Thyroid Disorders
KEY CONCEPTS
![]() Advances in the understanding of the structure and metabolism of thyroid hormones, and the molecular biology of thyroid hormone receptors have provided insight into the various abnormalities that give rise to hyperthyroidism and hypothyroidism.
Advances in the understanding of the structure and metabolism of thyroid hormones, and the molecular biology of thyroid hormone receptors have provided insight into the various abnormalities that give rise to hyperthyroidism and hypothyroidism.
![]() Thyrotoxicosis is most commonly caused by Graves’ disease, which is an autoimmune disorder in which thyroid-stimulating antibody (TSAb) directed against the thyrotropin receptor elicits the same biologic response as thyroid-stimulating hormone (TSH).
Thyrotoxicosis is most commonly caused by Graves’ disease, which is an autoimmune disorder in which thyroid-stimulating antibody (TSAb) directed against the thyrotropin receptor elicits the same biologic response as thyroid-stimulating hormone (TSH).
![]() Hyperthyroidism may be treated with antithyroid drugs such as methimazole (MMI) or propylthiouracil (PTU), radioactive iodine (RAI: sodium iodide-131 [131I]), or surgical removal of the thyroid gland; selection of the initial treatment approach is based on patient characteristics such as age, concurrent physiology (e.g., pregnancy), comorbidities (e.g., chronic obstructive lung disease), and convenience.
Hyperthyroidism may be treated with antithyroid drugs such as methimazole (MMI) or propylthiouracil (PTU), radioactive iodine (RAI: sodium iodide-131 [131I]), or surgical removal of the thyroid gland; selection of the initial treatment approach is based on patient characteristics such as age, concurrent physiology (e.g., pregnancy), comorbidities (e.g., chronic obstructive lung disease), and convenience.
![]() MMI and PTU reduce the synthesis of thyroid hormones and are similar in efficacy and overall adverse effects, but their dosing ranges differ by 10-fold.
MMI and PTU reduce the synthesis of thyroid hormones and are similar in efficacy and overall adverse effects, but their dosing ranges differ by 10-fold.
![]() Response to MMI and PTU is seen in 4 to 6 weeks with a maximal response in 4 to 6 months; treatment usually continues for 1 to 2 years, and therapy is monitored by clinical signs and symptoms and by measuring the serum concentrations of TSH and free thyroxine (T4).
Response to MMI and PTU is seen in 4 to 6 weeks with a maximal response in 4 to 6 months; treatment usually continues for 1 to 2 years, and therapy is monitored by clinical signs and symptoms and by measuring the serum concentrations of TSH and free thyroxine (T4).
![]() Subclinical hyperthyroidism is associated with cardiovascular mortality, especially in the elderly in whom treatment is recommended.
Subclinical hyperthyroidism is associated with cardiovascular mortality, especially in the elderly in whom treatment is recommended.
![]() Adjunctive therapy with β-blockers controls the adrenergic symptoms of thyrotoxicosis but does not correct the underlying disorder; iodine may also be used adjunctively in preparation for surgery and acutely for thyroid storm.
Adjunctive therapy with β-blockers controls the adrenergic symptoms of thyrotoxicosis but does not correct the underlying disorder; iodine may also be used adjunctively in preparation for surgery and acutely for thyroid storm.
![]() Many patients choose to have ablative therapy with 131I rather than undergo repeated courses of MMI or PTU treatment; most patients receiving RAI eventually become hypothyroid and require thyroid hormone supplementation.
Many patients choose to have ablative therapy with 131I rather than undergo repeated courses of MMI or PTU treatment; most patients receiving RAI eventually become hypothyroid and require thyroid hormone supplementation.
![]() Hypothyroidism is most often due to an autoimmune disorder known as Hashimoto’s thyroiditis.
Hypothyroidism is most often due to an autoimmune disorder known as Hashimoto’s thyroiditis.
![]() The drug of choice for replacement therapy in hypothyroidism is levothyroxine.
The drug of choice for replacement therapy in hypothyroidism is levothyroxine.
![]() Studies of combination therapy with levothyroxine and triiodothyronine have not shown reproducible benefits. This approach to treatment of hypothyroidism requires further study.
Studies of combination therapy with levothyroxine and triiodothyronine have not shown reproducible benefits. This approach to treatment of hypothyroidism requires further study.
![]() Monitoring of levothyroxine replacement therapy is achieved by observing clinical signs and symptoms and by measuring the serum TSH. An elevated TSH indicates underreplacement; a suppressed TSH indicates overreplacement.
Monitoring of levothyroxine replacement therapy is achieved by observing clinical signs and symptoms and by measuring the serum TSH. An elevated TSH indicates underreplacement; a suppressed TSH indicates overreplacement.
![]() There is controversy about the treatment of mild (subclinical) hypothyroidism. Generally, benefits of treatment are clearest in younger populations.
There is controversy about the treatment of mild (subclinical) hypothyroidism. Generally, benefits of treatment are clearest in younger populations.
![]() Despite the simplicity of the concept of correction of hypothyroidism with levothyroxine, many treated patients have iatrogenic hyperthyroidism or are underreplaced.
Despite the simplicity of the concept of correction of hypothyroidism with levothyroxine, many treated patients have iatrogenic hyperthyroidism or are underreplaced.
![]() Hypothyroidism during pregnancy should be treated to achieve TSH values that are normal, based on reference ranges for TSH derived from the pregnant population.
Hypothyroidism during pregnancy should be treated to achieve TSH values that are normal, based on reference ranges for TSH derived from the pregnant population.
Thyroid hormones affect the function of virtually every organ system. In a child, thyroid hormone is critical for normal growth and development. In an adult, the major role of thyroid hormone is to maintain metabolic stability. Substantial reservoirs of thyroid hormone in the thyroid gland and blood provide constant thyroid hormone availability. In addition, the hypothalamic–pituitary–thyroid axis is exquisitely sensitive to small changes in circulating thyroid hormone concentrations, and alterations in thyroid hormone secretion maintain peripheral free thyroid hormone levels within a narrow range. Patients seek medical attention for evaluation of symptoms due to abnormal thyroid hormone levels or because of diffuse or nodular thyroid enlargement.
THYROID HORMONE PHYSIOLOGY
Thyroid Hormone Synthesis
![]() The thyroid hormones thyroxine (T4) and triiodothyronine (T3) (Fig. 58-1) are formed within thyroglobulin (TG), a large glycoprotein synthesized in the thyroid cell. Because of the unique tertiary structure of this glycoprotein, iodinated tyrosine residues present in TG are able to bind together to form active thyroid hormones.1
The thyroid hormones thyroxine (T4) and triiodothyronine (T3) (Fig. 58-1) are formed within thyroglobulin (TG), a large glycoprotein synthesized in the thyroid cell. Because of the unique tertiary structure of this glycoprotein, iodinated tyrosine residues present in TG are able to bind together to form active thyroid hormones.1
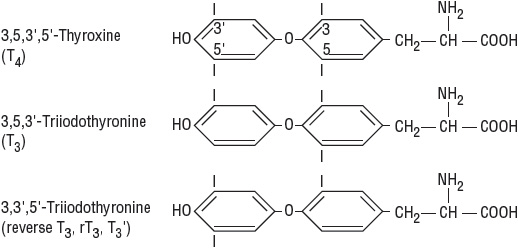
FIGURE 58-1 Structure of thyroid hormones.
Iodide is actively transported through the basolateral membrane via a Na+/I– symporter from the extracellular space into the thyroid follicular cell against an electrochemical gradient, driven by the coupled transport of sodium.2 Structurally related anions such as thiocyanate (SCN–), perchlorate (ClO4–), and pertechnetate (TcO4–) are competitive inhibitors of iodine transport.3 In addition, bromine, fluorine, and, under certain circumstances, lithium block iodide transport into the thyroid (Table 58-1). Inorganic iodide that enters the thyroid follicular cell is ushered through the cell to the apical membrane, where it is transported into the follicular lumen by pendrin, and possibly other transport proteins.4,5 Located on the luminal side of the apical membrane, thyroid peroxidase oxidizes iodide and covalently binds the organified iodide to tyrosine residues within TG (Fig. 58-2). It is interesting that although salivary glands and the gastric mucosa are able to actively transport iodide, they are unable to effectively incorporate iodide into proteins given the lack of similar oxidizing machinery.6 Similarly, when tyrosine molecules are iodinated on proteins other than TG, they lack the proper tertiary structure needed to allow the formation of active thyroid hormones.7
TABLE 58-1 Thyroid Hormone Synthesis and Secretion Inhibitors
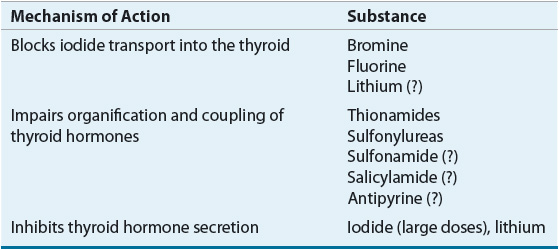
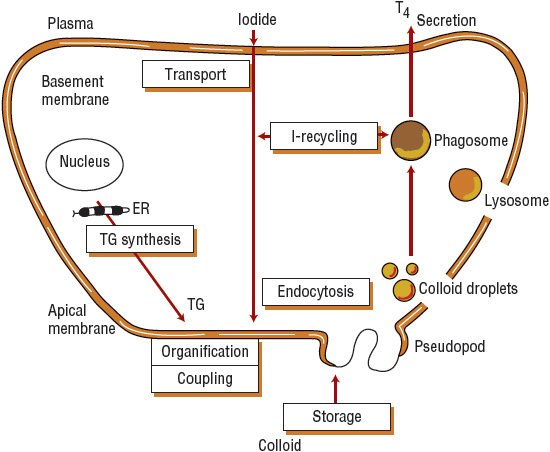
FIGURE 58-2 Thyroid hormone synthesis. Iodide is transported from the plasma, through the cell, to the apical membrane, where it is organified and coupled to the thyroglobulin (TG) synthesized within the thyroid cell. Hormone stored as colloid reenters the cell through endocytosis and moves back toward the basal membrane, where thyroxine (T4) is secreted.
The iodinated tyrosine residues monoiodotyrosine (MIT) and diiodotyrosine (DIT) combine to form iodothyronines (Fig. 58-3). Thus, two molecules of DIT combine to form T4, whereas MIT and DIT constitute T3. In addition to its role in iodine organification, the hemoprotein thyroid peroxidase also catalyzes the formation of iodothyronines (coupling).
