Alzheimer’s Disease
KEY CONCEPTS
![]() Alzheimer’s disease (AD) is the most common form of dementing illness, and the prevalence of AD increases with each decade of life.
Alzheimer’s disease (AD) is the most common form of dementing illness, and the prevalence of AD increases with each decade of life.
![]() The etiology of AD is unknown, and current pharmacotherapy neither cures nor arrests the pathophysiology.
The etiology of AD is unknown, and current pharmacotherapy neither cures nor arrests the pathophysiology.
![]() Neuritic plaques and neurofibrillary tangles are the pathologic hallmarks of AD; however, the definitive cause of this disease is yet to be determined.
Neuritic plaques and neurofibrillary tangles are the pathologic hallmarks of AD; however, the definitive cause of this disease is yet to be determined.
![]() AD affects multiple areas of cognition and is characterized by a gradual onset with a slow, progressive decline.
AD affects multiple areas of cognition and is characterized by a gradual onset with a slow, progressive decline.
![]() A thorough physical examination (including neurologic examination), as well as laboratory and imaging studies, is required to rule out other disorders and diagnose AD before considering drug therapy.
A thorough physical examination (including neurologic examination), as well as laboratory and imaging studies, is required to rule out other disorders and diagnose AD before considering drug therapy.
![]() Pharmacotherapy for AD focuses on impacting three domains: cognition, behavioral and psychiatric symptoms, and functional ability.
Pharmacotherapy for AD focuses on impacting three domains: cognition, behavioral and psychiatric symptoms, and functional ability.
![]() Nondrug therapy and social support for the patient and family are the primary treatment interventions for AD.
Nondrug therapy and social support for the patient and family are the primary treatment interventions for AD.
![]() Cholinesterase inhibitors and memantine are used to treat cognitive symptoms of AD; other medications have been suggested to be beneficial because of their potential preventive or cognitive effects.
Cholinesterase inhibitors and memantine are used to treat cognitive symptoms of AD; other medications have been suggested to be beneficial because of their potential preventive or cognitive effects.
![]() Appropriate management of vascular disease risk factors may reduce the risk for developing AD and may prevent the worsening of dementia in patients with AD.
Appropriate management of vascular disease risk factors may reduce the risk for developing AD and may prevent the worsening of dementia in patients with AD.
![]() A thorough behavioral assessment and plan with careful examination of environmental factors should be conducted before initiating drug therapy for behavioral symptoms.
A thorough behavioral assessment and plan with careful examination of environmental factors should be conducted before initiating drug therapy for behavioral symptoms.
“I now begin the journey that will lead me into the sunset of my life.”
Ronald Reagan
Alzheimer’s disease (AD), first characterized by Alois Alzheimer in 1907, is a gradually progressive dementia affecting cognition, behavior, and functional status. The exact pathophysiologic mechanisms underlying AD are not entirely known, and no cure exists.1 Although drugs may reduce AD symptoms for a time, the disease is eventually fatal.
AD profoundly affects the family as well as the patient. The need for supervision and assistance increases until the late stages of the disease, when AD patients become totally dependent on a caregiver for all of their basic needs. These are the all-too-common experiences of the millions of people in the United States who care for someone with AD. To address the growing AD crisis facing the United States, the first national strategic plan, the National Alzheimer’s Plan, was released in 2012 with the goals of coordinating efforts across the federal government to prevent and treat AD, increase public awareness, and improve the quality of care and support for patients and their caregivers.2
EPIDEMIOLOGY
![]() AD is the most common cause of dementia, accounting for 50% to 60% of cases of late-life cognitive dysfunction. Its prevalence among dementia patients increases to 80% if AD lesions in conjunction with other pathologic brain lesions are considered.3–5 Table 38-1 lists the most common types of dementia. Dementia can result from multiple etiologies. This chapter focuses exclusively on dementia of the Alzheimer’s type. However, the reader is encouraged to use the nonpharmacologic approaches and management of behavioral problems outlined in this chapter as a general treatment approach for other types of dementia that may share similar features with AD.
AD is the most common cause of dementia, accounting for 50% to 60% of cases of late-life cognitive dysfunction. Its prevalence among dementia patients increases to 80% if AD lesions in conjunction with other pathologic brain lesions are considered.3–5 Table 38-1 lists the most common types of dementia. Dementia can result from multiple etiologies. This chapter focuses exclusively on dementia of the Alzheimer’s type. However, the reader is encouraged to use the nonpharmacologic approaches and management of behavioral problems outlined in this chapter as a general treatment approach for other types of dementia that may share similar features with AD.
TABLE 38-1 Common Types of Dementia in Late Life

Approximately 5.4 million Americans have AD.4,5 By the year 2050, one in five people will be older than age 65 years, and the number of AD patients is projected to be 13.2 million (Fig. 38-1). Most cases present in persons older than age 65 years, but approximately 4% of cases occur in persons younger than age 65 years. Onset can be as early as age 30 years, resulting in the arbitrary age classifications of younger-onset (age less than 65 years) and late-onset (age 65 years and older).4
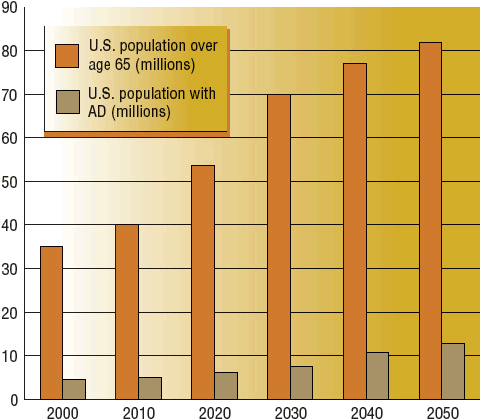
FIGURE 38-1 Our aging population. The percentage of U.S. population older than age 65 years and the percentage with AD projected from years 2000 to 2050. (Estimates based on data from references 1 and 134.)
