effects are mediated by sympathetic adrenergic receptors of the autonomic nervous system located in the vasculature, myocardium, and bronchioles. Postsynaptic adrenoceptors are located at or near the synaptic junction. These receptors can be activated by naturally circulating or exogenous catecholamines (e.g., norepinephrine, epinephrine, and phenylephrine), whereas presynaptic adrenoceptors are stimulated by locally released neurotransmitters (e.g., norepinephrine) and are controlled by a negative feedback mechanism.
Comparative receptor activities of endogenous and exogenously administered catecholamines are summarized in Table 13-3.6–15,52–54 Endogenous catecholamines are responsible for regulation of vascular and bronchiolar smooth muscle tone and myocardial contractility. These effects are mediated by sympathetic adrenergic receptors of the autonomic nervous system located in the vasculature, myocardium, and bronchioles. Postsynaptic adrenoceptors are located at or near the synaptic junction. These receptors can be activated by naturally circulating or exogenous catecholamines (e.g., norepinephrine, epinephrine, and phenylephrine), whereas presynaptic adrenoceptors are stimulated by locally released neurotransmitters (e.g., norepinephrine) and are controlled by a negative feedback mechanism.
TABLE 13-3 Adrenergic, Dopaminergic, and Vasopressin Receptor Pharmacology, and Organ Distribution

The signal transduction pathways associated with catecholamine and vasopressin-induced effects in the heart and blood vessels are illustrated in Figure 13-1.6–15,52–54 Agonists of β-adrenoceptors and dopamine (D1) receptors stimulate adenylate cyclase by a G-protein (Gs)-dependent mechanism (Fig. 13-1, top). Adenylate cyclase generates cyclic adenosine monophosphate (cAMP) from adenosine triphosphate (ATP). cAMP-dependent protein kinase A, which is activated by elevations in intracellular cAMP, phosphorylates target proteins to modify cellular function. Through these mechanisms, β1-adrenoceptor activation exerts positive inotropic and chronotropic effects in the heart, and β2-adrenoceptor and D1-receptor activation induces vascular smooth muscle relaxation. Agonists of α1-adrenoceptors stimulate phospholipase C-β (PLC-β) through a G-protein (Gq)-dependent process (Fig. 13-1, bottom). PLC-β produces inositol trisphosphate and diacylglycerol from cell membrane phosphatidylinositol bisphosphate. Diacylglycerol activates protein kinase C, an enzyme that phosphorylates several key proteins (e.g., extracellular signal-regulated kinases, c-Jun NH2-terminal kinases, and-mitogen-activated protein kinases) that modify cellular function (e.g., hypertrophy). Inositol trisphosphate elicits the release of calcium from intracellular stores, such as the sarcoplasmic reticulum. Calcium forms a complex with calmodulin, which then activates calcium–calmodulin-dependent protein kinases (CaMK). CaMKs phosphorylate target proteins to alter cellular function. Myosin light-chain kinase is an example of a CaMK. Its action of phosphorylating myosin light chain leads to vascular smooth muscle contraction.