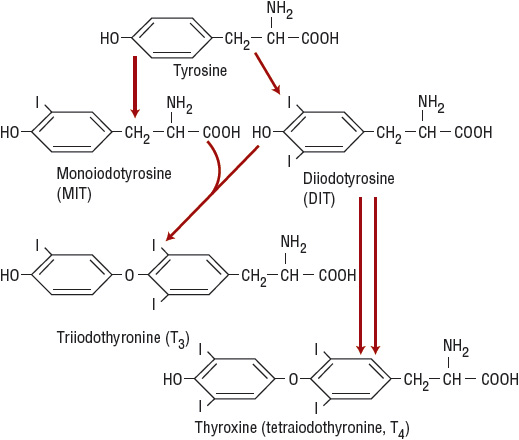
FIGURE 58-3 Scheme of coupling reactions. After tyrosine is iodinated to form monoiodotyrosine (MIT) or diiodotyrosine (DIT) (organification of the iodine), MIT and DIT combine to form triiodothyronine (T3) or two molecules of DIT combine to form thyroxine T4.
Iodine deficiency causes an increase in the MIT:DIT ratio in TG and leads to a relative increase in the production of T3.8 Because T3 is more potent than T4, the increase in T3 production in iodine-deficient areas may be beneficial. The thionamide drugs used to treat hyperthyroidism inhibit thyroid peroxidase and thus block thyroid hormone synthesis.
TG is stored in the follicular lumen and must reenter the cell, where the process of proteolysis liberates thyroid hormone into the bloodstream. Thyroid follicles active in hormone synthesis are identified histologically by columnar epithelial cells lining a follicular lumen, which is depleted of colloid. Inactive follicles are lined by cuboidal epithelial cells and are replete with colloid. Both iodide and lithium block the release of preformed thyroid hormone, through poorly understood mechanisms.
T4 and T3 are transported in the bloodstream primarily by three proteins: thyroxine-binding globulin (TBG), transthyretin (TTR), and albumin.9 It is estimated that 99.96% of circulating T4 and 99.5% of T3 are bound to these proteins. However, only the unbound (free) thyroid hormone is able to diffuse into the cell, elicit a biologic effect, and regulate thyroid-stimulating hormone (TSH; also known as thyrotropin) secretion from the pituitary. Multiple functions have been ascribed to these transport proteins, including (a) assuring minimal urinary loss of iodide, (b) providing a mechanism for uniform tissue distribution of free hormone, and (c) transport of hormone into the CNS.
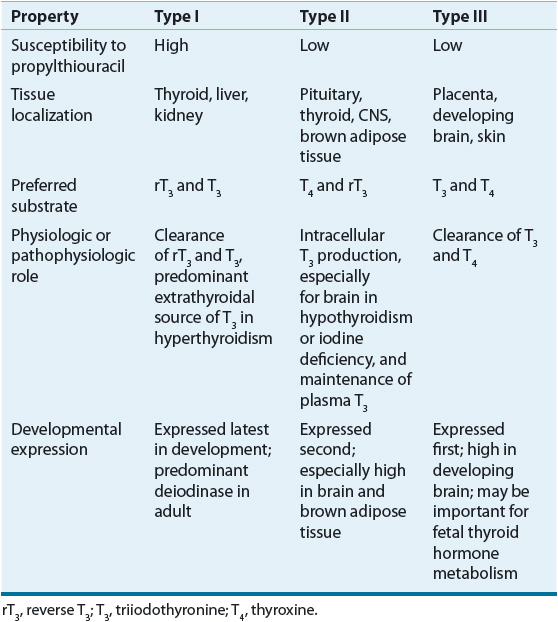
Whereas T4 is secreted solely from the thyroid gland, less than 20% of T3 is produced in the thyroid. The majority of T3 is formed from the breakdown of T4 catalyzed by the 5′-monodeiodinase enzymes found in extrathyroidal peripheral tissues. Because the binding affinity of nuclear thyroid hormone receptors (TRs) is 10 to 15 times higher for T3 than for T4, the deiodinase enzymes play a pivotal role in determining overall metabolic activity. Three different monodeiodinase enzymes are present in the body.10 Of the enzymes that catalyze 5′-monodeiodination, type I enzymes are present in peripheral tissues such as the liver and kidney, whereas type II enzymes are found in the CNS, pituitary, and thyroid. Type III enzymes, found in the placenta, skin, and developing brain, inactivate T4 and T3 by deiodinating the inner ring at the 5 position. The principal characteristics of these enzymes are listed in Table 58-2. T4 may also be acted on by the enzyme 5′-monodeiodinase to form reverse T3, but this accounts for a small component of hormone metabolism. Polymorphisms in the deiodinase genes may prove to be of clinical significance. For example, a polymorphism in the type I deiodinase leading to increased activity seems to be associated with an increased circulating ratio of free T3 to free T4.11 Reverse T3 has no known significant biologic activity. T3 is removed from the body by deiodinative degradation and through the action of sulfotransferase enzyme systems converting to T3 sulfate and 3,3-diiodothyronine sulfates, thus facilitating enterohepatic clearance. Thyronamines are derivatives of thyroid hormone that are present in low concentrations in human serum.12 The most studied thyronamine, 3-iodothyronamine, can theoretically be made from T4 by decarboxylation and deiodination. Administration of pharmacologic amounts of 3-iodothyronamine to animals has profound effects on temperature regulation and cardiac function, and shifts fuel metabolism from carbohydrates to lipids. However, a possible physiologic role for thyronamines has yet to be determined,13 although altered levels may be associated with some disease states.14
TABLE 58-2 Properties of Iodothyronine 5’-DeiodinaseIsoforms
The growth and function of the thyroid are stimulated by activation of the thyrotropin receptor by TSH.15 The receptor belongs to the family of G-protein–coupled receptors. The thyrotropin receptor is coupled to the α subunit of the stimulatory guanine-nucleotide–binding protein (Gsα), activating adenylate cyclase and increasing the accumulation of cyclic adenosine monophosphate. Through this mechanism, TSH stimulates the expression of Na+/I– symporter, TG, and thyroid peroxidase genes as well as increases apical iodide efflux. Somatic activating mutations in the receptor are commonly seen in autonomously functioning thyroid nodules.16 Rarely, germline-activating mutations of the TSH receptor have been reported in kindreds with Leclere’s syndrome, and thyrotoxicosis can result from germline-activating mutations in G-protein signaling in McCune-Albright syndrome.15 Conversely, thyrotropin resistance results from point mutations that prevent TSH binding, leading to abnormalities in the thyrotropin receptor–adenylate cyclase system and congenital hypothyroidism.17 Individuals with this abnormality have high levels of TSH but decreased TG levels and a normal or small gland.
Thyroid hormone nuclear receptors regulate the transcription of target genes in the presence of physiologic concentrations of T3.18 Unlike most other nuclear receptors, TRs also actively regulate gene expression in the absence of hormone, typically resulting in an opposite effect. TRs translocate from the cytoplasm to the nucleus, interact in the nucleus with T3, and target genes and other proteins required for basal and T3-dependent gene transcription. TRs exist in several isoforms, including TRβ1, TRβ2, and TRα1. Thyroid hormone has different actions in different tissues based on tissue-specific expression of the different TR isoforms. There is interest in developing thyroid hormone analogs that selectively activate specific TR isoforms. Such agents could theoretically have targeted desirable effects such as stimulating energy expenditure without having adverse effects on other tissues.19
The production of thyroid hormone is regulated in two main ways. First, thyroid hormone is regulated by TSH secreted by the anterior pituitary. The secretion of TSH is itself under negative feedback control by the circulating level of free thyroid hormone and the positive influence of hypothalamic thyrotropin-releasing hormone (TRH). Second, extrathyroidal deiodination of T4 to T3 is regulated by a variety of factors including nutrition, nonthyroidal hormones, ambient temperatures, drugs, and illness.
THYROTOXICOSIS
Thyrotoxicosis results when tissues are exposed to excessive levels of T4, T3, or both.20 In the National Health and Nutrition Examination Survey III, 0.7% of those surveyed who were not taking thyroid medications and had no history of thyroid disease had subclinical hyperthyroidism (TSH <0.1 milli–international unit/L, and T4 normal), and 0.5% had “clinically significant” hyperthyroidism (TSH <0.1 milli–international unit/L, and T4 >13.2 mcg/dL).21 The prevalence of suppressed TSH values peaked for people aged 20 to 39, declined in those 40 to 79, and increased again in those 80 or older. Abnormal TSH levels were more common among women than among men.
Elderly patients are also more likely to develop atrial fibrillation with thyrotoxicosis than younger patients. The frequency of bowel movements may increase, but frank diarrhea is unusual. For the elderly patient and for the patient with very severe disease, anorexia may be present as well. Palpitations are a prominent and distressing symptom, particularly in the patient with preexisting heart disease. Proximal muscle weakness is common and is noted on climbing stairs or in getting up from a sitting position. Women may note their menses are becoming scanty and irregular. Extremely thyrotoxic patients may have tachycardia, heart failure, psychosis, hyperpyrexia, and coma, a presentation described as thyroid storm.22
CLINICAL PRESENTATION Thyrotoxicosis
Differential Diagnosis
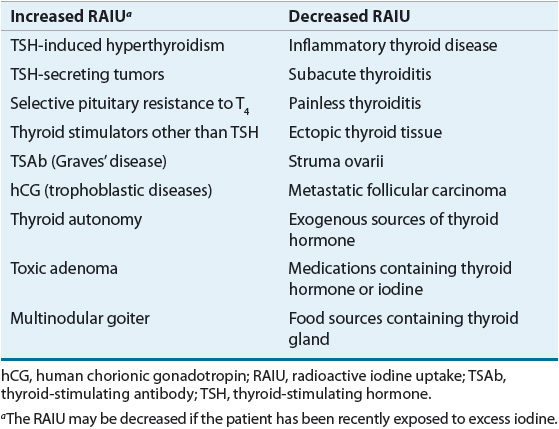
If the clinical history and examination do not provide pathognomonic clues to the etiology of the patient’s thyrotoxicosis, measurement of the RAIU is critical in the evaluation (Table 58-3). The normal 24-hour RAIU ranges from 10% to 30% with some regional variation that is due to differences in iodine intake. An elevated RAIU indicates endogenous hyperthyroidism, that is, the patient’s thyroid gland is actively overproducing T4, T3, or both. Conversely, a low RAIU in the absence of iodine excess indicates that high levels of thyroid hormone are not a consequence of thyroid gland hyperfunction but are likely due to thyroiditis or hormone ingestion. The importance of differentiating endogenous hyperthyroidism from other causes of thyrotoxicosis lies in the widely different prognosis and treatment of the diseases in these two categories. Therapy of thyrotoxicosis associated with thyroid hyperfunction is mainly directed at decreasing the rate of thyroid hormone synthesis, secretion, or both. Such measures are ineffective in treating thyrotoxicosis that is not the result of endogenous hyperthyroidism, because hormone synthesis and regulated hormone secretion are already at a minimum.