Increasing age is the greatest risk factor for AD, but AD is not a normal part of aging. The prevalence of AD increases exponentially with age, affecting approximately 4% of people <65 years, 6% of individuals age 65 to 74 years, 44% of those age 75 to 84 years, and 46% of persons age 85 years and older.4 Factors determining age of onset and rate of progression remain largely undefined.
Survival following AD diagnosis is typically 4 to 8 years, but may be as long as 20 years. It is the fifth leading cause of death for those age 65 years and older in the United States. AD may not cause death directly. The most common cause of death in patients with AD is pneumonia, possibly resulting from swallowing difficulties and immobility in the terminal stage of the disease.4 Those diagnosed with AD spend, on average, more years in the most severe stage of the disease than any other stage, and much of this time is spent in a nursing home.4
Etiology and Genetics
![]() The exact etiology of AD is unknown; however, several genetic and environmental factors have been explored as potential causes of AD. Dominantly inherited forms of AD account for less than 1% of cases.6,7 More than half of young-onset, dominantly inherited cases of AD can be attributed to alterations on chromosomes 1, 14, or 21. The majority and most aggressive young-onset cases are attributed to mutations of a gene located on chromosome 14, which produces a protein called presenilin 1.8 A structurally similar protein, presenilin 2, is produced by a gene on chromosome 1. Both presenilin 1 and presenilin 2 encode for membrane proteins that may be involved in amyloid precursor protein (APP) processing. Scientists have identified more than 160 mutations in presenilin genes, and these mutations appear to result in reduced activity of γ-secretase, an enzyme important in β-amyloid peptide (Aβ) formation.8 APP is encoded on chromosome 21. Only a small number of young-onset familial AD cases have been associated with mutations in the APP gene, resulting in overproduction of Aβ or an increase in the proportion of Aβ ending at residue 42.8
The exact etiology of AD is unknown; however, several genetic and environmental factors have been explored as potential causes of AD. Dominantly inherited forms of AD account for less than 1% of cases.6,7 More than half of young-onset, dominantly inherited cases of AD can be attributed to alterations on chromosomes 1, 14, or 21. The majority and most aggressive young-onset cases are attributed to mutations of a gene located on chromosome 14, which produces a protein called presenilin 1.8 A structurally similar protein, presenilin 2, is produced by a gene on chromosome 1. Both presenilin 1 and presenilin 2 encode for membrane proteins that may be involved in amyloid precursor protein (APP) processing. Scientists have identified more than 160 mutations in presenilin genes, and these mutations appear to result in reduced activity of γ-secretase, an enzyme important in β-amyloid peptide (Aβ) formation.8 APP is encoded on chromosome 21. Only a small number of young-onset familial AD cases have been associated with mutations in the APP gene, resulting in overproduction of Aβ or an increase in the proportion of Aβ ending at residue 42.8
Genetic susceptibility to late-onset AD is primarily linked to the apolipoprotein E (APOE) genotype.6–9 Thus far, the contribution of other candidate genes appears to be minor, although AD may be a heterogeneous disease resulting from complex interactions among multiple susceptibility genes and environmental factors. There are three major subtypes or alleles of APOE (e.g., *2, *3, and *4). Inheritance of the APOE*4 allele is believed to account for much of the genetic risk in late-onset AD. The mechanism through which APOE*4 confers an increased risk is unknown, although APOE*4 is associated with factors that may contribute to AD pathology, such as abnormalities in mitochondria, cytoskeletal dysfunction, and low glucose usage.5 The risk for AD is two- to threefold higher in individuals with one APOE*4 allele and 12-fold higher in individuals with two APOE*4 alleles compared to those with no APOE*4 alleles.9 Moreover, onset of symptoms occurs at a relatively younger age as compared with patients having zero or only one copy of APOE*4 in their genotype.9 The strength of association is not the same across all races however.1,4 The APOE*4 allele is not diagnostic or even essential for disease presence.
Genetic factors have been linked to both younger- and late-onset AD. Genetic explanatory factors continue to be investigated.6–8 Epigenetic modifications, particularly DNA methylation, have been reported in AD, and this is an emerging area of research that may help explain the pathologic complexity of AD and aging as a risk factor for the development of AD.10
Environmental and Other Factors
A number of environmental factors are associated with an increased risk of AD, including age, decreased reserve capacity of the brain (reduced brain size, low educational level, and reduced mental and physical activity in late life), head injury, Down’s syndrome, depression, mild cognitive impairment (MCI), and risk factors for vascular disease (hypercholesterolemia, hypertension, atherosclerosis, coronary heart disease, smoking, elevated homocysteine, obesity, metabolic syndrome, and diabetes).4,5,11 Whether these vascular risk factors are true causal risk factors for AD contributing to AD pathology, or whether they result in cerebrovascular pathology that, in turn, contributes to the symptoms of AD, remains to be established.