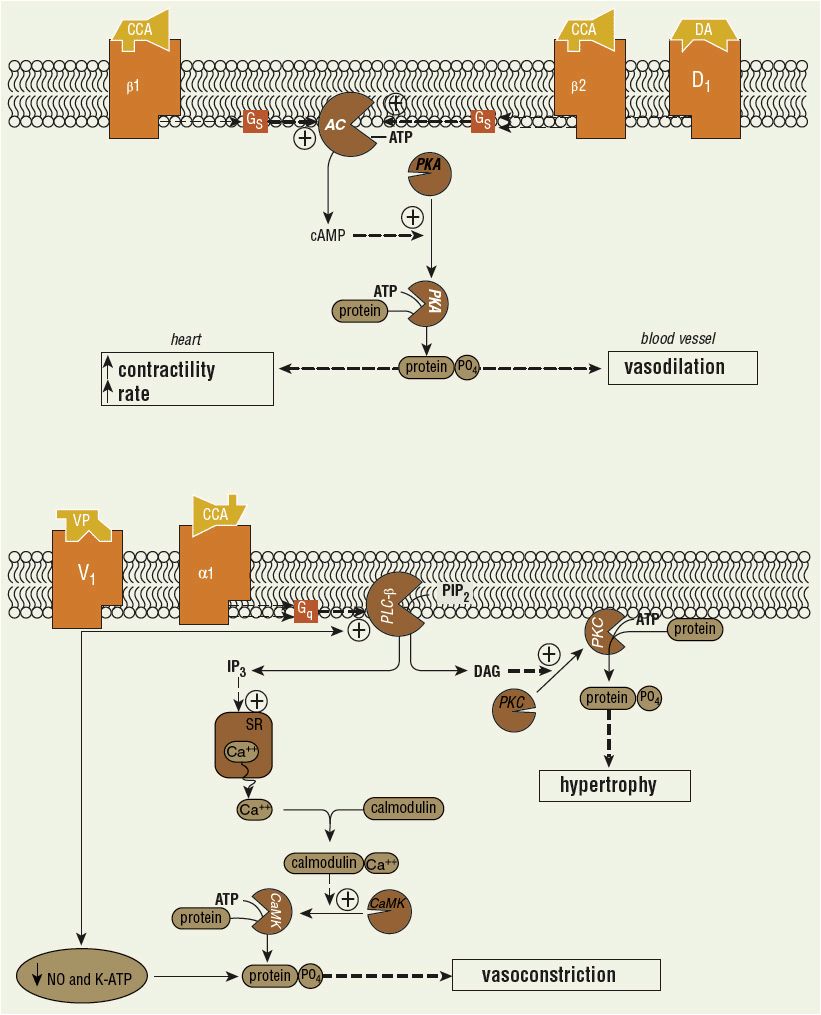
FIGURE 13-1 Signal transduction pathways in heart and blood vessels. Top: Catecholamine (CCA)-induced effects mediated in heart (β1) or vascular smooth muscle (β2, D1). (AC, adenylate cyclase; ATP, adenosine triphosphate; cAMP, cyclic adenosine monophosphate; PKA, cAMP-dependent protein kinase; +, stimulation.) Bottom: CCA (α1) and vasopressin (VP)-induced actions in vascular smooth muscle. (Ca++, calcium ion; CaMK, calcium/calmodulin-dependent protein kinase; DAG, diacylglycerol; IP3, inositol trisphosphate; NO, nitric oxide; PIP2, phosphatidylinositol bisphosphate; PKC, protein kinase C; PLC-β, phospholipase C-β; SR, sarcoplasmic reticulum.) These pathways have been extensively simplified, and denoted cellular effects represent one of many produced. (Figure based on data from references 6–15, 52–54.)
The normal heart contains primarily postsynaptic β1-receptors, which when stimulated cause increased rate and force of contraction. This effect is mediated by activation of adenylate cyclase and subsequent generation and accumulation of cAMP. Stimulation of postsynaptic cardiac α1-receptors causes a significant increase in contractility without an increase in rate, an effect mediated by PLC rather than adenylate cyclase. The increased contractility is more pronounced at lower heart rates and has a slower onset and longer duration in comparison with β1-mediated inotropic response. Presynaptic α2-adrenoceptors also are found in the heart and appear to be activated by norepinephrine released by the sympathetic nerve itself. Their activation inhibits further norepinephrine release from the nerve terminal.