TABLE 58-3 Differential Diagnosis of Thyrotoxicosis
Causes of Thyrotoxicosis Associated with Elevated RAIU
TSH-Induced Hyperthyroidism
To better understand these syndromes, we must first review TSH biosynthesis and secretion. TSH is synthesized in the anterior pituitary as separate α– and β-subunit precursors. The α subunits from luteinizing hormone (LH), follicle-stimulating hormone (FSH), human chorionic gonadotropin (hCG), and TSH are similar, whereas the β subunits are unique and confer immunologic and biologic specificity. Free β subunits are devoid of receptor binding and biologic activity and require combination with an α subunit to express their activity. Criteria for the diagnosis of TSH-induced hyperthyroidism include (a) evidence of peripheral hypermetabolism, (b) diffuse thyroid gland enlargement, (c) elevated free thyroid hormone levels, and (d) elevated or inappropriately “normal” serum immunoreactive TSH concentrations. Because the pituitary gland is extremely sensitive to even minimal elevations of free T4, a “normal” or elevated TSH level in any thyrotoxic patient indicates the inappropriate production of TSH.
TSH-Secreting Pituitary Adenomas
TSH-secreting pituitary tumors occur sporadically and release biologically active hormone that is unresponsive to normal feedback control.23 The mean age at diagnosis is around 40 years, with women being diagnosed more than men (8:7). These tumors may cosecrete prolactin or growth hormone; therefore, the patients may present with amenorrhea/galactorrhea or signs of acromegaly. Most patients present with classic symptoms and signs of thyrotoxicosis. Visual field defects may be present due to impingement of the optic chiasm by the tumor. Tumor growth and worsening visual field defects have been reported following antithyroid therapy because lowering of thyroid hormone levels is associated with loss of feedback inhibition from high thyroid hormone levels.
Diagnosis of a TSH-secreting adenoma should be made by demonstrating lack of TSH response to TRH stimulation, inappropriate TSH levels, elevated α-subunit levels, and radiologic imaging; given the lack of routine availability of TRH, the other three criteria are essential. Note that some small tumors are not identified by MRI. Moreover, 10% of “normal” individuals may have incidental pituitary tumors or other benign focal lesions noted on pituitary imaging.
Transsphenoidal pituitary surgery is the treatment of choice for TSH-secreting adenomas. Pituitary gland irradiation is often given following surgery to prevent tumor recurrence. Dopamine agonists and octreotide have been used to treat tumors, especially those that cosecrete prolactin.
Pituitary Resistance to Thyroid Hormone
Resistance to thyroid hormone is a rare condition that can be due to a number of molecular defects, including mutations in the TRβ gene. Pituitary resistance to thyroid hormone (PRTH) refers to selective resistance of the pituitary thyrotrophs to thyroid hormone.24 As nonpituitary tissues respond normally to thyroid hormone, patients experience the toxic peripheral effects of thyroid hormone excess. About 90% of patients studied have an appropriate increase in TSH in response to TRH; conversely, the TSH will be suppressed by T3 administration.
Patients with PRTH require treatment to reduce their elevated thyroid hormone levels. Determining the appropriate serum T4 level is difficult because TSH cannot be used to evaluate adequacy of therapy. Any reduction in thyroid hormone carries the risk of inducing thyrotroph hyperplasia. Ideally, agents that suppress TSH secretion could be used to treat these individuals. Glucocorticoids, dopaminergic drugs, somatostatin and its analogs, and thyroid hormone analogs with reduced metabolic activity have all been tried, but with relatively little benefit. β-Blocker therapy can also be used. Triiodothyroacetic acid (TRIAC), an agent that is devoid of thyromimetic properties on peripheral tissues, but blocks the secretion of TSH, has been used to treat this condition. However, it is not available in the United States. Given the ability of retinoid X receptor ligands to inhibit TSH production, drugs such as bexarotene may have therapeutic benefit in PRTH.25
Thyroid Stimulators Other than TSH
Graves’ Disease
![]() Graves’ disease is an autoimmune syndrome that usually includes hyperthyroidism, diffuse thyroid enlargement, exophthalmos, and, less commonly, pretibial myxedema and thyroid acropachy (Fig. 58-4).20,26 Graves’ disease is the most common cause of hyperthyroidism, with a prevalence estimated to be 3 per 1,000 population in the United States. Hyperthyroidism results from the action of TSAbs, which are directed against the thyrotropin receptor on the surface of the thyroid cell.27 When these immunoglobulins bind to the receptor, they activate downstream G-protein signaling and adenylate cyclase in the same manner as TSH. Autoantibodies that react with orbital muscle and fibroblast tissue in the skin are responsible for the extrathyroidal manifestations of Graves’ disease, and these autoantibodies are encoded by the same germline genes that encode for other autoantibodies for striated muscle and thyroid peroxidase.28 Clinically, the extrathyroidal disorders may not appear at the same time that hyperthyroidism develops.
Graves’ disease is an autoimmune syndrome that usually includes hyperthyroidism, diffuse thyroid enlargement, exophthalmos, and, less commonly, pretibial myxedema and thyroid acropachy (Fig. 58-4).20,26 Graves’ disease is the most common cause of hyperthyroidism, with a prevalence estimated to be 3 per 1,000 population in the United States. Hyperthyroidism results from the action of TSAbs, which are directed against the thyrotropin receptor on the surface of the thyroid cell.27 When these immunoglobulins bind to the receptor, they activate downstream G-protein signaling and adenylate cyclase in the same manner as TSH. Autoantibodies that react with orbital muscle and fibroblast tissue in the skin are responsible for the extrathyroidal manifestations of Graves’ disease, and these autoantibodies are encoded by the same germline genes that encode for other autoantibodies for striated muscle and thyroid peroxidase.28 Clinically, the extrathyroidal disorders may not appear at the same time that hyperthyroidism develops.
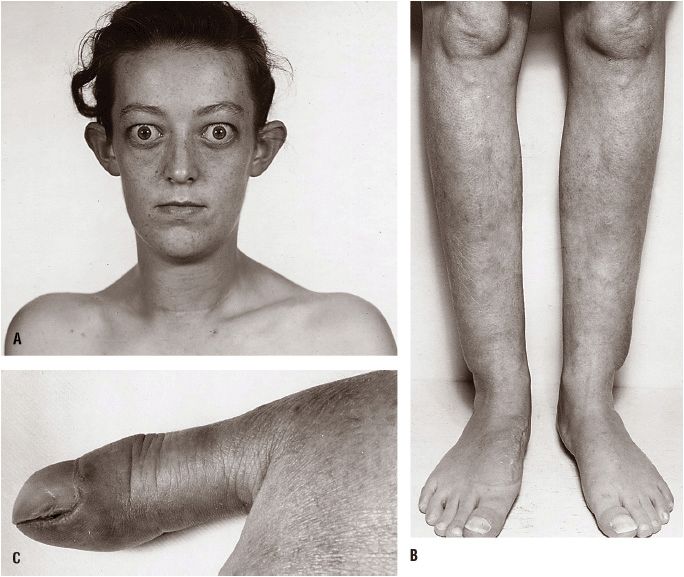
FIGURE 58-4 Features of Graves’ disease. A. Facial appearance in Graves’ disease; lid retraction, periorbital edema, and proptosis are marked. B. Thyroid dermopathy over the lateral aspects of the shins. C. Thyroid acropachy. (Reproduced with permission from Fauci AS, Kasper DL, Longo DL, et al., eds. Harrison’s Principles of Internal Medicine, 16th ed. New York: McGraw-Hill, 2005:2114.)
There is now compelling evidence that heredity predisposes the susceptible individual to development of clinically overt autoimmune thyroid disease in the setting of appropriate environmental and hormonal triggers. A role for gender in the emergence of Graves’ disease is suggested by the fact that hyperthyroidism is approximately eight times more common in women than in men. Other lines of evidence support a role for heredity. First, there is a well-recognized clustering of Graves’ disease within some families. Twin studies in Graves’ disease have revealed that a monozygotic twin has a 35% likelihood of ultimately developing the disease compared with a 3% likelihood for a dizygotic twin, resulting in estimation that 79% of the predisposition to Graves’ disease is genetic.29 Second, the occurrence of other autoimmune diseases, including Hashimoto’s thyroiditis, is also increased in families of patients with Graves’ disease. Third, several studies have demonstrated an increased frequency of certain human leukocyte antigens (HLAs) in patients with Graves’ disease. Differing HLA associations have been identified in the various ethnic groups studied. In whites, for example, the relative risk of Graves’ disease in carriers of the HLA-DR3 haplotype is between 2.5 and 5, whereas lesser associations have been reported for HLA-B8 and the HLA-DQA*0501 allele.30 As with other autoimmune conditions, certain polymorphisms of the T-cell immunoregulatory protein CTLA-4 have also been associated with Graves’ disease. Despite these statistical associations, however, even detailed molecular genetic linkage studies have failed to identify specific genes responsible for the disease.31
The thyroid gland is diffusely enlarged in the majority of patients and is commonly 40 to 60 g (two to three times the normal size). The surface of the gland is either smooth or bosselated, and the consistency varies from soft to firm. For patients with severe disease, a thrill may be felt and a systolic bruit may be heard over the gland, reflecting the increased intraglandular vascularity typical of hyperplasia. Whereas the presence of any of the extrathyroidal manifestations of this syndrome, including exophthalmos, thyroid acropachy, or pretibial myxedema, in a thyrotoxic patient is pathognomonic of Graves’ disease, most patients can be diagnosed on the basis of their history and examination of their diffuse goiter (see Fig. 58-4). An important clinical feature of Graves’ disease is the occurrence of spontaneous remissions, albeit uncommon. The abnormalities in TSAb production may decrease or disappear over time in many patients.
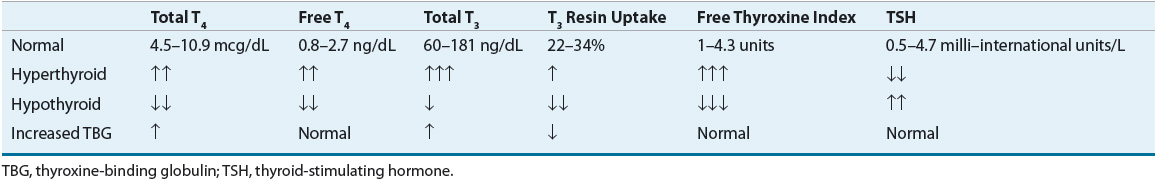
The results of laboratory tests in thyrotoxic Graves’ disease include an increase in the overall hormone production rate with a disproportionate increase in T3 relative to T4 (Table 58-4). In an occasional patient, the disproportionate overproduction of T3 is exaggerated, with the result that only the serum T3 concentration is increased (T3 toxicosis). The saturation of TBG is increased due to the elevated levels of serum T4 and T3, which is reflected in elevated values for the T3 resin uptake. As a result, the concentrations of free T4, free T3, and the free T4 and T3 indices are increased to an even greater extent than are the measured serum total T4 and T3 concentrations. The TSH level will be suppressed or undetectable due to negative feedback by elevated levels of thyroid hormone at the pituitary.
