Chapter 33 Tumors of the sweat glands
Apocrine nevus
Clinical features
Apocrine nevus as defined by an excess of normal apocrine glands (apocrine hamartoma, hamartomatous apocrine gland hyperplasia) is a very rare and clinically heterogeneous condition. Most often it presents with a fleshy axillary swelling.1–4 Erythematous or brown nodules on the neck, chest, and inguinal region, a plaque on the cheek, and multiple papules on the chest have also been documented.5–10 Lesions are present at birth or develop in adulthood. Hyperhidrosis is not usually a feature. There is generally no underlying systemic disease although one patient with a background of focal dermal hypoplasia (Goltz syndrome) and another with axillary apocrine carcinoma have been documented.4,5,11 In addition, the development of syringocystadenoma papilliferum has been described within apocrine nevi.12,13
Apocrine hidrocystoma and apocrine cystadenoma
Clinical features
Apocrine hidrocystoma is an uncommon cystic lesion and is most often solitary.1–3 Despite its apocrine derivation it is rare at sites rich in normal apocrine glands.3 It is usually found on the head and neck, commonly affecting the cheek (Fig. 33.1).2,3 Multiple lesions have also been documented.4–8 Those on the face are sometimes known as the Robinson variant and these present most often in middle-aged females.4,5,9 Multiple apocrine hidrocystomas are a feature of ectodermal dysplasia (Schöpf-Schulz-Passarge syndrome) and focal dermal hypoplasia (Goltz syndrome).10–13 Similar lesions on the eyelids are also known as Moll’s gland cysts.6,7 Rarely, it may present on the chest, shoulder, axilla, umbilicus, prepuce, vulva, penis, and the finger.14–23 Penile variants are now thought at least in part to represent median raphe rather than true apocrine cysts. The cyst shows an equal sex incidence and arises most often in the middle aged.15 Exceptionally, it has been described in childhood.24
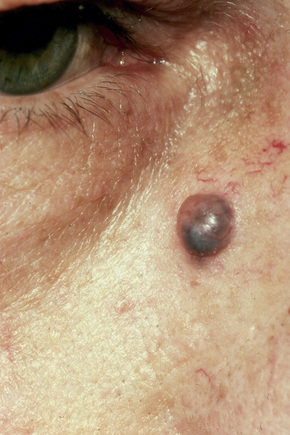