The incidence of AD rises with increasing age and AD may develop in individuals over the course of decades,4 suggesting that AD is a disease most people are in the process of developing throughout adulthood. The debate about whether dementia is a distinct disease or part of aging remains unresolved. An in-depth discussion of the aging—AD controversy is not possible in this chapter; it is reviewed elsewhere.12–14
PATHOPHYSIOLOGY
![]() The signature lesions in AD are amyloid plaques and neurofibrillary tangles (NFTs) located in the cortical areas and medial temporal lobe structures of the brain.3 Along with these lesions, degeneration of neurons and synapses, as well as cortical atrophy occurs. Plaques and NFTs may also be present in other diseases, even in normal aging, but at least in younger demographics there tends to be a higher burden of plaques and NFTs in AD-affected subjects than there is in age-matched controls. Several mechanisms have been proposed to explain changes in the brain that result in symptoms of AD, including misfolding of proteins (Aβ aggregation and deposition leading to the formation of plaques and hyperphosphorylation of tau protein leading to NFT development); synaptic failure and depletion of neurotrophin and neurotransmitters; and mitochondrial dysfunction (oxidative stress, impaired insulin signaling in the brain, vascular injury, inflammatory processes, loss of calcium regulation, and defects in cholesterol metabolism).3
The signature lesions in AD are amyloid plaques and neurofibrillary tangles (NFTs) located in the cortical areas and medial temporal lobe structures of the brain.3 Along with these lesions, degeneration of neurons and synapses, as well as cortical atrophy occurs. Plaques and NFTs may also be present in other diseases, even in normal aging, but at least in younger demographics there tends to be a higher burden of plaques and NFTs in AD-affected subjects than there is in age-matched controls. Several mechanisms have been proposed to explain changes in the brain that result in symptoms of AD, including misfolding of proteins (Aβ aggregation and deposition leading to the formation of plaques and hyperphosphorylation of tau protein leading to NFT development); synaptic failure and depletion of neurotrophin and neurotransmitters; and mitochondrial dysfunction (oxidative stress, impaired insulin signaling in the brain, vascular injury, inflammatory processes, loss of calcium regulation, and defects in cholesterol metabolism).3
Amyloid Cascade Hypothesis
Amyloid plaques are extracellular lesions found in the brain and cerebral vasculature. Plaques largely consist of Aβ. Aβ peptides consisting of 36 to 43 amino acids are produced via processing of a larger protein, APP. Aβ42 is less common than other Aβ peptides, but is prone to aggregation and plaque formation.3 The amyloid cascade hypothesis states that there is an imbalance between the production and clearance of Aβ peptides resulting in aggregation that causes accumulation of Aβ ultimately leading to AD.3 Studies on younger-onset AD and Down’s syndrome led to the formulation of the amyloid cascade hypothesis. Recent versions of the amyloid cascade hypothesis assume Aβ that is not sequestered in plaques actually drives the disease.3 Even so, the amyloid cascade hypothesis seems most applicable in cases of younger-onset, autosomal dominant AD. It is not clear whether it is reasonable to etiologically extrapolate to the late-onset form (which afflicts the vast majority of those affected). Whether individuals with late-onset AD also carry genetic variations that promote a primary Aβ amyloidosis remains to be shown. If this turns out not to be the case, the possibility that amyloidosis in late-onset AD is secondary to a more upstream event will require consideration. Before this conceptual conundrum is laid to rest, however, the amyloid cascade hypothesis will likely undergo a therapy-based practical test. If treatments that efficiently reduce Aβ production or remove brain Aβ fail to arrest disease progression, it would argue amyloidosis is not the primary pathology in most of those with AD.
Neurofibrillary Tangles
As Aβ was being identified in plaques, other researchers showed that NFTs are commonly found in the cells of the hippocampus and cerebral cortex in persons with AD and are composed of abnormally hyperphosphorylated tau protein. Tau protein provides structural support to microtubules, the cell’s transportation and skeletal support system.3 When tau filaments undergo abnormal phosphorylation at a specific site, they cannot bind effectively to microtubules, and the microtubules collapse. Without an intact system of microtubules, the cell cannot function properly and eventually dies. The density of the NFTs correlates with the severity of the dementia.3 NFTs are found in other dementing illnesses besides AD, and may represent a common method by which various inciting factors culminate in cell death.3
Inflammatory Mediators
Inflammatory or immunologic paradigms are often viewed as a corollary of the amyloid cascade hypothesis. Certainly, brain amyloid deposition associates with local inflammatory and immunologic alterations. This led some to propose that inflammation is relevant to AD neurodegeneration.3 Inflammatory/immunologic hypotheses argue that although Aβ may have direct neurotoxicity, at least some of its toxicity might actually be an indirect consequence of an Aβ protofibril-induced microglia activation and astrocyte recruitment. This inflammatory response may represent an attempt to clear amyloid deposition. However, it is also associated with release of cytokines, nitric oxide, and other radical species, and complement factors that can both injure neurons and promote ongoing inflammation.3 Indeed, levels of multiple cytokines and chemokines are elevated in AD brains, and certain proinflammatory gene polymorphisms are reported to be associated with AD.3
Consistent with these molecular observations are epidemiologic data suggesting that exposure to nonsteroidal antiinflammatory drugs (NSAIDs) may reduce AD risk.15,16 However, multiple prospective short duration trials of NSAIDs in AD prevention and of NSAIDs as AD treatment have been disappointing.3,17
The Cholinergic Hypothesis
Multiple neuronal pathways are destroyed in AD. Neuronal damage can be seen in conjunction with plaque structures.3 Widespread cell dysfunction or degeneration results in a variety of neurotransmitter deficits, with cholinergic abnormalities being the most prominent.3 Loss of cholinergic activity correlates with AD severity. In the late stage of AD, the number of cholinergic neurons is reduced, and there is loss of nicotinic receptors in the hippocampus and cortex. Presynaptic nicotinic receptors control the release of acetylcholine, as well as other neurotransmitters important for memory and mood, including glutamate, serotonin, and norepinephrine.3
The discovery of vast cholinergic cell loss led to the development of a cholinergic hypothesis of the pathophysiology of AD. The cholinergic hypothesis targeted cholinergic cell loss as the source of memory and cognitive impairment in AD. Consequently, it was presumed that increasing cholinergic function would improve symptoms of memory loss. This approach is flawed because cholinergic cell loss appears to be a secondary consequence of Alzheimer’s pathology, not the disease-producing event, and cholinergic neurons are only one of many neuronal pathways destroyed in AD. Simple addition of acetylcholine cannot compensate for the loss of neurons, receptors, and other neurotransmitters lost during the course of the illness. Thus the goal is to minimize or improve symptoms through augmentation of neurotransmission at remaining synapses.