Both presynaptic and postsynaptic adrenoceptors are present in the vasculature. Postsynaptic α1– and α2-receptors mediate vasoconstriction, whereas postsynaptic β2-receptors induce vasodilation. Presynaptic α2-receptors inhibit norepinephrine release in the vasculature, also promoting vasodilation. Presynaptic β1-adrenoceptors promote neurotransmitter release. Stimulation of peripheral D1-receptors produces renal, coronary, and mesenteric vasodilation and a natriuretic response. Stimulation of D2-receptors inhibits norepinephrine release from sympathetic nerve endings, sequesters prolactin and aldosterone, and may induce nausea and vomiting. D1– and D2-receptor stimulation also suppresses peristalsis and may precipitate ileus.
![]() Vasopressin-induced vasoconstriction occurs through a variety of direct and indirect mechanisms.52,53 Stimulation of vascular vasopressin (V)1 receptors causes vasoconstriction by receptor-coupled activation of PLC and calcium release from intracellular stores via secondary messengers similar to α1-adrenergic stimulation (Fig. 13-1, bottom). Vasopressin also directly inhibits vascular potassium-sensitive ATP channels to produce vasoconstriction (Fig. 13-1, bottom). V1-receptor stimulation inhibits the actions of IL-1β and thereby facilitates vasoconstriction. Vasopressin also increases the activity of adrenergic receptors. The greatest vasoconstriction occurs in the skin and soft tissue, skeletal muscle, fat tissue, pancreas, and thyroid gland. In contrast, vasopressin causes vasodilation in the cerebral, pulmonary, coronary, and selected renal vascular beds by enhancing endothelial NO release through V1-receptor stimulation in these tissues.52–54 Vasopressin has minimal to no inotropic or chronotropic effects.
Vasopressin-induced vasoconstriction occurs through a variety of direct and indirect mechanisms.52,53 Stimulation of vascular vasopressin (V)1 receptors causes vasoconstriction by receptor-coupled activation of PLC and calcium release from intracellular stores via secondary messengers similar to α1-adrenergic stimulation (Fig. 13-1, bottom). Vasopressin also directly inhibits vascular potassium-sensitive ATP channels to produce vasoconstriction (Fig. 13-1, bottom). V1-receptor stimulation inhibits the actions of IL-1β and thereby facilitates vasoconstriction. Vasopressin also increases the activity of adrenergic receptors. The greatest vasoconstriction occurs in the skin and soft tissue, skeletal muscle, fat tissue, pancreas, and thyroid gland. In contrast, vasopressin causes vasodilation in the cerebral, pulmonary, coronary, and selected renal vascular beds by enhancing endothelial NO release through V1-receptor stimulation in these tissues.52–54 Vasopressin has minimal to no inotropic or chronotropic effects.
V2 receptors located in the kidneys are responsible for the antidiuretic properties of vasopressin.52–54 Stimulation of V2 receptors facilitates integration of aquaporins into the luminal cell membrane of distal tubules and collecting duct capillaries to increase permeability and thus retain intravascular volume. However, vasopressin stimulation of V1 receptors causes vasoconstriction of efferent arterioles and relative vasodilation of afferent arterioles to increase glomerular perfusion pressure and filtration rate to enhance urine production.
Vasopressin rapidly increases serum cortisol concentration by stimulating V3 receptors in the pituitary gland to enhance the release of adrenocorticotropic hormone (ACTH).52–54 Cortisol helps regulate the proinflammatory state associated with sepsis and increases blood pressure through several mechanisms, including inhibition of inducible nitric oxide synthase (iNOS) to reduce NO production, reversal of adrenergic receptor desensitization, and increased intravascular volume through retention of sodium and water.