It presents as an intradermal, moderately firm, dome-shaped, translucent, blue, bluish-black or purple cystic nodule measuring up to about 1 cm across. Giant variants measuring up to 7.0 cm in diameter are exceptionally encountered.25–27 Apocrine hidrocystoma is not associated with a familial incidence. Although solitary apocrine hidrocystoma is said not to show seasonal variation, multiple lesions in some patients worsen in summer or with excessive heat and decrease during the winter months.4,5 Apocrine hidrocystoma is an occasional feature of nevus sebaceus.28
Histological features
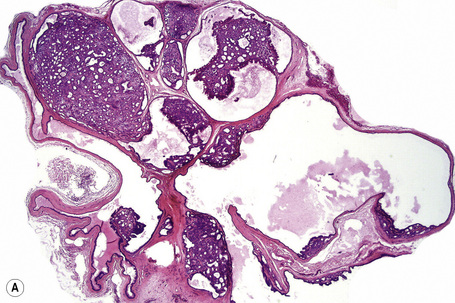
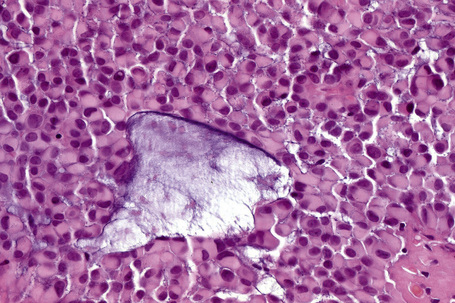
Apocrine hidrocystoma consists of a large unilocular or multilocular cystic space situated within the dermis (Fig. 33.2).3 A fibrous pseudocapsule is often present. Typically, the cystic spaces are lined by a double layer of epithelial cells: an outer layer of flattened vacuolated myoepithelial cells and an inner layer of tall columnar cells with eosinophilic cytoplasm and basally located, round or oval vesicular nuclei. Ultrastructural observations have confirmed the presence of myoepithelial in addition to secretory cells.29 Decapitation secretion is usually present (Fig. 33.3). Diastase-resistant periodic acid-Schiff (PAS)-positive granules may be evident in the cytoplasm of the inner lining cells and occasionally iron or melanin is also demonstrable.3,15–17,30 In about 50% of lesions, numerous papillary projections are seen growing into the central cavity. Occasionally, the cyst cavity is partially replaced by a papillary or adenomatous proliferation (apocrine cystadenoma) (Fig. 33.4).31
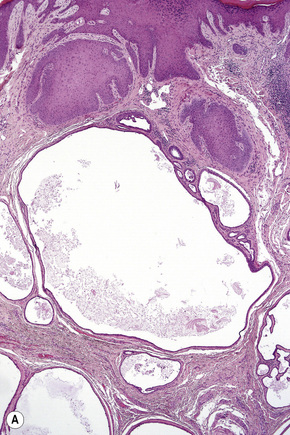
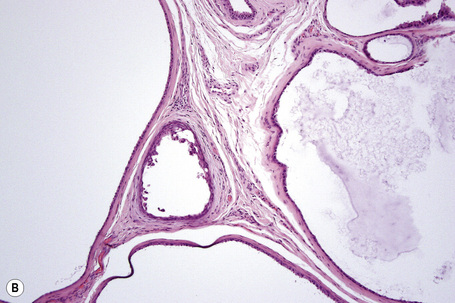
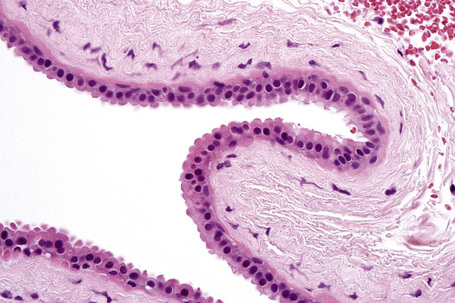
Fig. 33.3 Apocrine hidrocystoma: high-power view of lining epithelium showing decapitation secretion.
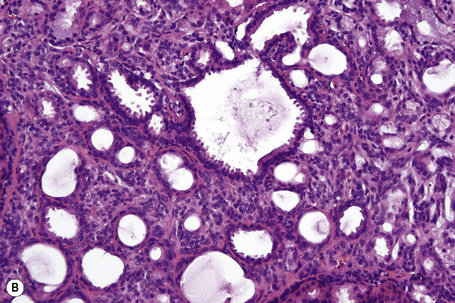
Moll’s gland cyst is lined in part by apocrine-type epithelium and elsewhere by keratinizing squamous epithelium.32
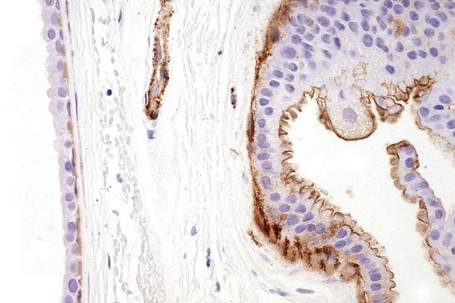
The myoepithelial layer can be highlighted with smooth muscle actin (SMA) and p63 immunohistochemistry (Fig. 33.5). Staining with S-100 protein is variable.