TABLE 58-4 Thyroid Function Test Results in Di. erent Thyroid Conditions
For the patient with manifest disease, measurement of the serum free T4 concentration (or total T4 and T3 resin uptake), total T3, and the TSH value will confirm the diagnosis of thyrotoxicosis. If the patient is not pregnant or lactating, a 24-hour RAIU should be obtained if there is any diagnostic uncertainty, for example, recent onset of symptoms or other factors suggestive of thyroiditis. An increased RAIU documents that the thyroid gland is inappropriately utilizing the iodine to produce more thyroid hormone at a time when the patient is thyrotoxic.
Thyrotoxic periodic paralysis is a rare complication of hyperthyroidism commonly observed in Asian and Hispanic populations.32 It presents as recurrent proximal muscle flaccidity ranging from mild weakness to total paralysis. The paralysis may be asymmetric and usually involves muscle groups that are strenuously exercised before the attack. Cognition and sensory perception are spared, whereas deep tendon reflexes are markedly diminished commonly. The condition is characterized by hypokalemia and low urinary potassium excretion. Hypokalemia results from a sudden shift of potassium from extracellular to intracellular sites rather than reduced total body potassium. High-carbohydrate loads and exercise provoke the attacks. Treatment includes correcting the hyperthyroid state, potassium administration, spironolactone to conserve potassium, and propranolol to minimize intracellular shifts. Some patients with this condition have a mutation in the inwardly rectifying potassium channel Kir2.6.33
Trophoblastic Diseases
hCG is a stimulator of the TSH receptor and may cause hyperthyroidism.34 The basis for the thyrotropic effect of hCG is the structural similarity of hCG to TSH (similar α subunits and unique β subunits). For patients with hyperthyroidism caused by trophoblastic tumors, serum hCG levels usually exceed 300 units/mL and always exceed 100 units/mL. The mean peak hCG level in normal pregnancy is 50 units/mL. On a molar basis, hCG has only 1/10,000 the activity of pituitary TSH in mouse bioassays. Nevertheless, this thyrotropic activity may be very substantial for patients with trophoblastic tumors, whose serum hCG concentrations may reach 2,000 units/mL.
Thyroid Autonomy
Toxic Adenoma
An autonomous thyroid nodule is a discrete thyroid mass whose function is independent of pituitary and TSH control.35 The prevalence of toxic adenoma ranges from about 2% to 9% of thyrotoxic patients, and depends on iodine availability and geographic location. Toxic adenomas are benign tumors that produce thyroid hormone. They arise from gain-of-function somatic mutations of the TSH receptor or, less commonly, the Gsα protein; more than a dozen TSH receptor mutations have been described.16 These nodules may be referred to as toxic adenomas, or “hot” nodules, because of their persistent uptake on a radioiodine thyroid scan, despite suppressed uptake in the surrounding nonnodular gland (Fig. 58-5). The amount of thyroid hormone produced by an autonomous nodule is mass related. Therefore, hyperthyroidism usually occurs with larger nodules (i.e., those >3 cm in diameter). Older patients (>60 years) are more likely (up to 60%) to be thyrotoxic from autonomous nodules than are younger patients (12%). There are many reports of isolated elevation of serum T3 for patients with autonomously functioning nodules. Therefore, if the T4 level is normal, a T3 level must be measured to rule out T3 toxicosis. If autonomous function is suspected but the TSH is normal, the diagnosis can be confirmed by a failure of the autonomous nodule to decrease its iodine uptake during exogenous T3 administration sufficient to suppress TSH. Surgical resection, thionamides, percutaneous ethanol injection, and radioactive iodine (RAI) ablation are treatment options, but since thionamides do not halt the proliferative process in the nodule, definitive therapies are recommended.36 Ethanol ablation may be associated with pain and damage to surrounding extrathyroidal tissues, limiting its acceptance in the United States. It has been hypothesized that sublethal radiation doses received by the surrounding nonnodular thyroid tissue during RAI therapy of toxic nodules may lead to induction of thyroid cancer, and excess thyroid cancer mortality has recently been associated with RAI therapy of toxic nodular disease. Thus, an autonomously functioning nodule, if not large enough to cause thyrotoxicosis, can often be observed conservatively without therapy.
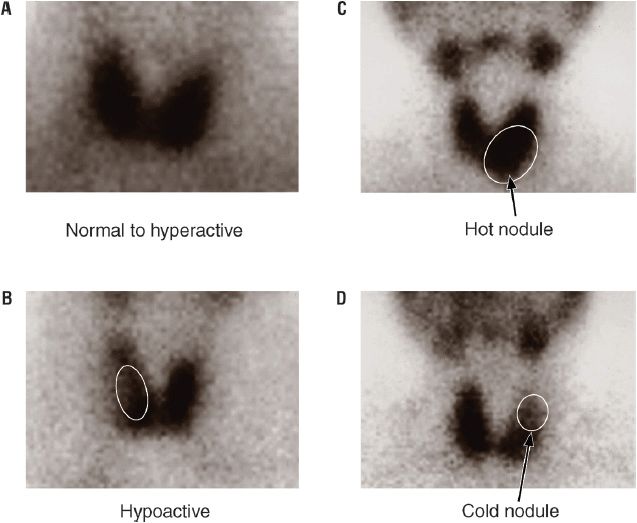
FIGURE 58-5 Radioiodine thyroid scans. A. Normal or increased thyroid uptake of iodine-125 (125I). B. Thyroid with marked decrease in 125I uptake in a large palpable mass. C. Increased 125I uptake isolated to a single nodule, the “hot nodule.” D. Decreased thyroid 125I uptake in an isolated region, the “cold nodule.” (Reproduced with permission from Molina PE. Endocrine Physiology, 2nd ed. New York: McGraw-Hill, 2006:90. Images courtesy of Dr. Luis Linares, Memorial Medical Center, New Orleans, LA.)
Multinodular Goiters
In multinodular goiters (MNGs), follicles with autonomous function coexist with normal or even nonfunctioning follicles. The pathogenesis of MNG is thought to be similar to that of toxic adenoma: diffuse hyperplasia caused by goitrogenic stimuli, leading to mutations and clonal expansion of benign neoplasms.37 The functional status of the nodule(s) depends on the nature of the underlying mutations, whether activating such as TSH receptor mutations or inhibitory such as ras mutations. Thyrotoxicosis in a MNG occurs when a sufficient mass of autonomous follicles generates enough thyroid hormone to exceed the needs of the patient. It is not surprising that this type of hyperthyroidism develops insidiously over a period of several years and predominantly affects older individuals with long-standing goiters. The patient’s complaints of weight loss, depression, anxiety, and insomnia may be attributed to old age. Any unexplained chronic illness in an elderly patient presenting with a MNG calls for the exclusion of hidden thyrotoxicosis.38 Third-generation TSH assays and T3 suppression testing may be useful in detecting subclinical hyperthyroidism.
A thyroid scan will show patchy areas of autonomously functioning thyroid tissue intermixed with hypofunctioning areas. When the patient is euthyroid, therapy is based on the need to reduce goiter size due to mass-related symptoms such as dysphagia. Doses of thyroid hormone sufficient to suppress TSH levels may slow goiter growth or cause some degree of shrinkage, but, in general, suppression therapy for nodular disease is inadequate to address mass effect.39 The preferred treatment for toxic MNG is RAI or surgery. Surgery is usually selected for younger patients and patients in whom large goiters impinge on vital organs. Alternatively, percutaneous injection of 95% ethanol has also been used to destroy single or multinodular adenomas with a 5-year success rate approaching 80%.
Causes of Thyrotoxicosis Associated with Suppressed RAIU
Inflammatory Thyroid Disease
Subacute Thyroiditis Painful subacute (granulomatous or de Quervain) thyroiditis often develops after a viral syndrome, but rarely has a specific virus been identified in thyroid parenchyma.40 A genetic predisposition exists, with markedly higher risk for developing subacute thyroiditis for patients with HLA-Bw35. Systemic symptoms often accompany the syndrome, including fever, malaise, and myalgia, in addition to those symptoms due to thyrotoxicosis. Typically, patients complain of severe pain in the thyroid region, which often extends to the ear on the affected side.41 With time, the pain may migrate from one side of the gland to the other. On physical examination, the thyroid gland is firm and exquisitely tender. Signs of thyrotoxicosis are present.
Thyroid function tests typically run a triphasic course. Initially, serum T4 levels are elevated due to release of preformed thyroid hormone from disrupted follicles. The 24-hour RAIU during this time is less than 2% due to thyroid inflammation and TSH suppression by the elevated T4 level. As the disease progresses, intrathyroidal hormone stores are depleted, and the patient may become mildly hypothyroid with an appropriately elevated TSH level. During the recovery phase, thyroid hormone stores are replenished, and serum TSH concentration gradually returns to normal. Recovery is generally complete within 2 to 6 months. Most patients remain euthyroid, and recurrences of painful thyroiditis are extremely rare. The patient with painful thyroiditis should be reassured that the disease is self-limited and is unlikely to recur. Thyrotoxic symptoms may be relieved with β-blockers. Aspirin (650 mg orally every 6 hours) will usually relieve the pain. Occasionally, prednisone (20 mg orally three times a day) must be used to suppress the inflammatory process. Antithyroid drugs are not indicated because they will not be effective as they do not decrease the release of preformed thyroid hormone.
Painless Thyroiditis Since its description in 1975, painless (silent, lymphocytic) thyroiditis has been recognized as a common cause of thyrotoxicosis and may represent up to 15% of cases of thyrotoxicosis in North America.42 In the setting of development of lymphocytic thyroiditis during the first 12 months after the end of pregnancy, the condition is also called postpartum thyroiditis.43 The etiology is not fully understood and may be heterogeneous, but evidence indicates that autoimmunity underlies most cases. There is an increased frequency of HLA-DR3 and DR5 in patients with painless thyroiditis; nonendocrine autoimmune diseases are also more common. Histologically, diffuse lymphocytic infiltration is generally identified. The triphasic course of this illness mimics that of subacute thyroiditis. Most patients present with mild thyrotoxic symptoms. Lid retraction and lid lag are present, but exophthalmos is absent. The thyroid gland may be diffusely enlarged, but thyroid tenderness is absent.