Altered Adrenoceptor Function: Implications for Critically Ill Patients
![]() Most of the work describing receptor function and associated clinical pharmacology has been performed in either animal models or human volunteers. In critically ill septic patients, derangements in adrenergic receptor activity may result in resistance to exogenously administered catecholamine.6–15,52,53 This “desensitization” frequently is characterized by myocardial and vascular hyporesponsiveness to high dosages of inotropes and vasopressor agents. Prolonged exposure of vascular endothelial tissue to vasopressor drugs (α-adrenergic agonists) or endogenous catecholamines may promote additional receptor downregulation. Increased endogenous catecholamine concentrations have been reported in endotoxemic and other critically ill patients, suggesting an acquired adrenergic receptor defect and desensitization of adrenergic receptors and alteration in voltage-sensitive calcium channels. The problem in critically ill patients may be related to decreased receptor activity or density. However, in patients with septic shock, catecholamine concentrations are even higher, so abnormalities in adrenergic receptor function are greater, with associated reductions in the concentrations of intracellular signal transduction mediators. The worsened receptor abnormality may be explained by defects distal to the receptor site, such as uncoupling of adrenergic receptors from adenylate cyclase or PLC, or dysfunction in the regulatory G-protein unit of signal transduction pathways.
Most of the work describing receptor function and associated clinical pharmacology has been performed in either animal models or human volunteers. In critically ill septic patients, derangements in adrenergic receptor activity may result in resistance to exogenously administered catecholamine.6–15,52,53 This “desensitization” frequently is characterized by myocardial and vascular hyporesponsiveness to high dosages of inotropes and vasopressor agents. Prolonged exposure of vascular endothelial tissue to vasopressor drugs (α-adrenergic agonists) or endogenous catecholamines may promote additional receptor downregulation. Increased endogenous catecholamine concentrations have been reported in endotoxemic and other critically ill patients, suggesting an acquired adrenergic receptor defect and desensitization of adrenergic receptors and alteration in voltage-sensitive calcium channels. The problem in critically ill patients may be related to decreased receptor activity or density. However, in patients with septic shock, catecholamine concentrations are even higher, so abnormalities in adrenergic receptor function are greater, with associated reductions in the concentrations of intracellular signal transduction mediators. The worsened receptor abnormality may be explained by defects distal to the receptor site, such as uncoupling of adrenergic receptors from adenylate cyclase or PLC, or dysfunction in the regulatory G-protein unit of signal transduction pathways.
In addition to catecholamines, circulating inflammatory cytokines may be partly responsible for distal alterations.52,53 Macrophage-derived IL-1 and TNF-α produce impaired coupling of β-adrenoceptors to adenylate cyclase. Patients with septic shock exhibit impaired β-adrenergic receptor stimulation of cAMP associated with myocardial hyporesponsiveness to various vasopressors and inotropes. However, increased chronotropic sensitivity to β-adrenergic stimulation with hypersensitivity of the adenylate cyclase system to isoproterenol stimulation also has been reported in animal models of bacteremia and endotoxemia. In the presence of intrinsic myocardial dysfunction and increased metabolic demands, this dysfunctional adrenergic system is incapable of mobilizing functional cardiac reserve to maintain adequate myocardial performance.49,52,53
IL-1 and TNF-α suppress gene expression of α1-adrenoceptors, resulting in fewer receptor proteins. Overproduction of NO by iNOS directly contributes to vasodilation by cyclic guanosine monophosphate-mediated smooth muscle relaxation. NO indirectly produces vasodilation by combining with superoxide to form peroxynitrite, a highly toxic reactive species that causes endothelial dysfunction, uncoupling of α1-adrenoceptors to PLC, and deactivation of catecholamines. The result of sepsis-induced inflammation is a system that promotes adrenergic receptor dysfunction to accentuate vasodilation and shock.6–15,49,52,53
![]() Functional α1-adrenergic receptor changes occur at various stages of sepsis; thus, adrenoceptor sensitivity may be time dependent during progression of sepsis to septic shock. The findings are not always consistent in various animal models of sepsis and in critically ill septic patients. Time-dependent alterations in the production of NO, a potent vasodilator, may explain the apparent differences in vascular reactivity to phenylephrine during the phases of endotoxemia. This finding suggests that the clinical response to vasopressors and possibly inotropic agents is variable during the stages of hemodynamic, myocardial, and peripheral vascular derangements of septic shock. β-adrenergic receptor changes are present within 24 to 48 hours of septic shock. In summary, α– and β-adrenergic receptor derangements may vary among patients and during each bacteremic insult; therefore, doses of catecholamines vary among patients and during the insult.6–15,52,53 For these reasons, these drugs should be dosed to clinical end points and not to arbitrary maximal doses. High dosages are frequently required.