Differential diagnosis
There is often difficulty distinguishing between apocrine hidrocystoma and eccrine hidrocystoma. This results largely from atrophy of the epithelial lining of the apocrine hidrocystoma due to cyst distension by excessive secretions. Eccrine hidrocystoma is thought to derive from cystic dilatation of a sweat duct.33 Whether such a cyst is of eccrine or apocrine derivation is a moot point, since the two ductal systems are generally thought to be identical.4,5 Some authors have proposed the alternative term ductal hidrocystoma to reflect this possible dual histogenesis.5 Recently, however, apocrine ducts have been reported as expressing human milk fat globulin 1 (HMFG-1) whereas eccrine ducts are negative for this protein.33 This may help separate true apocrine duct hidrocystoma from that of eccrine derivation. In any event, in hidrocystomas where decapitation secretion is unapparent, S-100 protein and α-SMA immunohistochemistry will readily resolve the problem since eccrine hidrocystoma is negative for both antibodies.33–35 In addition, the luminal epithelial layer in apocrine hidrocystoma expresses keratins K7, K8, and K18 whereas in eccrine hidrocystoma the luminal layer expresses K1, K5, K10, and K14.33,35
Hybrid epidermoid and apocrine cyst
Syringocystadenoma papilliferum
Clinical features
Syringocystadenoma papilliferum is usually a solitary lesion, which may be present at birth or develop in childhood, and most commonly occurs on the scalp.1–5 It can also be found on the face, neck, trunk, and rarely the lower limbs (Fig. 33.6).3 Surprisingly, it is not often present in the axilla, a site where apocrine glands are abundant. Rarely, lesions have been described on the eyelid, breast, arm, thigh, popliteal fossa, vulva, and scrotum.6–16 Scalp involvement is commonly associated with nevus sebaceous (Fig. 33.7).17–19 Syringocystadenoma papilliferum is thus found in between 5% and 19% of cases of nevus sebaceus, sometimes in association with trichilemmoma.20,21 It has also been described in association with nevus comedonicus and in a patient with focal dermal hypoplasia (Goltz syndrome).22,23
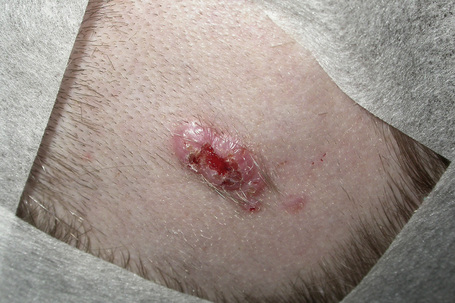
Fig. 33.6 Syringocystadenoma papilliferum: ulcerated, scaly plaque just prior to surgery.
By courtesy of J.C. Pascual, MD, Alicante, Spain.
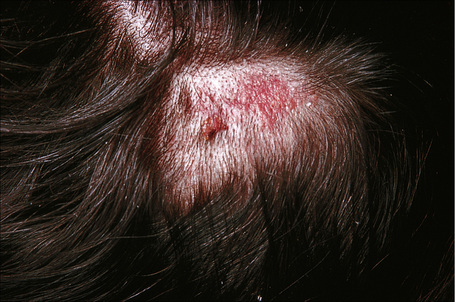
Fig. 33.7 Syringocystadenoma papilliferum: scalp tumor, which has arisen within a nevus sebaceus.
By courtesy of the Institute of Dermatology, UMDS, London, UK.
Syringocystadenoma papilliferum most often presents as a gray or dark-brown papillary or rather warty, sometimes crusted, excrescence with a moist appearance. Less commonly, multiple small papules, occasionally in a linear or segmental distribution sometimes following Blaschko’s lines are seen.3,24–29 Lesions may be excoriated due to pruritus, and those that develop on the scalp sometimes bleed due to the trauma of hair brushing or combing. Occasionally, there is central umbilication with drainage of serosanguinous secretions.6 Multifocal disease is exceptional.30
Occasionally, syringocystadenoma papilliferum has been described in association with apocrine hidrocystoma, apocrine cystadenoma, hidradenoma papilliferum, tubular apocrine adenoma, apocrine poroma, mixed tubulopapillary hidradenoma, as well as apocrine nevus.18,31–36 It may also present with condyloma accuminatum, apocrine acrosyringeal keratosis, papillary eccrine hidradenoma, cutaneous horn, verrucous carcinoma, verrucous cyst, giant comedone, and poroma folliculare.37–45
Pathogenesis and histological features
Although syringocystadenoma papilliferum is generally classified within the apocrine group, the results of electron microscopy, enzyme histochemistry, and immunocytochemistry are conflicting, variably offering support for both eccrine and apocrine derivation/differentiation.3 Perhaps the tumor is truly of apocrine histogenesis or derived from an undifferentiated pluripotential cell as originally postulated by Lever in the 1960s.