The 24-hour RAIU will typically be suppressed to less than 2% during the thyrotoxic phase of painless thyroiditis. Anti-TG and antithyroid peroxidase antibody (anti-TPOAb) levels are elevated in more than 50% of patients. Patients with mild hyperthyroidism and painless thyroiditis should be reassured that they have a self-limited disease, although patients with postpartum thyroiditis may experience recurrence of the disease with subsequent pregnancies. As with other thyrotoxic syndromes, adrenergic symptoms may be ameliorated with propranolol or metoprolol. Antithyroid drugs, which inhibit new hormone synthesis, are not indicated because they do not decrease the release of preformed thyroid hormone. A small proportion of patients may have recurrent episodes of thyroiditis,44 or may develop permanent hypothyroidism.45
Ectopic Thyroid Tissue
Struma Ovarii Struma ovarii is a teratoma of the ovary that contains differentiated thyroid follicular cells and is capable of making thyroid hormone.46 This extremely rare cause of thyrotoxicosis is suggested by the absence of thyroid enlargement in a thyrotoxic patient with a suppressed RAIU in the neck and no findings to suggest thyroiditis. The diagnosis is established by localizing functioning thyroid tissue in the ovary with whole-body RAI (sodium iodide-131 [131I]) scanning. Interestingly, struma ovarii without associated hyperthyroidism is much more common than struma ovarii associated with hyperthyroidism. Because the tissue is neoplastic and potentially malignant, combined surgical and radioiodine treatment of malignant struma ovarii for both monitoring and therapy of relapse is the recommended treatment.
Thyroid Cancer In widely metastatic differentiated papillary or follicular carcinomas with relatively well-preserved function, sufficient thyroid hormone can be synthesized and secreted to produce thyrotoxicosis.47 In most instances, a previous diagnosis of thyroid malignancy has been made. The diagnosis can be confirmed by whole-body 131I scanning. Treatment with 131I is generally effective at ablating functioning thyroid metastases.
Exogenous Sources of Thyroid Hormone or Iodine
Exogenous Thyroid Hormone Thyrotoxicosis factitia was described in the recent American Thyroid Association guidelines on the management of hyperthyroidism as “all causes of hyperthyroidism due to ingestion of thyroid hormone.”48 This category includes hyperthyroidism produced by the intentional ingestion of exogenous thyroid hormone.49 Obesity is the most common nonthyroidal disorder for which thyroid hormone is inappropriately used, but thyroid hormone has been used for almost every conceivable problem from menstrual irregularities and infertility to hypercholesterolemia and baldness. There is little evidence to suggest that treatment with thyroid hormone is beneficial for such conditions in euthyroid individuals. Obviously, thyrotoxicosis factitia can also occur when too large a dose of thyroid hormone is employed for conditions in which it is likely to be beneficial, such as differentiated thyroid carcinoma. In addition to this iatrogenic cause, accidental ingestion such as may occur with pediatric ingestion or pharmacy error is also possible. Rarely, thyrotoxicosis factitia is caused by the purposeful and secretive ingestion of thyroid hormone by disturbed patients (usually with a medical background) who wish to obtain attention or lose weight.
Thyroid hormone may also be accidentally ingested in food sources. Reports of thyrotoxicosis in Minnesota and Nebraska in 1980s were attributed to ingestion of ground beef contaminated by bovine thyroid glands.50,51 More recently thyrotoxicosis due to porcine thyroid tissue in meat products has been reported in Spain and Uruguay.52,53
Thyrotoxicosis factitia should be suspected in a thyrotoxic patient without evidence of increased hormone production, thyroidal inflammation, or ectopic thyroid tissue. The RAIU uptake is at low levels because the patient’s thyroid gland function is suppressed by the exogenous thyroid hormone. Measurement of plasma TG is a valuable laboratory aid in the diagnosis of thyrotoxicosis factitia. TG is normally secreted in small amounts by the thyroid gland; however, when thyroid hormone is taken orally, TG levels tend to be lower than the normal range. In other entities characterized by a low RAIU, such as thyroiditis, leakage of preformed thyroid hormone results in elevated TG levels. If a history of thyroid hormone ingestion is elicited or deduced, exogenous thyroid hormone should be withheld for between 4 and 6 weeks, and thyroid function tests repeated to document that the euthyroid state has been restored. Rarely, thyroid hormone analogs or metabolites may be the drug of abuse, specific detection of which may be difficult with standard thyroid hormone assays. For example, tiratricol (TRIAC), an endogenous metabolite of T3 that has been used for weight loss and paradoxically by body builders, will suppress TSH at high enough doses and may cross-react in many T3 immunoassays; thus, thyrotoxicosis factitia due to tiratricol abuse may be misinterpreted as T3 toxicosis.54
Medications Containing Iodine Amiodarone may induce thyrotoxicosis (2% to 3% of patients), overt hypothyroidism (5% of patients), subclinical hypothyroidism (25% of patients), or euthyroid hyperthyroxinema, depending on the underlying thyroid pathology or lack thereof.55 Because amiodarone contains 37% iodine by weight, approximately 6 mg/day of iodine is released for each 200 mg of amiodarone, 1,000 times greater than the recommended daily amount of iodine of 150 mcg/day. As a result of this iodine overload, iodine-exacerbated thyroid dysfunction commonly occurs among those patients with preexisting thyroid disease: thyrotoxicosis in patients with hyperthyroidism or euthyroid nodular autonomy and hypothyroidism in patients with autoimmune thyroid disease. In contrast to hyperthyroidism with increased synthesis of thyroid hormone induced by amiodarone (type I), destructive thyroiditis with loss of TG and thyroid hormones also occurs (type II), typically among individuals with otherwise normal glands. The two types of amiodarone-induced thyrotoxicosis may be differentiated using color flow Doppler ultrasonography. Such distinction is critically important, given the therapeutic implications of the two syndromes: type I amiodarone-induced hyperthyroidism responds somewhat to thionamides, whereas type II may respond to glucocorticoids or iopanoic acid.55 Unfortunately, however, the latter agent is no longer available. Obviously, RAI therapy is inappropriate in type I due to the drug-induced iodine excess, and in type II due to lack of increased hormone synthesis. The manifestations of amiodarone-induced thyrotoxicosis may be atypical symptoms such as ventricular tachycardia and exacerbation of underlying chronic obstructive pulmonary disease, both of which are even more significant given the severe underlying cardiac pathology that led to the use of the drug in the first place. Amiodarone also directly interferes with type I 5′-deiodinase, leading to reduced conversion of T4 to T3 and hyperthyroxinemia without thyrotoxicosis.
TREATMENT
Hyperthyroidism
Desired Outcomes
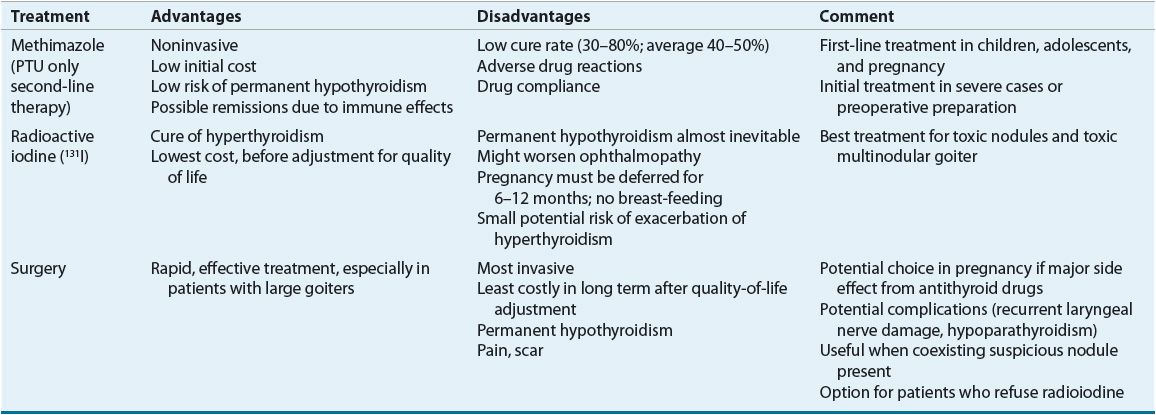
![]() Three common treatment modalities are used in the management of hyperthyroidism: surgery, antithyroid medications, and RAI (Table 58-5). The overall therapeutic objectives are to eliminate the excess thyroid hormone and minimize the symptoms and long-term consequences of hyperthyroidism. Therapy must be individualized based on the type and severity of hyperthyroidism, patient age and gender, existence of nonthyroidal conditions, and response to previous therapy.56,57 Clinical guidelines for the treatment of hyperthyroidism have been published.48
Three common treatment modalities are used in the management of hyperthyroidism: surgery, antithyroid medications, and RAI (Table 58-5). The overall therapeutic objectives are to eliminate the excess thyroid hormone and minimize the symptoms and long-term consequences of hyperthyroidism. Therapy must be individualized based on the type and severity of hyperthyroidism, patient age and gender, existence of nonthyroidal conditions, and response to previous therapy.56,57 Clinical guidelines for the treatment of hyperthyroidism have been published.48
TABLE 58-5 Treatments for Hyperthyroidism Caused by Graves’ Disease
Nonpharmacologic Therapy
Surgical removal of the hypersecreting thyroid gland became feasible in 1923 when Plummer discovered that iodine reduced the gland’s vascularity, making this definitive procedure possible. Surgery should be considered for patients with a large thyroid gland (>80 g), severe ophthalmopathy, and a lack of remission on antithyroid drug treatment. In case of cosmetic issues or pressure symptoms, the choice in MNG stands between surgery, which is still the first choice, and radioiodine therapy if uptake is adequate (hot). In addition to surgery, the solitary nodule, whether hot or cold, can be treated with percutaneous ethanol injection therapy. If hot, radioiodine is the therapy of choice.58 Traditional preparation of the patient for thyroidectomy includes methimazole (MMI) or propylthiouracil (PTU) until the patient is biochemically euthyroid (usually 6 to 8 weeks), followed by the addition of iodides (500 mg/day) for 10 to 14 days before surgery to decrease the vascularity of the gland. Iodine supplementation in iodine-deficient areas of the country may lead to a greater reduction in remnant volume in nontoxic goiter.59 Propranolol for several weeks preoperatively and 7 to 10 days after surgery has also been used to maintain a pulse rate of less than 90 beats/min. Combined pretreatment with propranolol and 10 to 14 days of potassium iodide also has been advocated.
The overall complication rate when surgery is performed for MNG by an experienced endocrine surgeon is low.60 If subtotal thyroidectomy, or an operation that attempts to maintain euthyroidism, is performed for Graves’ disease, there is a risk of recurrence of hyperthyroidism that is directly related to remnant size.61 Near total thyroidectomy is generally recognized as the procedure of choice for patients with Graves’ disease.48 The complication rates of surgery for Graves’ disease are low when surgery is performed by a high-volume thyroid surgeon, and include hypoparathyroidism (up to 2%), and recurrent laryngeal nerve injury (up to 1%). Formal cost-effective analysis indicates that a total thyroidectomy may be the most cost-effective method for managing hyperthyroidism when considering outcomes in quality-adjusted life-years.62
Pharmacologic Therapy
Antithyroid Medications
![]() Thiourea Drugs Two drugs within this category, MMI and PTU, are approved for the treatment of hyperthyroidism in the United States.63 They are classified as thioureylenes (thionamides), which incorporate an N–C–S=N group into their ring structures.