Functional α1-adrenergic receptor changes occur at various stages of sepsis; thus, adrenoceptor sensitivity may be time dependent during progression of sepsis to septic shock. The findings are not always consistent in various animal models of sepsis and in critically ill septic patients. Time-dependent alterations in the production of NO, a potent vasodilator, may explain the apparent differences in vascular reactivity to phenylephrine during the phases of endotoxemia. This finding suggests that the clinical response to vasopressors and possibly inotropic agents is variable during the stages of hemodynamic, myocardial, and peripheral vascular derangements of septic shock. β-adrenergic receptor changes are present within 24 to 48 hours of septic shock. In summary, α– and β-adrenergic receptor derangements may vary among patients and during each bacteremic insult; therefore, doses of catecholamines vary among patients and during the insult.6–15,52,53 For these reasons, these drugs should be dosed to clinical end points and not to arbitrary maximal doses. High dosages are frequently required.
Relative Deficiencies of Vasopressin and Cortisol
![]() Endogenous arginine vasopressin, a peptide hormone also known as antidiuretic hormone, is important for osmoregulation under normal physiologic conditions. Vasopressin is produced in the hypothalamus, stored in the posterior pituitary, and released from magnocellular neurons of the hypothalamus.54 Increased serum osmolality and hypovolemia are the major stimuli for vasopressin release.54 Other stimuli commonly associated with shock are dopamine, histamine, angiotensin II, prostaglandins, pain, hypoxia, acidosis, hypotension, hypercarbia, and α1-adrenergic receptor stimulation. Vasopressin release is inhibited by NO, natriuretic peptides, γ-aminobutyric acid, β-adrenergic receptor stimulation, and α2-adrenergic receptor stimulation.54
Endogenous arginine vasopressin, a peptide hormone also known as antidiuretic hormone, is important for osmoregulation under normal physiologic conditions. Vasopressin is produced in the hypothalamus, stored in the posterior pituitary, and released from magnocellular neurons of the hypothalamus.54 Increased serum osmolality and hypovolemia are the major stimuli for vasopressin release.54 Other stimuli commonly associated with shock are dopamine, histamine, angiotensin II, prostaglandins, pain, hypoxia, acidosis, hypotension, hypercarbia, and α1-adrenergic receptor stimulation. Vasopressin release is inhibited by NO, natriuretic peptides, γ-aminobutyric acid, β-adrenergic receptor stimulation, and α2-adrenergic receptor stimulation.54
Normal serum vasopressin concentrations are <4 pg/mL.54 Serum vasopressin concentrations are elevated with hypotension. Vasopressin response in septic shock is biphasic. During the first 8 hours of septic shock requiring catecholamine adrenergic therapy, serum concentrations of vasopressin are appropriately high to help maintain blood pressure and organ perfusion. Thereafter, serum vasopressin concentrations decline dramatically over the next 96 hours to physiologically normal but inappropriately low values, resulting in a state of “relative deficiency.” In contrast, serum vasopressin concentrations remain elevated in patients with cardiogenic shock. Administration of vasopressin at 0.01 to 0.06 units/min produces concentrations similar to those observed in early septic shock and other hypotensive states; however, vasopressin concentrations do not correlate with blood pressure.54 Administration of vasopressin improves arterial pressure while minimizing the dose of catecholamine vasopressors.54
The mechanism of vasopressin insufficiency in septic shock is not well understood. Neurohypophyseal stores in the posterior lobe of the pituitary gland are depleted during septic shock, likely as a result of excessive and continuous baroreceptor stimulation that eventually exhausts the limited vasopressin secretory stores. In addition, secretion of vasopressin is inhibited by enhanced endothelial production of NO, high circulating concentrations of adrenergic agonists (both endogenous and exogenous), and tonic inhibition by stretch receptors in response to volume replacement and mechanical ventilation.54
![]() As with vasopressin, during sepsis a state of “relative adrenal insufficiency” is produced by continuous activation of the hypothalamic–pituitary–adrenal axis by IL-1, IL-6, and TNF-α that causes depletion of cortisol in the adrenal glands.55 Administration of corticosteroids improves arterial pressure while minimizing the dose of catecholamine vasopressors.56,57 Current proposed mechanisms of the vasoconstrictor effect of corticosteroids include increasing the number and stimulating the function of α1– and β-adrenergic receptors and attenuating the production of inflammatory mediators responsible for vasodilation.