Deletion at 9q22 (PTCH) and at 9p21 (p16) have been identified in a subset of cases of syringocystadenoma papilliferum.46
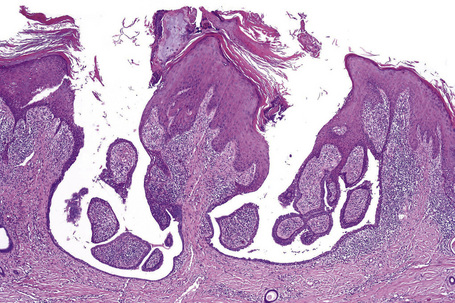
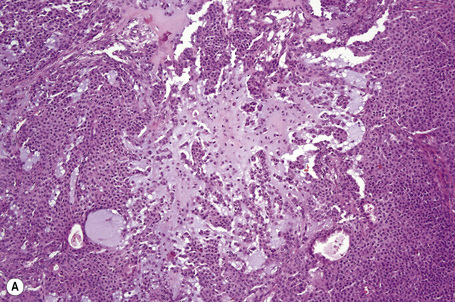
Syringocystadenoma papilliferum has a characteristic and readily recognizable appearance. On low-power examination it appears as an invagination from the overlying epidermis or else has an exophytic configuration (Fig. 33.8). Central to the diagnosis are superficially located epithelium-covered papillae, which communicate with ductlike structures in the deeper aspect of the lesion. At the surface, residual squamous epithelium is often hyperplastic and may show hyperkeratosis and parakeratosis. Superficially, the villi can be covered by stratified squamous epithelium, but this soon gives way to a typical double-layered epithelium consisting of an inner zone of small cells with scant cytoplasm and oval hyperchromatic nuclei, and an outer zone of tall columnar cells with abundant eosinophilic cytoplasm and fairly large vesicular nuclei (Fig. 33.9). Decapitation secretion is often a feature. The glandular spaces are also lined by a double layer of epithelium. The papillary processes are supported by a fibrovascular core, which typically contains large numbers of plasma cells (Fig. 33.10). Although the histological features of syringocystadenoma papilliferum are quite classic and characteristic, the morphological spectrum appears to be broad and there may be at least some morphological overlap with tubular apocrine adenoma.47 Syringocystadenoma papilliferum can show areas reminiscent of classic tubular apocrine adenoma as well as apocrine hidrocystoma and clear cell syringoma, and a tumor with focal sebaceous differentiation has also been reported to arise within a nevus sebaceous.32–34,47–50
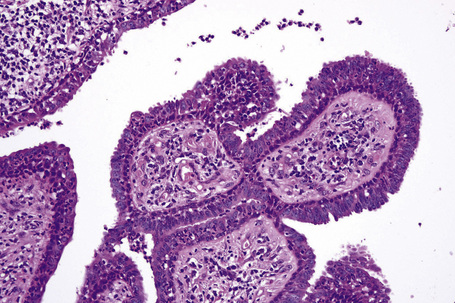
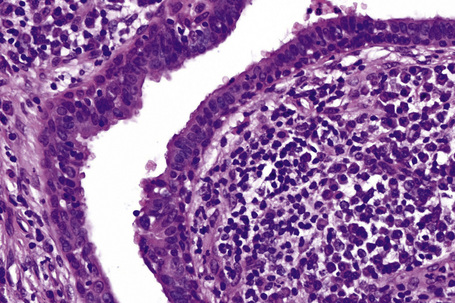
Syringocystadenoma papilliferum expresses AE1/AE3, CAM 5.2, epithelial membrane antigen (EMA), and carcinoembryonic antigen (CEA).12,33,51,52 The inner layer is positive for SMA.12 The results of markers of apocrine differentiation are variable. Some authors have found gross cystic disease fluid protein 15 (GCDFP-15) and HMFG-1 negative.10,39 Others have found GCDFP-15 and/or HMFG-2 present.12,51
Syringocystadenocarcinoma papilliferum
Clinical features
Only very rare cases of syringocystadenocarcinoma have been described.1–6 These have included three in situ variants.1,5,6–8 There are insufficient cases to allow for meaningful clinical data other than documenting that they have been described particularly on the scalp (three cases) but the chest, breast, back, and perianal region have also been affected (one case each). The sex incidence is equal and patients have been middle aged or elderly (range 47–74 years). Patients presented with verrucous nodules or large plaques, usually of many years’ duration. When documented, an episode of sudden rapid growth and tumor fixation to the underlying tissues has been evidence of malignant transformation. Occasionally, an association with nevus sebaceous has been documented.8–10
Thus far, these tumors appear to be low grade, only one case having metastasized to regional lymph nodes.3
Histological features
Rarely, the development of mucinous adenocarcinoma or apocrine ductal carcinoma presenting in a syringocystadenoma papilliferum which had arisen in a nevus sebaceus has been documented.9,10
The tumor epithelial cells express AE1/AE3, EMA, and CEA.6 HMFG-2 and GCDFP are variably positive.5,6
Hidradenoma papilliferum
Clinical features
Hidradenoma papilliferum (papillary hidradenoma) in the vast majority of cases has been described in females.1–9 Almost all cases have been reported in white women.5 Patients are generally young or middle-aged adults (range 20–89 years).6,7 There are only very rare documented cases reported in males and some authors would regard such lesions as representing apocrine papillary cystadenomas.7,10,11 The same might also be said for at least some examples of so-called ectopic hidradenoma papilliferum which have been described on the eyelid, nose, cheek, axilla, upper and lower limbs, chest, back, and external auditory canal.12–17 The examples arising on the eyelid and external auditory meatus are likely derived from the gland of Moll and ceruminous gland, respectively.