Other Neurotransmitter Abnormalities
Although the cholinergic system has received particular attention in AD pharmaceutical research, deficits also exist in other neuronal pathways. For example, serotonergic neurons of the raphe nuclei and noradrenergic cells of the locus ceruleus are lost, while monoamine oxidase type B activity is increased. Monoamine oxidase type B is found predominantly in the brain and in platelets, and is responsible for metabolizing dopamine. In addition, abnormalities appear in glutamate pathways of the cortex and limbic structures, where a loss of neurons leads to a focus on excitotoxicity models as possible contributing factors to AD pathology.
Glutamate is the major excitatory neurotransmitter in the cortex and hippocampus. Many neuronal pathways essential to learning and memory use glutamate as a neurotransmitter, including the pyramidal neurons (a layer of neurons with long axons carrying information out of the cortex), hippocampus, and entorhinal cortex. Glutamate and other excitatory amino acid neurotransmitters have been implicated as potential neurotoxins in AD.18 Dysregulated glutamate activity is thought to be one of the primary mediators of neuronal injury after stroke or acute brain injury. Although intimately involved in cell injury, the role of excitatory amino acids in AD is as yet unclear; however, blockade of N-methyl-D-aspartate (NMDA) receptors decreases activity of glutamate in the synapse and may hypothetically lessen the degree of cellular injury in AD.
Brain Vascular Disease and High Cholesterol
There is growing evidence of a causal association between cardiovascular disease and its risk factors and the incidence of AD. Cardiovascular risk factors that are also risk factors for dementia include hypertension, elevated low-density lipoprotein cholesterol, low high-density lipoprotein cholesterol, and diabetes.19 Brain vascular disease may augment the cognitive impairment observed for a given amount of AD pathology in the brain. Dysfunctional blood vessels may impair nutrient delivery to neurons and reduce clearance of Aβ from the brain.3 Vascular disease may accelerate amyloid deposition and increase amyloid toxicity to neurons.3 Midlife diastolic hypertension is adversely associated with AD, while late-life hypertension may show an inverse association with AD.20 Diabetes may increase the risk of dementia through factors related to “metabolic syndrome” (dyslipidemia and hypertension), effects of potentially toxic glucose metabolites on the brain and vasculature, and through insulin itself.19 Disturbances in insulin-signaling pathways, both in the periphery and the brain, have been linked to AD. Insulin may also regulate the metabolism of Aβ and tau protein.3
Research has found multiple links between cholesterol and AD. APOE is synthesized in the liver, central nervous system, and cerebrospinal fluid (CSF) and is responsible for transporting cholesterol in the blood through the brain. It is carried by low-density lipoprotein into neurons and binds to NFTs. APOE*4 is associated with increasing deposition of Aβ and is thought to act as an accelerating modulator in vascular dementia. Elevated cholesterol levels in brain neurons may alter membrane functioning and result in the cascade leading to plaque formation and AD.
Other Mechanisms
Other hypotheses proposed to explain AD pathogenesis include oxidative stress, mitochondrial dysfunction, and loss of estrogen. Each of these mechanisms may contribute to AD pathogenesis, but the extent of the contribution is uncertain. There is a growing body of evidence of a role for oxidative stress and the accumulation of free radicals in the brain of AD patients.3 Some epidemiologic studies suggest vitamin E, and possibly the combination of vitamin E and vitamin C, may reduce AD risk while others do not.3 Mitochondrial dysfunction may result in disruption of energy metabolism in the neuron.3,21,22 The role of estrogen in cognitive aging and dementia continues to be an active area of investigation. Despite convincing evidence that estrogens affect the brain in ways that would be expected to improve cognitive aging and reduce the risk of AD, the results of clinical studies have been largely disappointing.21 A single common mechanism for producing AD does not exist. Regardless of the source, however, the features remain the same: degeneration of neurons in higher brain areas; accumulation of NFTs and amyloid plaques; profound destruction of cholinergic pathways; and an insidious dementia, slowly progressive until death.