As with vasopressin, during sepsis a state of “relative adrenal insufficiency” is produced by continuous activation of the hypothalamic–pituitary–adrenal axis by IL-1, IL-6, and TNF-α that causes depletion of cortisol in the adrenal glands.55 Administration of corticosteroids improves arterial pressure while minimizing the dose of catecholamine vasopressors.56,57 Current proposed mechanisms of the vasoconstrictor effect of corticosteroids include increasing the number and stimulating the function of α1– and β-adrenergic receptors and attenuating the production of inflammatory mediators responsible for vasodilation.
The use of corticosteroids for treatment of septic shock has been a topic of controversy for many years. Meta-analyses of early studies of steroids in patients with sepsis demonstrated a lack of benefit and potential harm in sepsis and septic shock.56,57 Interest in corticosteroid use is renewed because of the increased awareness of adrenocortical insufficiency in critically ill patients with septic shock.55 Relative adrenal insufficiency has been defined as a random cortisol concentration <10 mcg/dL (278 nmol/L) or an increase of <9 mcg/dL (250 nmol/L) following a dose of synthetic ACTH irrespective of the initial serum cortisol concentration.58 Although absolute insufficiency is rare, relative adrenocortical insufficiency is present in 50% to 70% of patients with septic shock and is associated with a poor outcome.59–62
An elevated random cortisol concentration (>34 mcg/dL) is an independent predictor of mortality.58 Mortality is further increased if ACTH response is <9 mcg/dL, suggesting that the risk of mortality is greatest in situations of adrenal gland “fatigue” (i.e., degree of stress is not matched by sufficient cortisol production by the adrenal glands despite operating at maximal functional capacity).
Clinical Pharmacology of Vasopressors and Inotropes
![]()
![]() The receptor selectivity of clinically used, catecholamine-based vasopressors and inotropes and hemodynamic effects are listed in Table 13-4.6–15,52–54,63,64 In general, these drugs are fast acting, with short durations of action. As such, these drugs are given as continuous infusions and titrated rapidly to predetermined effects.6 Vasopressin is administered as a replacement dosage of 0.01 to 0.04 units/min and should not be titrated.54 Careful monitoring and calculation of infusion rates are advised for all vasopressors because dosing adjustments are made frequently, and varying admixtures and concentrations are used in volume-restricted patients.
The receptor selectivity of clinically used, catecholamine-based vasopressors and inotropes and hemodynamic effects are listed in Table 13-4.6–15,52–54,63,64 In general, these drugs are fast acting, with short durations of action. As such, these drugs are given as continuous infusions and titrated rapidly to predetermined effects.6 Vasopressin is administered as a replacement dosage of 0.01 to 0.04 units/min and should not be titrated.54 Careful monitoring and calculation of infusion rates are advised for all vasopressors because dosing adjustments are made frequently, and varying admixtures and concentrations are used in volume-restricted patients.
TABLE 13-4 Receptor Pharmacology and Adverse Events of Selected Inotropic and Vasopressor Agents Used in Septic Shocka