Thiourea Drugs Two drugs within this category, MMI and PTU, are approved for the treatment of hyperthyroidism in the United States.63 They are classified as thioureylenes (thionamides), which incorporate an N–C–S=N group into their ring structures.
Mechanism of Action PTU and MMI share several mechanisms to inhibit the biosynthesis of thyroid hormone.20 These drugs serve as preferential substrates for the iodinating intermediate of thyroid peroxidase and divert iodine away from potential iodination sites in TG. This prevents subsequent incorporation of iodine into iodotyrosines and ultimately iodothyronine (“organification”). Second, they inhibit coupling of MIT and DIT to form T4 and T3. The coupling reaction may be more sensitive to these drugs than the iodination reaction. Experimentally, these drugs exhibit immunosuppressive effects, although the clinical relevance of this finding is unclear. For patients with Graves’ disease, antithyroid drug treatment has been associated with lower TSAb titers and restoration of normal suppressor T-cell function. However, ClO4–, which has a different mechanism of action, also decreases TSAbs, suggesting that normalization of the thyroid hormone level may itself improve the abnormal immune function. PTU inhibits the peripheral conversion of T4 to T3. This effect is acutely dose related and occurs within hours of PTU administration. MMI does not have this effect. Over time, depletion of stored hormone and lack of continuing synthesis of thyroid hormone results in the clinical effects of these drugs.
Pharmacokinetics Both antithyroid drugs are well absorbed (80% to 95%) from the GI tract, with peak serum concentrations about 1 hour after ingestion. The plasma half-life ranges of PTU and MMI are 1 to 2.5 and 6 to 9 hours, respectively, and are not appreciably affected by thyroid status. Urinary excretion is about 35% for PTU and less than 10% for MMI. These drugs are actively concentrated in the thyroid gland, which may account for the disparity between their relatively short plasma half-lives and the effectiveness of once-daily dosing regimens even with PTU. Approximately 60% to 80% of PTU is bound to plasma albumin, whereas MMI is not protein bound. MMI readily crosses the placenta and appears in breast milk. Older studies suggested that PTU crosses the placental membranes only one tenth as well as MMI; however, these studies were done in the course of therapeutic abortion early in pregnancy. Newer studies show little difference between fetal concentrations of PTU and MMI, and both are associated with elevated TSH in about 20% and low T4 in about 7% of fetuses.64
![]() Dosing and Monitoring PTU is available as 50 mg tablets, and MMI as 5 and 10 mg tablets. MMI is approximately 10 times more potent than PTU. Initial therapy with PTU ranges from 300 to 600 mg daily, usually in three or four divided doses. MMI is given in two or three divided doses totaling 30 to 60 mg/day. Although the traditional recommendation is for divided doses, evidence exists that both drugs can be given as single daily doses. Patients with severe hyperthyroidism may require larger initial doses, and some may respond better at these larger doses if the dose is divided. The maximal blocking doses of PTU and MMI are 1,200 and 120 mg daily, respectively. Once the intrathyroidal pool of thyroid hormone is reduced and new hormone synthesis is sufficiently blocked, clinical improvement should ensue. Usually within 4 to 8 weeks of initiating therapy, symptoms will diminish and circulating thyroid hormone levels will return to normal. At this time the tapering regimen can be started. Changes in dose for each drug should be made on a monthly basis, because the endogenously produced T4 will reach a new steady-state concentration in this interval. Typical ranges of daily maintenance doses for PTU and MMI are 50 to 300 and 5 to 30 mg, respectively.
Dosing and Monitoring PTU is available as 50 mg tablets, and MMI as 5 and 10 mg tablets. MMI is approximately 10 times more potent than PTU. Initial therapy with PTU ranges from 300 to 600 mg daily, usually in three or four divided doses. MMI is given in two or three divided doses totaling 30 to 60 mg/day. Although the traditional recommendation is for divided doses, evidence exists that both drugs can be given as single daily doses. Patients with severe hyperthyroidism may require larger initial doses, and some may respond better at these larger doses if the dose is divided. The maximal blocking doses of PTU and MMI are 1,200 and 120 mg daily, respectively. Once the intrathyroidal pool of thyroid hormone is reduced and new hormone synthesis is sufficiently blocked, clinical improvement should ensue. Usually within 4 to 8 weeks of initiating therapy, symptoms will diminish and circulating thyroid hormone levels will return to normal. At this time the tapering regimen can be started. Changes in dose for each drug should be made on a monthly basis, because the endogenously produced T4 will reach a new steady-state concentration in this interval. Typical ranges of daily maintenance doses for PTU and MMI are 50 to 300 and 5 to 30 mg, respectively.
If the objective of therapy is to induce a long-term remission, the patient should remain on continuous antithyroid drug therapy for 12 to 24 months. Antithyroid drug therapy induces permanent remission rates of 10% to 98%, with an overall average of about 40% to 50%.65 This is much higher than the remission rate seen with propranolol alone, which is reported to range from 22% to 36%. Patient characteristics for a favorable outcome include older patients (>40 years), low T4:T3 ratio (<20), a small goiter (<50 g), short duration of disease (<6 months), no previous history of relapse with antithyroid drugs, duration of therapy 1 to 2 years or longer, and low TSAb titers at baseline or a reduction with treatment.20 A recent study provides preliminary evidence that a new assay that has better specificity for detection of antibodies that stimulate the TSH receptors, without detecting coexistent blocking antibodies, may be a useful predictor of remission of Graves’ disease.66
It is important that patients be followed every 6 to 12 months after remission occurs. If a relapse occurs, alternate therapy with RAI is preferred to a second course of antithyroid drugs. Relapses seem to plateau after about 5 years and eventually 5% to 20% of patients will develop spontaneous hypothyroidism. There has been interest in whether concurrent administration of T4 with thionamide therapy for thyrotoxicosis and subclinical hyperthyroidism can reduce autoantibodies directed toward the thyroid gland and improve remission rate. In a Japanese study, adjunctive treatment with T4 was associated with a 20-fold reduction in the recurrence rate of Graves’ disease compared with the recurrence rate seen for patients treated with antithyroid drugs alone.67 Attempts to reproduce these results in American and European patients with Graves’ disease have failed to show any delay or reduction in the recurrence of Graves’ disease with T4 administration.68
![]() Subclinical hyperthyroidism is defined as the finding of a serum TSH below the lower limit of the reference range combined with free T4 and T3 concentrations that are normal. Subclinical hyperthyroidism is associated with an increased risk of atrial fibrillation, and may be associated with increased all-cause mortality. Some studies show an increased risk of hip fractures in postmenopausal women with subclinical hyperthyroidism. Most practitioners agree that treatment of older patients (greater than 65 years) with TSH values below 0.1 milli–international unit/L is reasonable. In patients who are younger or have TSH values of 0.1 to 0.4 milli–international unit/L a decision whether to treat the patient for mild hyperthyroidism or to monitor thyroid function may depend on his or her cardiovascular risk factors and bone health.69
Subclinical hyperthyroidism is defined as the finding of a serum TSH below the lower limit of the reference range combined with free T4 and T3 concentrations that are normal. Subclinical hyperthyroidism is associated with an increased risk of atrial fibrillation, and may be associated with increased all-cause mortality. Some studies show an increased risk of hip fractures in postmenopausal women with subclinical hyperthyroidism. Most practitioners agree that treatment of older patients (greater than 65 years) with TSH values below 0.1 milli–international unit/L is reasonable. In patients who are younger or have TSH values of 0.1 to 0.4 milli–international unit/L a decision whether to treat the patient for mild hyperthyroidism or to monitor thyroid function may depend on his or her cardiovascular risk factors and bone health.69
Adverse Effects Minor adverse reactions to MMI and PTU have an overall incidence of 5% to 25% depending on the dose and the drug, whereas major adverse effects occur in 1.5% to 4.6% of patients receiving these drugs.63 Pruritic maculopapular rashes (sometimes associated with vasculitis based on skin biopsy), arthralgias, and fevers occur in up to 5% of patients and may occur at greater frequency with higher doses and in children. Rashes often disappear spontaneously but, if persistent, may be managed with antihistamines.
Perhaps one of the most common side effects is a benign transient leukopenia characterized by a white blood cell (WBC) count of less than 4,000/mm3. This condition occurs in up to 12% of adults and 25% of children, and sometimes can be confused with mild leukopenia seen in Graves’ disease. This mild leukopenia is not a harbinger of the more serious adverse effect of agranulocytosis, so therapy can usually be continued. If a minor adverse reaction occurs with one antithyroid drug, the alternate thiourea may be tried, but cross-sensitivity occurs for about 50% of patients.63
Agranulocytosis is one of the serious adverse effects of thiourea drug therapy and is characterized by fever, malaise, gingivitis, oropharyngeal infection, and a granulocyte count less than 250/mm3.63 These drugs are concentrated in granulocytes, and this reaction may represent a direct toxic effect rather than hypersensitivity. This toxic reaction has occurred with both thioureas, and the incidence varies from 0.5% to 6%. It is higher for patients over age 40 receiving a MMI dose greater than 40 mg/day or the equivalent dose of PTU, and is linked to HLA class II genes containing the DRB1*08032 allele.70 Agranulocytosis usually develops in the first 3 months of therapy. Because the onset is sudden, routine monitoring has not been recommended. Colony-stimulating factors have been used with some success to restore cell counts to normal, but it is unclear how effective this form of therapy is compared with routine supportive care.71 Peripheral lymphocytes obtained from patients with PTU-induced agranulocytosis undergo transformation in the presence of other thioamides, suggesting that these severe reactions are immunologically mediated and patients should not receive other thionamides. Aplastic anemia has been reported with MMI and may be associated with an inhibitor to colony-forming units. Once antithyroid drugs are discontinued, clinical improvement is seen over several days to weeks. Patients should be counseled to discontinue therapy and contact their physician when flu-like symptoms such as fever, malaise, or sore throat develop.
Arthralgias and a lupus-like syndrome (sometimes in the absence of antinuclear antibodies) have been reported in 4% to 5% of patients. This generally occurs after 6 months of therapy. Uncommonly, polymyositis, presenting as proximal muscle weakness and elevated creatine phosphokinase, has been reported with PTU administration. GI intolerance is also reported to occur in 4% to 5% of patients. Patients receiving interferon products for hepatitis C or other disorders may develop hyperthyroidism or hypothyroidism along with liver enzyme abnormalities.72 Hypoprothrombinemia is a rare complication of thionamide therapy. Patients who have experienced a major adverse reaction to one thiourea drug should not be converted to the alternate drug because of cross-sensitivity.