CLINICAL PRESENTATION OF ALZHEIMER’S DISEASE
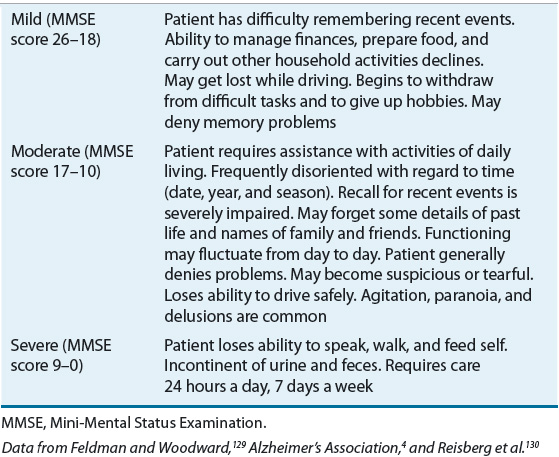
![]() The onset of AD is almost imperceptible, without abrupt changes in cognition or function. Deficits occur progressively over time, affecting multiple areas of cognition.4,22 For treatment and assessment purposes, it is helpful to divide AD symptoms into two basic categories: cognitive symptoms and noncognitive (behavioral) symptoms. Cognitive symptoms are present throughout the illness, whereas behavioral symptoms are less predictable. Table 38-2 summarizes the stages of AD.
The onset of AD is almost imperceptible, without abrupt changes in cognition or function. Deficits occur progressively over time, affecting multiple areas of cognition.4,22 For treatment and assessment purposes, it is helpful to divide AD symptoms into two basic categories: cognitive symptoms and noncognitive (behavioral) symptoms. Cognitive symptoms are present throughout the illness, whereas behavioral symptoms are less predictable. Table 38-2 summarizes the stages of AD.
TABLE 38-2 Stages of Alzheimer’s Disease
Diagnosis
A family member often first brings memory complaints to the attention of a primary care clinician. Up to 50% of patients who meet criteria for dementia are not given a diagnosis in the primary care setting, leading some to believe that an appropriate screening tool may be helpful in aiding diagnosis and leading to earlier treatment.23,24 Despite the phenomenon of underdiagnosis, the United States Preventative Services Task Force concluded that there are insufficient data to recommend for or against cognitive screening for AD, because it could not be determined if the benefits outweigh the risks.23 Screening is being promoted as part of the Medicare Annual Wellness Visit by the Alzheimer’s Association (AA).25
Until recently the only way to confirm a clinical diagnosis of AD was through direct examination of brain tissue at autopsy or biopsy. Several criteria have been used in clinical practice and research for the detection and diagnosis of dementia, including the Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition (DSM-5) criteria,26 the Agency for Healthcare Research and Quality (AHRQ) Guidelines,27 the American Academy of Neurology Guidelines,28 the National Institute of Neurological Disorders and Stroke (NINDS) criteria,29 and the National Institute of Neurological and Communicative Disorders and Stroke (NINCDS), and the Alzheimer’s Disease and Related Disorders Association (ADRDA) Criteria.30 In 2011, revisions to the NINCDS and ADRDA Criteria for the clinical diagnosis of AD were recommended by the National Institute on Aging (NIA) and the AA25,31 DSM-5 provides criteria for diagnosis of minor and major neurocognitive disorders, with specific criteria for neurocognitive disorders due to AD.32 The new NIA-AA criteria view AD as a spectrum beginning with a preclinical phase progressing to increasingly severe clinical stages of AD. Three workgroups formulated diagnostic criteria for the dementia phase,33 the symptomatic, predementia phase (MCI),34 and the asymptomatic, preclinical phase of AD.35 At this time, AD is still primarily a clinical diagnosis, but this is expected to change in coming years as brain imaging, CSF, and other AD biomarkers are validated and available for routine clinical use. The patient’s examination should suggest that cognitive decline from a previously higher baseline has occurred. The history should corroborate this, and further indicate that cognitive decline has reached the point where changes in social or occupational functioning are present. It is possible to administer a sophisticated exam that defines cognitive domain strengths and weaknesses and enables a neuroanatomic localization of the observed deficits. When approached in this way, the exam can indicate a pattern of cognitive decline that is consistent with AD, and assist with rendering a diagnosis that is as much a diagnosis of inclusion as it is of exclusion.
CLINICAL PRESENTATION Alzheimer’s Disease
Objectively defining social or occupational dysfunction can prove tricky in the older patient who may be retired, and who may also lead a socially restricted lifestyle for reasons of frailty. For such patients, the minimal requirement is to establish a change in activities of daily living. Early on, this usually involves a change in instrumental activities of daily living (handling finances, organizing medications) rather than basic activities of daily living (hygiene, dressing). Some AD subspecialists use a detailed, standardized, semistructured interview of a nonpatient informant as the most critical piece of the diagnostic evaluation.36
![]() For patients who meet criteria for dementia (whether the underlying cause is ultimately felt to be AD or not), current recommendations from the American Academy of Neurology include a neuroimaging study (computed tomography or magnetic resonance imaging), as well as a serologic evaluation that includes blood cell counts, serum electrolytes, liver function tests, a test of thyroid function, and a vitamin B12 level.28 When circumstances suggest AD is not the leading entity on the differential diagnosis, other neurologic tests such as CSF analysis or electroencephalogram can occasionally be justified. Neuropsychological testing is also optional, but can prove quite useful for the diagnosis of AD by helping to establish a neuroanatomical localization for the patient’s cognitive deficits.