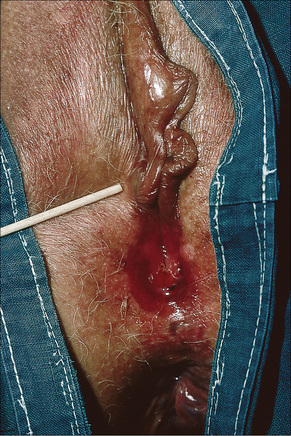
The tumor presents as a small (1–2 cm in diameter), solitary, usually asymptomatic papule or nodule in a vulval, perineal or perianal location.4 Very occasionally pain, tenderness, pruritus, burning, discharge or bleeding may be encountered.6 Most often it affects the labium majus, but on occasions it has been described as involving the lateral aspect of the labium minus, the interlabial sulcus, the clitoris, posterior fourchette, and mons pubis (Fig. 33.11).6 Lesions are round, solid or cystic and sometimes umbilicated or ulcerated.5
Pathogenesis and histological features
Hidradenoma papilliferum arises from apocrine glands or possibly the anogenital mammary-like glands.18
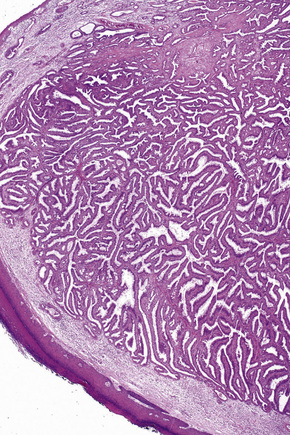
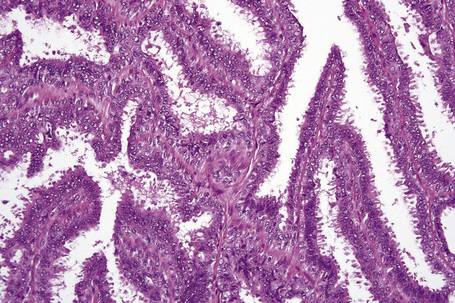
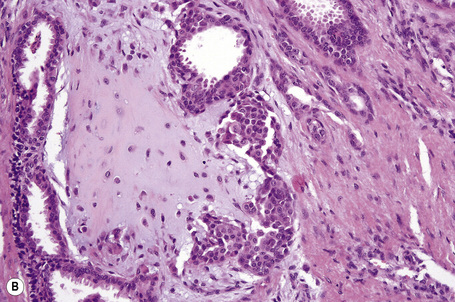
The epidermis may be normal, acanthotic or ulcerated. The tumor forms a fairly well-demarcated nodule in the dermis or lamina propria and may sometimes show foci of continuity with the overlying epithelium (Fig. 33.12).4,19 The lesion consists primarily of epithelium-covered papillary processes that project into cystic spaces. The epithelial lining is typically double layered, comprising inner small myoepithelial cells with oval hyperchromatic nuclei and outer tall columnar cells with eosinophilic cytoplasm, sometimes manifesting decapitation secretion (Fig. 33.13). Prominent oxyphilic metaplasia of the epithelial cells, in areas showing mild nuclear pleomorphism, may be a focal feature.8,9 Occasionally, the lining is only one cell thick (columnar). Diastase-resistant, (PAS)-positive intracytoplasmic granules are usually present. The presence of normal mitotic activity has no sinister implication (Fig. 33.14).20 The larger villi have a fibrous core in which occasional ductular structures may be identified, sometimes forming a cribriform pattern. Often the fibrous tissue surrounding the tumor is compressed to form a pseudocapsule. An inflammatory cell component is not a significant feature although aggregates of lymphocytes and plasma cells have been described in the stroma of ectopic lesions.14 Rare observations include a focally solid growth pattern composed of small monomorphous cells with lumen formation, a spindle cell population as well as areas resembling sclerosing adenosis in the breast.8 Uncommonly, focal sebaceous differentiation may be a feature.4,17
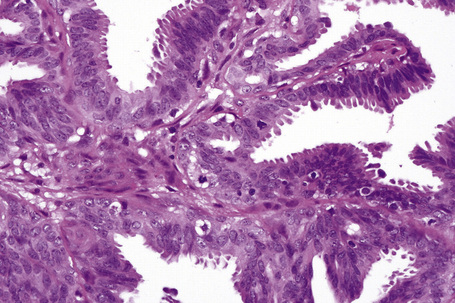
Exceptionally rarely, a malignant variant may be encountered including intraductal carcinoma.21–25 In the single example encountered by the authors, focal areas showing an extensive infiltrative growth pattern accompanied by marked nuclear pleomorphism and conspicuous, sometimes abnormal, mitotic activity were identified against a background of typical benign morphology. The outcome in this case is unfortunately not known. Coincidental hidradenoma papilliferum in a patient with vulval Paget’s disease has been described.7,26
Ultrastructurally, hidradenoma papilliferum shows features of apocrine differentiation.27