Older reports suggested that congenital skin defects (aplasia cutis) may be caused by MMI and carbimazole, although a registry review from the Netherlands could not find an association between maternal use of these drugs and skin defects.73 However, more recently, several serious congenital malformations including tracheoesophageal fistulas and choanal atresia have been observed with MMI and carbimazole but not PTU use during pregnancy.74,75 Thus, in the past, PTU was considered the drug of choice throughout pregnancy for women with hyperthyroidism, because of concerns about the possible teratogenic effects of MMI. However, currently heightened concerns about the greater hepatotoxicity of PTU than of MMI have led to the recommendation that PTU no longer be considered a first-line drug, except during the first trimester of pregnancy.
Hepatotoxicity can be seen with both MMI and PTU with a prevalence of approximately 1.3%. In mice, MMI undergoes epoxidation of the C-4,5 double bond by cytochrome P450 enzymes, and after being hydrolyzed, the resulting epoxide is decomposed to form N-methylthiourea, a proximate toxicant.76 At moderate doses, some authors have found that initial enzyme elevations eventually normalize in most patients with continued therapy. PTU-induced subclinical liver injury is common and is usually transient and asymptomatic.77 Thus, it has generally been thought that therapy with PTU may be continued with caution in the absence of symptoms and hyperbilirubinemia. However, a 1997 literature review documented 49 cases of hepatotoxicity. Twenty-eight cases were associated with PTU use, and 21 cases were associated with MMI use. The hepatotoxicity was associated with seven deaths and three deaths in the PTU and MMI groups, respectively.78 There did not appear to be a relationship between the dose or duration of thionamide treatment and outcome. During the past 20 years of PTU use in the United States, 22 adults developed severe hepatotoxicity leading to 9 deaths and 5 liver transplants. The risk of this complication was greater in children (1:2,000) than in adults (1:10,000).79 A recent reanalysis of data reported to the FDA from 1982 to 2008 found that toxicity in children was generally related to higher doses of PTU and that toxicity in both children and adults was associated with therapy lasting more than 4 months in duration.80 In light of such evidence, it has been recommended by the American Thyroid Association and the FDA that PTU not be considered as first-line therapy in either adults or children.81 One of three exceptions includes the first trimester of pregnancy, when the risk of MMI-induced embryopathy may exceed that of PTU-induced hepatotoxicity. Other exceptions include intolerance to MMI and thyroid storm.
Iodides Iodide was the first form of drug therapy for Graves’ disease. Its mechanism of action is to acutely block thyroid hormone release, inhibit thyroid hormone biosynthesis by interfering with intrathyroidal iodide utilization (the Wolff-Chaikoff effect), and decrease the size and vascularity of the gland. This early inhibitory effect provides symptom improvement within 2 to 7 days of initiating therapy, and serum T4 and T3 concentrations may be reduced for a few weeks. Despite the reduced release of T4 and T3, thyroid hormone synthesis continues at an accelerated rate, resulting in a gland rich in stored hormones. The normal and hyperfunctioning thyroid soon escapes from this inhibitory effect within 1 to 2 weeks by decreasing the active transfer of iodide into the gland. Iodides are often used as adjunctive therapy to prepare a patient with Graves’ disease for surgery, to acutely inhibit thyroid hormone release and quickly attain the euthyroid state in severely thyrotoxic patients with cardiac decompensation, or to inhibit thyroid hormone release following RAI therapy. However, large doses of iodine may exacerbate hyperthyroidism or indeed precipitate hyperthyroidism in some previously euthyroid individuals (Jod-Basedow disease).82 This Jod-Basedow phenomenon is most common in iodine-deficient areas, particularly for patients with preexisting nontoxic goiter. Iodide is contraindicated in toxic MNG.
Potassium iodide is available either as a saturated solution (SSKI), which contains 38 mg of iodide per drop, or as Lugol’s solution, which contains 6.3 mg of iodide per drop. The typical starting dose of SSKI is 3 to 10 drops daily (120 to 400 mg) in water or juice. There is no documented advantage to using doses in excess of 6 to 8 mg/day. When used to prepare a patient for surgery, it should be administered 7 to 14 days preoperatively. As an adjunct to RAI, SSKI should not be used before, but rather 3 to 7 days after RAI treatment, so that the radioactive iodide can concentrate in the thyroid. The most frequent toxic effects with iodide therapy are hypersensitivity reactions (skin rashes, drug fever, rhinitis, and conjunctivitis), salivary gland swelling, “iodism” (metallic taste, burning mouth and throat, sore teeth and gums, symptoms of a head cold, and sometimes stomach upset and diarrhea), and gynecomastia.
Other compounds containing organic iodide have also been used therapeutically for hyperthyroidism. These include various radiologic contrast media that share a triiodoaminobenzene and monoaminobenzene ring with a propionic acid chain (e.g., iopanoic acid and sodium ipodate). The effect of these compounds is a result of the iodine content inhibiting thyroid hormone release as well as competitive inhibition of 5′-monodeiodinase conversion related to their structures, which resemble thyroid analogs. Unfortunately, these extremely useful agents are no longer available in the United States
![]() Adrenergic Blockers Because many of the manifestations of hyperthyroidism are mediated by β-adrenergic receptors, β-blockers (especially propranolol) have been used widely to ameliorate thyrotoxic symptoms such as palpitations, anxiety, tremor, and heat intolerance. Although β-blockers are quite effective for symptom control, they have no effect on the urinary excretion of calcium, phosphorus, hydroxyproline, creatinine, or various amino acids, suggesting a lack of effect on peripheral thyrotoxicosis and protein metabolism. Furthermore, β-blockers neither reduce TSAb nor prevent thyroid storm. Propranolol and nadolol partially block the conversion of T4 to T3, but this contribution to the overall therapeutic effect is small in magnitude. Inhibition of conversion of T4 to T3 is mediated by D-propranolol, which is devoid of β-blocking activity, and L-propranolol, which is responsible for the antiadrenergic effects, has little effect on the conversion.
Adrenergic Blockers Because many of the manifestations of hyperthyroidism are mediated by β-adrenergic receptors, β-blockers (especially propranolol) have been used widely to ameliorate thyrotoxic symptoms such as palpitations, anxiety, tremor, and heat intolerance. Although β-blockers are quite effective for symptom control, they have no effect on the urinary excretion of calcium, phosphorus, hydroxyproline, creatinine, or various amino acids, suggesting a lack of effect on peripheral thyrotoxicosis and protein metabolism. Furthermore, β-blockers neither reduce TSAb nor prevent thyroid storm. Propranolol and nadolol partially block the conversion of T4 to T3, but this contribution to the overall therapeutic effect is small in magnitude. Inhibition of conversion of T4 to T3 is mediated by D-propranolol, which is devoid of β-blocking activity, and L-propranolol, which is responsible for the antiadrenergic effects, has little effect on the conversion.
β-Blockers are usually used as adjunctive therapy with antithyroid drugs, RAI, or iodides when treating Graves’ disease or toxic nodules; in preparation for surgery; or in thyroid storm. The only conditions for which β-blockers are primary therapy for thyrotoxicosis are those associated with thyroiditis. The dose of propranolol required to relieve adrenergic symptoms is variable, but an initial dose of 20 to 40 mg four times daily is effective (heart rate <90 beats/min) for most patients. Younger or more severely toxic patients may require as much as 240 to 480 mg/day because there seems to be an increased clearance rate for these patients. β-Blockers are contraindicated for patients with decompensated heart failure unless it is caused solely by tachycardia (high output). Nonselective agents and those lacking intrinsic sympathomimetic activity should be used with caution for patients with asthma and bronchospastic chronic obstructive lung disease. β-Blockers that are cardioselective and have intrinsic sympathomimetic activity may have a slight margin of safety in these situations. Other patients in whom contraindications exist are those with sinus bradycardia, those receiving monoamine oxidase inhibitors or tricyclic antidepressants, and those with spontaneous hypoglycemia. β-Blockers may also prolong gestation and labor during pregnancy. Other side effects include nausea, vomiting, anxiety, insomnia, light-headedness, bradycardia, and hematologic disturbances.
Antiadrenergic agents such as centrally acting sympatholytics and calcium channel antagonists may have some role in the symptomatic treatment of hyperthyroidism. These drugs might be useful when contraindications to β-blockade exist. When compared with nadolol 40 mg twice daily, clonidine 150 mcg twice daily reduced plasma catecholamines, whereas nadolol increased both epinephrine and norepinephrine after 1 week of treatment. Diltiazem 120 mg given every 8 hours reduced heart rate by 17%; fewer ventricular extrasystoles were noted after 10 days of therapy, and diltiazem has been shown to be comparable to propranolol in lowering heart rate and blood pressure.
![]() Radioactive Iodine Although other radioisotopes have been used to ablate thyroid tissue, 131 I is considered to be the agent of choice for Graves’ disease, toxic autonomous nodules, and toxic MNGs.83 RAI is administered as a colorless and tasteless liquid that is well absorbed and concentrates in the thyroid. 131 I is a β– and αg-emitter with a tissue penetration of 2 mm and a half-life of 8 days. Other organs take up 131I, but the thyroid gland is the only organ in which organification of the absorbed iodine takes place. Initially, RAI disrupts hormone synthesis by incorporating into thyroid hormones and TG. Over a period of weeks, follicles that have taken up RAI and surrounding follicles develop evidence of cellular necrosis, breakdown of follicles, development of bizarre cell forms, nuclear pyknosis, and destruction of small vessels within the gland, leading to edema and fibrosis of the interstitial tissue. Pregnancy is an absolute contraindication to the use of RAI since radiation will be delivered to the fetal tissue, including the fetal thyroid.
Radioactive Iodine Although other radioisotopes have been used to ablate thyroid tissue, 131 I is considered to be the agent of choice for Graves’ disease, toxic autonomous nodules, and toxic MNGs.83 RAI is administered as a colorless and tasteless liquid that is well absorbed and concentrates in the thyroid. 131 I is a β– and αg-emitter with a tissue penetration of 2 mm and a half-life of 8 days. Other organs take up 131I, but the thyroid gland is the only organ in which organification of the absorbed iodine takes place. Initially, RAI disrupts hormone synthesis by incorporating into thyroid hormones and TG. Over a period of weeks, follicles that have taken up RAI and surrounding follicles develop evidence of cellular necrosis, breakdown of follicles, development of bizarre cell forms, nuclear pyknosis, and destruction of small vessels within the gland, leading to edema and fibrosis of the interstitial tissue. Pregnancy is an absolute contraindication to the use of RAI since radiation will be delivered to the fetal tissue, including the fetal thyroid.