For patients who meet criteria for dementia (whether the underlying cause is ultimately felt to be AD or not), current recommendations from the American Academy of Neurology include a neuroimaging study (computed tomography or magnetic resonance imaging), as well as a serologic evaluation that includes blood cell counts, serum electrolytes, liver function tests, a test of thyroid function, and a vitamin B12 level.28 When circumstances suggest AD is not the leading entity on the differential diagnosis, other neurologic tests such as CSF analysis or electroencephalogram can occasionally be justified. Neuropsychological testing is also optional, but can prove quite useful for the diagnosis of AD by helping to establish a neuroanatomical localization for the patient’s cognitive deficits.
Almost any medication can contribute to cognitive impairment in vulnerable individuals, but certain classes of medication are more commonly implicated. Benzodiazepines and other sedative hypnotics, anticholinergics, opioid analgesics, antipsychotics, and anticonvulsants have been associated with cognitive impairment.37,38 NSAIDs, histamine H2-receptor antagonists, digoxin, amiodarone, antihypertensives, and corticosteroids have been implicated in cases of delirium.37 Because medications are a reversible cause of cognitive symptoms, medication review and management are essential.
Efforts to define the role of other AD diagnostic tests are ongoing. Positron emission tomography scanning may reveal a pattern of hypometabolism typical of AD, but by itself the diagnostic accuracy of positron emission tomography scanning still lags behind that of the clinical examination and history.39 APOE genotyping by itself is also insufficient to make or break a diagnosis of AD, but demonstrating an APOE*4 allele in a suspected patient increases the specificity of the diagnosis and can help predict which patients with MCI are most likely to progress to a diagnosis of AD over the next several years.40 Unless the patient developed dementia prior to age 60 years and also had a parent that developed AD before age 60 years, presenilin 1, presenilin 2, or APP genotyping is usually not indicated.
Mild Cognitive Impairment
It has long been recognized that aging individuals experience changes in cognitive function. MCI constitutes a syndromic designation that categorizes patients with cognitive complaints insufficient to warrant a diagnosis of dementia. The NIA-AA diagnostic criteria specifically address the diagnosis of MCI.34 Persons diagnosed with MCI carry a 10% to 15% chance per year of progressing to an AD diagnosis.41 What clinicians are likely seeing in most people with MCI is the initial manifestation of a progressive degenerative dementia that will eventually meet AD diagnostic criteria.34,41 However, it is important to note that not everyone meeting MCI criteria will have AD. As the MCI designation is increasingly applied, MCI criteria continue to evolve.34,41
TREATMENT
Desired Outcomes
![]() The primary goal of treatment in AD is to symptomatically treat cognitive difficulties and preserve patient function as long as possible. Secondary goals include treating the psychiatric and behavioral sequelae. Current AD treatments have not been shown to prolong life, cure AD, or halt or reverse the pathophysiologic processes of the disorder.36
The primary goal of treatment in AD is to symptomatically treat cognitive difficulties and preserve patient function as long as possible. Secondary goals include treating the psychiatric and behavioral sequelae. Current AD treatments have not been shown to prolong life, cure AD, or halt or reverse the pathophysiologic processes of the disorder.36
General Treatment Approach
Clinical trials have consistently demonstrated modest benefits of early and continuous treatment with cholinesterase inhibitors.42 Memantine added in moderate to severe disease may also provide benefit. Following this approach allows for maximal maintenance of cognition and activities of daily living. A symptomatic approach is used to treat behavioral symptoms as they arise.
Provision of education to the patient and family at the time of diagnosis, including discussion of the course of illness, realistic expectations of treatment, and the importance of legal and financial planning, are essential to appropriate treatment.
Nonpharmacologic Therapy
![]() AD has a profound effect on both the patient and family, so appropriate treatment is needed. Nonmedication interventions are the current primary interventions for management of AD, and medications should be used in the context of multimodal interventions. Behavioral and psychiatric symptoms are among the most challenging and distressing symptoms of the disease and may be the determining factor in a family’s decision to seek institutional care. Symptoms such as sleep disturbances, wandering, urinary incontinence, agitation, and aggression in patients with dementia are best managed using behavioral interventions rather than medications whenever possible.42–44
AD has a profound effect on both the patient and family, so appropriate treatment is needed. Nonmedication interventions are the current primary interventions for management of AD, and medications should be used in the context of multimodal interventions. Behavioral and psychiatric symptoms are among the most challenging and distressing symptoms of the disease and may be the determining factor in a family’s decision to seek institutional care. Symptoms such as sleep disturbances, wandering, urinary incontinence, agitation, and aggression in patients with dementia are best managed using behavioral interventions rather than medications whenever possible.42–44
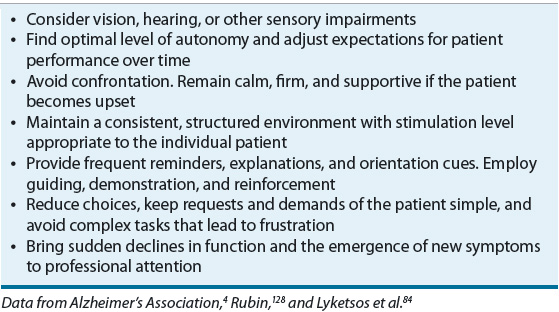
Upon initial diagnosis, the patient and caregiver should be educated on the course of illness, prognosis, available treatments, legal decisions, and quality-of-life issues. Caregiving strategies, including stress-management techniques and support group options, should also be discussed. Caregiver education and support programs have been shown to improve caregiver skill, knowledge, confidence, and quality of life, and, in some cases, delay time to nursing home placement.45,46 Table 38-3 lists basic principles of care for the AD patient. The general approach to nonmedication strategies for behavioral symptoms is to identify the symptom, identify causative factors, and adapt the caregiving environment to remedy the situation.4 Environmental triggers may include noise, glare, an insecure space, and too much background distraction, including television. Personal discomfort may also trigger behaviors, so it is important to monitor for pain, hunger, thirst, constipation, full bladder, fatigue, infections, skin irritation, comfortable temperature, fears, and frustrations.4 Medical comorbidity is a major source of functional and cognitive impairment in patients with AD, so general health maintenance is warranted.4 Interventions should redirect the patient’s attention rather than be confrontational and should specifically address known triggers. Creating a calm environment and removing stressors and triggers is key. Other nonpharmacological approaches include exercise, light therapy, music therapy, reminiscence therapy, aroma therapy, relaxation techniques, validation therapy, massage and touch therapy, and multisensory stimulation.47 Caregivers should be referred to support services, such as the AA, for assistance in developing nonpharmacologic strategies for managing difficult behaviors.