Immunohistochemically, the epithelial cells express low molecular weight keratin, EMA, CEA, HMFG, and GCDFP-15.7,28 Estrogen and to a lesser extent progesterone receptors are positive, and androgen receptor is expressed in up to 20% of tumors.28–30 The myoepithelial cells express S-100 protein and SMA.28
Human papillomavirus (HPV) types 16, 31, 33, 53, and 56 have been detected in a subset of anogenital lesions but no definite causal role in the pathogenesis of this tumor has been established as yet and the significance of this finding is unclear.8,31
Tubular apocrine adenoma
Clinical features
Tubular apocrine adenoma (apocrine adenoma, tubulopapillary hidradenoma, papillary tubular adenoma) is a rare benign tumor which shows a female predominance (2:1) and a wide age distribution (18–78 years).1–12 The scalp is most commonly affected although lesions have been described at a variety of other sites including the face, eyelid, axilla, leg, and genitalia.3,5,8–11,13,14 The last, however, may represent an adenoma of the anogenital mammary-like glands.10 Those that present on the scalp often arise in a background of nevus sebaceus and are sometimes associated with syringocystadenoma papilliferum.12,15–19 The tumor generally presents as a dermal nodule 1–2 cm in diameter or pedunculated lesion, frequently of many years’ duration, particularly those developing within a nevus sebaceus. The lesion is benign and recurrence following excision is uncommon.
Histological features
Histologically, tubular apocrine adenoma presents most often as a circumscribed intradermal nodule although in some cases the subcutaneous fat is involved. Sometimes the tumor communicates with the epidermis through ductlike structures or dilated follicular infundibula (Fig. 33.15). As mentioned above, there may be continuity with a syringocystadenoma papilliferum or an organoid nevus. It is composed of variably sized, well-formed tubules lined by a double or multilayered epithelial cell layer comprising cuboidal or columnar forms with abundant eosinophilic cytoplasm and uniform round to oval nuclei (Fig. 33.16). There is no pleomorphism and mitoses are scanty. In those tubules showing glandular differentiation, the inner lining cells often show decapitation secretion while the outer layer is composed of flattened myoepithelial cells. Cystic change is common and in many tumors intraluminal papillae are present although usually these are devoid of a fibrovascular core. True papillae are, however, sometimes seen although these are generally evident in lesions associated with a syringocystadenoma papilliferum. The tumor has a well-developed connective tissue stroma in which only small numbers of chronic inflammatory cells are present. Areas of follicular and/or sebaceous differentiation may rarely be encountered.20
The luminal surface of the tubular lining epithelial cells shows strong expression of EMA and CEA (Fig. 33.17).7,11,12,17 The cytoplasm is also sometimes weakly EMA positive.12 HMFG-1 and GCDFP may be present.11 The myoepithelial cells can be highlighted with SMA or S-100 protein immunohistochemistry.11,12,17
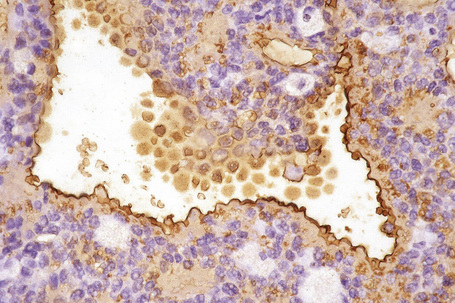
Fig. 33.17 Apocrine tubular adenoma: the luminal aspect of the tubules shows striking EMA positivity.