β-Blockers may be given any time without compromising RAI therapy, accounting for their role as a mainstay of adjunctive therapy to RAI treatment. If iodides are administered, they should be given 3 to 7 days after RAI to prevent interference with the uptake of RAI in the thyroid gland. Because thyroid hormone levels will transiently increase following RAI treatment due to release of preformed thyroid hormone, patients with cardiac disease and elderly patients are often treated with thionamides prior to RAI ablation. For patients with underlying cardiac disease, it may be necessary to reinstitute antithyroid drug therapy following RAI ablation. The standard practice is to withdraw the thionamide 4 to 6 days prior to RAI treatment and to reinstitute it 4 days after therapy is concluded. Administering antithyroid drug therapy following RAI treatment may result in a higher rate of posttreatment recurrence or persistent hyperthyroidism.83 Pretreatment with PTU may lead to higher rates of treatment failure, but this does not appear to be the case with MMI pretreatment.85 Use of lithium, as adjunctive therapy to RAI therapy, has multiple benefits of increasing the cure rate, shortening the time to cure, and preventing posttherapy increase in thyroid hormone levels.86 Lithium is likely to achieve these effects by increasing RAI retention in the thyroid and inhibiting thyroid hormone release from the gland.
Corticosteroid administration will blunt and delay the rise in antibodies to the TSH receptor, TG, and thyroid peroxidase while reducing T3 and T4 concentrations following RAI. Bartalena et al. found no progression in ophthalmopathy for patients receiving prednisone after RAI (0% worsened) compared with 3% worsening in those receiving MMI, and 5% worsening in those receiving RAI alone.87 Theoretically, if shared thyroidal and orbital antigen is involved in the pathogenesis of Graves’ ophthalmopathy, antigen released with RAI treatment could aggravate preexisting eye disease. There is some disagreement as to what degree of ophthalmopathy should be considered a contraindication to RAI. However, in those with moderate or severe orbitopathy it seems reasonable to delay RAI until the patient’s eye disease has been stable.
Destruction of the gland attenuates the hyperthyroid state, and hypothyroidism commonly occurs months to years following RAI.20,88 The goal of therapy is to destroy overactive thyroid cells, and a single dose of 4,000 to 8,000 rad results in a euthyroid state in 60% of patients at 6 months or less. The remaining 40% become euthyroid within 1 year, requiring two or more doses. It is advisable that a second dose of RAI be given 6 months after the first RAI treatment if the patient remains hyperthyroid. Variables that predict an unsuccessful outcome of RAI include gender (men are less likely to develop hypothyroidism), race, the size of the thyroid (euthyroidism is less likely in large glands), severity of disease, and perhaps a higher level of TSAb. In a recent study predictors of successful treatment with RAI included higher ablative dose, female gender, lower free T4 levels at diagnosis, and absence of a palpable goiter.84 The acute, short-term side effects of 131I therapy are minimal and include mild thyroidal tenderness and dysphagia. Concern over increased risk of mutations and congenital defects now appears to be unfounded because long-term followup studies have not revealed increased risk for these complications.89 In studies examining the risk of malignancies after RAI therapy, there seems to be a small but significant increase in the risk of cancer of the small bowel and thyroid.89 Although RAI is very effective in the treatment of hyperthyroidism, long-term followup from Great Britain suggests that among patients with hyperthyroidism treated with RAI, mortality from all causes and mortality resulting from cardiovascular and cerebrovascular disease and fracture are increased.90
A common approach to Graves’ hyperthyroidism is to administer a single dose of 5 to 15 mCi (80 to 200 μCi/g of tissue).83 The optimal method for determining 131I treatment doses for Graves’ hyperthyroidism is unknown, and techniques have varied from a fixed dose to more elaborate calculations based on gland size, iodine uptake, and iodine turnover. In a trial of 88 patients with Graves’ disease, no difference in outcome was seen among high or low, fixed or adjusted doses.91 Thyroid glands estimated to weigh >80 g may require larger doses of RAI. Larger doses are likely to induce hypothyroidism and are seldom given outside the United States due to the imposition of stringent safety restrictions. For example, in the United Kingdom, a nursery school teacher is advised to stay out of school for 3 weeks following a 15 mCi dose of 131I.88
Evaluation of Therapeutic Outcomes: Hyperthyroidism
After therapy (surgery, thionamides, or RAI) for hyperthyroidism has been initiated, patients should be evaluated on a monthly basis until they reach a euthyroid condition. Clinical signs of continuing thyrotoxicosis (tachycardia, weight loss, and heat intolerance, among others) or the development of hypothyroidism (bradycardia, weight gain, and lethargy, among others) should be noted. β-Blockers may be used to control symptoms of thyrotoxicosis until the definitive treatment has returned the patient to a euthyroid state. Once T4 replacement is initiated, the goal is to maintain both the free T4 level and the TSH concentration in the normal range. Once a stable dose of T4 is identified, the patient may be followed up every 6 to 12 months.
Finally, a common, potentially confusing clinical situation should be mentioned. Why are the TSH concentrations suppressed for some patients who are clinically hypothyroid and who have a low free T4 level? For patients with long-standing hyperthyroidism, the pituitary thyrotrophs responsible for making TSH become atrophic. The average amount of time required for these cells to resume normal functioning is 6 to 8 weeks.92 Therefore, if a thyrotoxic patient has his or her free T4 concentration lowered rapidly, before the thyrotrophs resume normal function, a period of “transient central hypothyroidism” will be observed.
Special Conditions
Graves’ Disease and Pregnancy
Inappropriate production of hCG is a cause of abnormal thyroid function tests during the first half of pregnancy, and hCG can cause either subclinical (normal T4, suppressed TSH) or overt hyperthyroidism.93,94 This is because the homology of hCG and TSH leads to hCG-mediated stimulation through the TSH receptor. A recent study showed that at hCG concentrations greater than 400 international units/mL, TSH levels were invariably suppressed and free T4 levels were generally above the normal range. Most patients with hCG greater than 200 international units/mL did not have symptoms of hyperthyroidism.95 The variability of the thyrotropic potency of hCG is believed to depend on its carbohydrate composition.
Hyperthyroidism during pregnancy is almost solely caused by Graves’ disease, with approximately 0.1% to 0.4% of pregnancies affected. Although the increased metabolic rate is usually well tolerated in pregnant women, two symptoms suggestive of hyperthyroidism during pregnancy are failure to gain weight despite good appetite and persistent tachycardia. There is no increase in maternal mortality or morbidity in well-controlled patients; however, postpartum thyroid storm has been reported in about 20% of untreated individuals. Fetal loss is also more common, due to the facts that spontaneous abortion and premature delivery are more common in untreated pregnant women, as are low-birth-weight infants and eclampsia. Transplacental passage of TSAb may occur, causing fetal as well as neonatal hyperthyroidism.96 An uncommon cause of hyperthyroidism is molar pregnancy; women present with a large-for-dates uterus and evacuation of the uterus is the preferred management approach.97
Because RAI is contraindicated in pregnancy and surgery is usually not recommended (especially during the first trimester), antithyroid drug therapy is usually the treatment of choice. MMI readily crosses the placenta and appears in breast milk.
PTU is considered the drug of choice during the first trimester of pregnancy, with the lowest possible doses used to maintain the maternal T4 level in the high-normal range.79,81 During this period the risk of MMI-associated embryopathy is believed to outweigh that of PTU-associated hepatotoxicity. To prevent fetal goiter and suppression of fetal thyroid function, PTU is usually prescribed in daily doses of 300 mg or less and tapered to 50 to 150 mg daily after 4 to 6 weeks. PTU doses of less than 200 mg daily are unlikely to produce fetal goiter.98 During the second and third trimesters, when the critical period of organogenesis is complete, MMI is thought to be the drug of choice because of the greater risk of hepatotoxicity with PTU.79,81 Thionamide doses should be adjusted to maintain free T4 within 10% of the upper normal limit of the nonpregnant reference range.93 During the last trimester, TSAbs fall spontaneously, and some patients will go into remission so that antithyroid drug doses may be reduced. A rebound in maternal hyperthyroidism occurs in about 10% of women postpartum and may require more intensive treatment than in the last trimester of pregnancy.96 For example, a recent study of patients who were euthyroid after thionamide discontinuation and subsequently became pregnant showed a relative risk of 4.26 for relapse of hyperthyroidism occurring 4 to 8 months after delivery.99
Recently, two very comprehensive guidelines have been published by the American Thyroid Association and the Endocrine Society regarding the management of thyroid disease during pregnancy.100,101
Neonatal and Pediatric Hyperthyroidism
Following delivery, some babies of hyperthyroid mothers will be hyperthyroid due to placental transfer of TSAbs, which stimulates thyroid hormone production in utero and postpartum.102 This is likely if the maternal TSAb titers were quite high. The disease is usually expressed 7 to 10 days postpartum and treatment with antithyroid drugs (PTU 5 to 10 mg/kg/day or MMI 0.5 to 1 mg/kg/day) may be needed for as long as 8 to 12 weeks until the antibody is cleared (immunoglobulin G half-life is about 2 weeks). Iodide (potassium iodide one drop per day or Lugol’s solution one to three drops per day) and sodium ipodate may be used for the first few days to acutely inhibit hormone release.
Childhood hyperthyroidism has classically been managed with either PTU or MMI. Long-term followup studies suggest that this form of therapy is quite acceptable, with 25% of a cohort experiencing remission every 2 years.103 Again, current recommendations suggest use of MMI as a first-line agent in both adults and children.
Thyroid Storm
Thyroid storm is a life-threatening medical emergency characterized by decompensated thyrotoxicosis, high fever (often >39.4°C [>103°F]), tachycardia, tachypnea, dehydration, delirium, coma, nausea, vomiting, and diarrhea.22 Although Graves’ disease and less commonly toxic nodular goiter are usually the underlying thyrotoxic pathology,104 at least two cases of subacute thyroiditis leading to thyroid storm have been reported.105
Precipitating factors for thyroid storm include infection, trauma, surgery, RAI treatment, and withdrawal from antithyroid drugs. Although the duration of clinical decompensation lasts for an average duration of 72 hours, symptoms may persist up to 8 days. With aggressive treatment, the mortality rate has been lowered to 20%. The following therapeutic measures should be instituted promptly: (a) suppression of thyroid hormone formation and secretion, (b) antiadrenergic therapy, (c) administration of corticosteroids, and (d) treatment of associated complications or coexisting factors that may have precipitated the storm. Specific agents used in thyroid storm are outlined in Table 58-6. PTU in large doses may be the preferred thionamide because, in addition to interfering with the production of thyroid hormones, it also blocks the peripheral conversion of T4 to T3. However, β-blockers and corticosteroids will serve the same purpose. A theoretical advantage of MMI is that it has a longer duration of action. If patients are unable to take medications orally, the tablets can be crushed into suspension and instilled by gastric or rectal tube106 or given IV.107 Iodides, which rapidly block the release of preformed thyroid hormone, should be administered after thionamide is initiated to inhibit iodide utilization by the overactive gland. If iodide is administered first, it could theoretically provide substrate to produce even higher levels of thyroid hormone.
TABLE 58-6 Drug Dosages Used in the Management of Thyroid Storm