TABLE 38-3 Basic Principles of Care for the Alzheimer’s Patient
The caregiver must be prepared to face the changes in life that will occur, and acceptance of this does not come easily. Denial on the part of the patient and rationalization on the part of the family are common. The clinician should encourage the family to address legal and financial matters and designate a durable power of attorney for execution of financial and medical decisions once the patient is incompetent. The caregiver will need to address issues such as respite services to provide time for rest, relaxation, and conduct of personal business. Eventually, the caregiver will need to face critical and difficult questions with respect to institutionalization. Local resources, such as the AA, can provide detailed information regarding support services. Table 38-4 lists this and other referral sources for caregivers.
TABLE 38-4 Resources for Caregivers of Persons with Alzheimer’s Disease

Education, communication, and planning are key nonpharmacologic components of caring for a patient with AD. Preparation in the early stages of illness may lessen some of the caregiver stress as the illness progresses.
Pharmacologic Therapy
Pharmacotherapy for Cognitive Symptoms
![]() Table 38-5 presents a treatment algorithm for managing cognitive symptoms in AD. Cholinesterase inhibitors and NMDA-receptor antagonists are indicated for treatment of AD. The latest treatment guidelines recommend the use of cholinesterase inhibitors for AD, with no preference for a specific agent.48–50 Donepezil, rivastigmine, and galantamine are indicated in mild to moderate AD, while donepezil is also indicated in severe disease. Memantine is indicated for moderate to severe AD; current evidence does not support its use in earlier stages of the disease.51 Additional benefit may be achieved when memantine is added to cholinesterase inhibitor therapy in moderate to severe AD.52 There is no evidence supporting combination therapy of more than one cholinesterase inhibitor. No head-to-head trials comparing memantine monotherapy to cholinesterase inhibitor therapy have been conducted to date.
Table 38-5 presents a treatment algorithm for managing cognitive symptoms in AD. Cholinesterase inhibitors and NMDA-receptor antagonists are indicated for treatment of AD. The latest treatment guidelines recommend the use of cholinesterase inhibitors for AD, with no preference for a specific agent.48–50 Donepezil, rivastigmine, and galantamine are indicated in mild to moderate AD, while donepezil is also indicated in severe disease. Memantine is indicated for moderate to severe AD; current evidence does not support its use in earlier stages of the disease.51 Additional benefit may be achieved when memantine is added to cholinesterase inhibitor therapy in moderate to severe AD.52 There is no evidence supporting combination therapy of more than one cholinesterase inhibitor. No head-to-head trials comparing memantine monotherapy to cholinesterase inhibitor therapy have been conducted to date.
TABLE 38-5 Treatment Options for Cognitive Symptoms in Alzheimer’s Disease

Disagreement exists about how best to determine effectiveness of treatments for AD. Selection of qualitative versus quantitative assessment may bias a clinician’s impression of response. Subtle changes are often detected only by psychometric testing. Because no standard has been suggested to define the effectiveness of medications for AD, great variation exists between clinicians, and the duration of treatment ranges from months to years. Realistic expectations for treatment success may include slowed decline in behavioral, functional, and cognitive abilities and delayed long-term care placement.53
Unfortunately, clinical trials have failed to provide answers to key questions in treating AD patients. Information from clinical trials is insufficient to know if a cholinesterase inhibitor dose–response relationship exists, or if additional cognitive improvement may be gained by increasing to the maximum tolerated dose, rather than continuing with the usual recommended daily dosage. Guidance in extrapolating data related to changes in cognition is needed, so that a reasonable duration of clinical treatment with cholinesterase inhibitors and NMDA-antagonists can be determined. One concern is that those who respond to treatment may lose the benefits of that treatment once the medication is stopped.54,55 Moreover, gaps in treatment have been linked with worse outcomes in open-label trials;56,57 however, in a more recent large observational study there was no increased risk of institutionalization or death associated with gaps in cholinesterase inhibitor therapy.58 Regardless, dosing regimens should be simplified and patient/caregiver preferences considered in an effort to improve adherence and persistence.