Ultrastructurally, the tubules are lined by cuboidal to columnar epithelial cells with conspicuous luminal microvilli and sometimes showing apical pinching or frank decapitation secretion. The cytoplasm contains prominent Golgi, conspicuous mitochondria, and lipid-rich secretory vacuoles. The outer layer shows features of myoepithelial cells.12
Differential diagnosis
Some tumors, particularly those arising in a background of an organoid nevus, develop in association with a syringocystadenoma papilliferum. Those that are wholly intradermal differ from syringocystadenoma papilliferum by the absence of true papillae with fibrovascular cores and by the absence of a plasma cell-rich inflammatory cell infiltrate. There may, however, be a morphological continuum between the two entities and reliable separation is not always possible.21
Tubular apocrine adenoma can be distinguished from papillary eccrine adenoma in many cases by the presence of apocrine decapitation secretion and the common location on the scalp, especially when developing in association with syringocystadenoma papilliferum or organoid nevus. In some cases, however, the distinction is difficult or impossible (Figs 33.18, 33.19). Occasional tumors show features of both lesions.11 This has led some authors to suggest the alternative terms tubulopapillary hidradenoma and papillary tubular adenoma.11,12,22
Adenoma and adenocarcinoma of the anogenital mammary-like glands
The anogenital mammary-like glands combine the features of eccrine, apocrine, and mammary glands.1–3 They are present in greatest concentration in the vulval interlabial sulcus. A range of tumors reminiscent of their mammary counterparts including epithelial hyperplasia, adenoma, fibroadenoma, phyllodes tumor, and in situ as well as invasive ductal carcinoma have been documented.4–24 Pseudoangiomatous stromal hyperplasia (PASH) has also been documented.25,26
Human papillomavirus (HPV) has been detected in a single case of invasive ductal carcinoma.27
Nipple adenoma
Clinical features
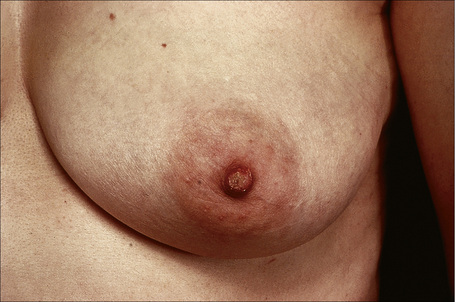
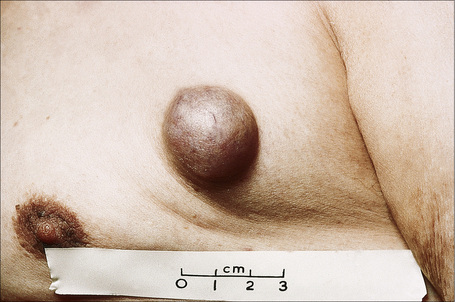
Nipple adenoma (erosive adenomatosis, florid papillomatosis, superficial papillary adenomatosis) is a benign tumor which most often presents in middle-aged females with a peak incidence in the fifth decade.1–8 Rarely, however, girls may be affected and there are exceptional reports of the condition in males.9–13 Patients present with erythematous, scaly or crusted and sometimes eroded lesions clinically mistaken for eczematous dermatitis or Paget’s disease (Fig. 33.20).1,6, 14 Pruritus, irritation, pain, and burning are variable complaints.9 An 0.5–1.5 cm tumor nodule and/or an increase in size of the nipple are sometimes present.4,8 Some patients complain of nipple discharge or bleeding.