In natural disease progression studies, scores on the Alzheimer’s Disease Assessment Scale—Cognition (ADAS-cog) have been shown to worsen (increase) by an average of less than or equal to five points over 1 year in mild dementia and 7 to 11 points annually in moderate dementia. Based on these findings, the general consensus is that a four-point change in the ADAS-cog represents a clinically significant change.42 Therefore, if a pharmacotherapeutic agent decreases the ADAS-cog score by four points, one could think of this as having delayed progression of disease symptoms by 6 months. The usefulness of the ADAS-cog in clinical practice is limited because of the time required for administration; it is much more practical to assess changes in disease severity using the Mini-Mental Status Examination (MMSE). An untreated patient has an average decline of two to four points in MMSE score per year. Successful treatment would reflect a decline of less than two points a year. It is reasonable to change to a different cholinesterase inhibitor if the decline in MMSE score is greater than two to four points after 1 year with the initial agent.59
![]() Cholinesterase Inhibitors In the early 1980s, researchers began to examine means to enhance cholinergic activity in patients with AD by inhibiting the hydrolysis of acetylcholine through reversible inhibition of cholinesterase. Tacrine was the first such drug to be examined in a systematic fashion. However, tacrine was fraught with significant side effects, including hepatotoxicity, which severely limited its usefulness. Tacrine is no longer available in the US market, having been replaced by safer, more tolerable cholinesterase inhibitors. The newer cholinesterase inhibitors donepezil, rivastigmine, and galantamine show similar symptomatic improvement in cognitive, global, and functional outcomes in patients with mild to moderate AD, and duration of benefit varies from 3 to 12 months.60–62
Cholinesterase Inhibitors In the early 1980s, researchers began to examine means to enhance cholinergic activity in patients with AD by inhibiting the hydrolysis of acetylcholine through reversible inhibition of cholinesterase. Tacrine was the first such drug to be examined in a systematic fashion. However, tacrine was fraught with significant side effects, including hepatotoxicity, which severely limited its usefulness. Tacrine is no longer available in the US market, having been replaced by safer, more tolerable cholinesterase inhibitors. The newer cholinesterase inhibitors donepezil, rivastigmine, and galantamine show similar symptomatic improvement in cognitive, global, and functional outcomes in patients with mild to moderate AD, and duration of benefit varies from 3 to 12 months.60–62
The mechanism of action differs slightly between drugs in this class.59 Donepezil specifically and reversibly inhibits acetylcholinesterase. Rivastigmine inhibits both butyrylcholinesterase and acetylcholinesterase. Galantamine is a selective, competitive, reversible acetylcholinesterase inhibitor and also enhances the action of acetylcholine on nicotinic receptors. The clinical relevance of these differences is unknown.
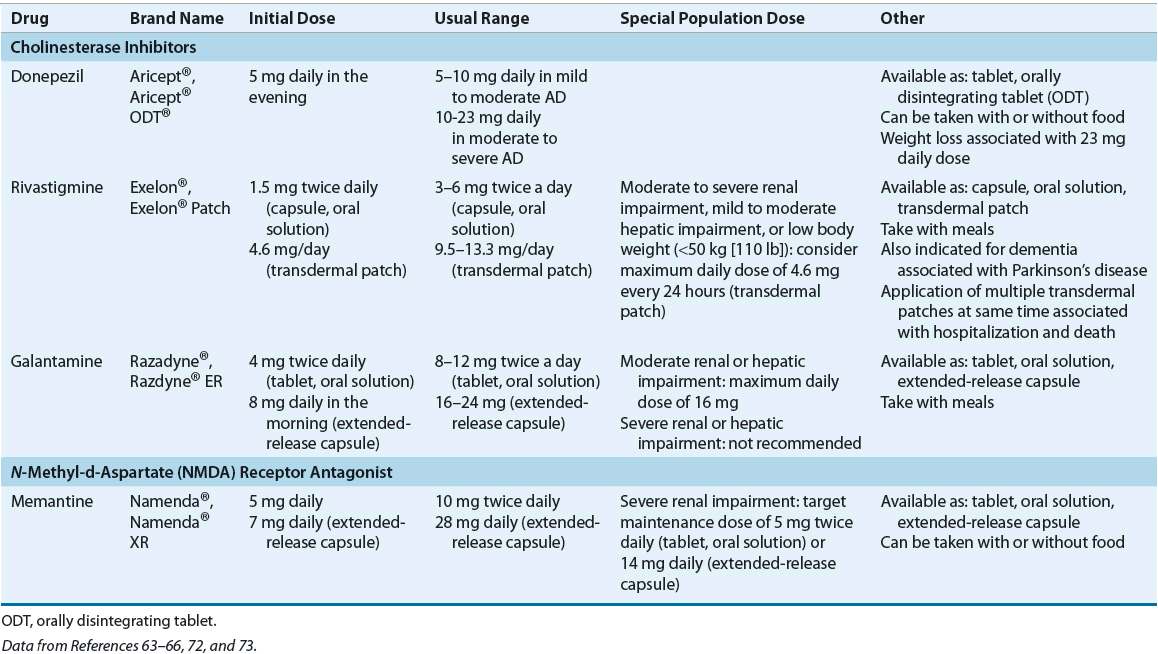
Choice of cholinesterase inhibitor therapy for an individual patient is based on ease of use, patient preference, cost, and safety issues, such as potential for drug interactions.48,49 Pharmacokinetic properties should also be considered, as rivastigmine and galantamine have short half-lives (1.5 and 7 hours, respectively) as compared to donepezil (70 hours). As such, if rivastigmine or galantamine treatment is interrupted for several days or longer, the patient should be restarted at the lowest dose and titrated to the current dose. This is true for all formulations of these drugs, including the Exelon® patch.63–66 Dosing strategies for cholinesterase inhibitors and memantine are summarized in Table 38-6.
TABLE 38-6 Dosing of Drugs Used for Cognitive Symptoms