Histological features
The tumor is unencapsulated and consists of adenomatous and papillary areas in varying proportion.1 It usually communicates with the surface epithelium where cysts lined by an admixture of squamous and columnar epithelium are sometimes evident.1 The glandular spaces are lined by tall columnar eosinophilic cells which invariably show decapitation secretion. A myoepithelial cell layer is present. Intraluminal papillomatosis is generally evident and giant cells are sometimes seen.1,9 Normal mitoses may be present. The papillae are devoid of a fibrovascular core and cytological atypia is absent. The stroma can be fibrotic or hyalinized and in some tumors this compresses the epithelium to give rise to a pseudoinfiltrative growth pattern.1 A plasma cell-rich inflammatory cell infiltrate is sometimes evident in the surrounding connective tissue.
Syringomatous adenoma of the nipple
Clinical features
Syringomatous adenoma of the nipple is a rare tumor presenting as a firm unilateral mass of few centimeters on the breast predominantly affecting the nipple and subareolar area.1–9 Development within a supernumerary breast has recently been reported.10 It is a tumor of adulthood with a peak incidence in the fourth decade and a strong female predilection.1–10 Presentation in males is exceptional.9 Syringomatous adenoma of the nipple is a locally aggressive tumor with potential for recurrence if inadequately excised.8,9 However, no distant metastasis or disease-related mortality has been documented and complete excision with negative margins appears to be curative.8,9
Histological features
The histological features are reminiscent of sclerosing sweat duct carcinoma to which it may be closely related. The tumor shows an infiltrative growth pattern within dermis, along nipple ducts, and extending into breast parenchyma.9,10 Infiltration of smooth muscle is a frequent feature and perineural infiltration may rarely be observed. The tumor is composed of small, well-formed ducts and basaloid epithelial strands within a fibrous stroma showing only little cytological atypia.9,10 Tubular and squamous differentiation may be seen and keratocysts as well as dystrophic calcification are common.
Apocrine poroma
Clinical features
Apocrine poroma (poroma with divergent differentiation, complex poroma-like adnexal adenoma, sebocrine adenoma, sebaceous and apocrine adenoma, and poroma with sebaceous differentiation) is an uncommon tumor, which presents as an often slowly growing flesh-colored, erythematous papule, nodule or plaque.1–10 There is no site predilection, lesions having been described on the lip, cheek, eyelid, nose, abdomen, back, and limbs.3,11,12 No cases presenting on the palms and soles have been documented to date. A wide age range may be involved (19–76 years). The sexes are affected equally.
Histological features
Apocrine poroma – in common with its eccrine counterpart – is composed of anastomosing trabeculae, displaying multiple points of origin from the epidermis and located largely in the papillary and upper reticular dermis.3 The individual cells are small and uniform with scanty cytoplasm and round to oval nuclei united by inconspicuous intercellular bridges. Foci of ductal differentiation with a well-developed eosinophilic cuticle are present. An example showing follicular infundibular origin in a patient with nevoid basal cell carcinoma has been reported.4
An infrequent feature is the presence of sebaceous cells, singly and in clusters with bubbly cytoplasm and crenated nuclei. Sebaceous ductlike tubular or cystic structures lined by squamous epithelium with an eosinophilic, scalloped cuticle and containing eosinophilic debris with pyknotic nuclei may also be present.3 In some examples, hair germlike structures manifest as small collections of basaloid cells with peripheral palisading, and perifollicular sheathlike connective tissue are seen.3,7,9 Occasional reports have described tubules lined by cells with intensely eosinophilic cytoplasm reminiscent of apocrine epithelium.3,7 Although frank decapitation has generally been absent, it is well illustrated in the series of Yamamoto and coworkers.6
Wholly intraepidermal hidroacanthoma simplex-like and largely intradermal variants have been documented.5,9 In addition, there is a report of an example associated with trichoblastoma.10
A case presenting on the areola has also been documented.13 In it, however, there was no evidence of sebaceous, follicular or apocrine differentiation. Although close proximity to the follicular infundibula was demonstrated, it is unclear whether the tumor might not have been better regarded as eccrine poroma.13
A metaplastic or sarcomatoid carcinoma has recently been shown to arise within an apocrine poroma (sarcomatoid apocrine porocarcinoma).11
Apocrine carcinoma
Clinical features
Apocrine adenocarcinoma is rare and most documented cases have affected the axilla.1–18 Occasionally, the tumor may present at a variety of other sites including the scalp, eyelid (Moll’s gland carcinoma), ear (ceruminous gland adenocarcinoma), anogenital region, chest, lip, and wrist, in descending order of frequency.2,14,18–30 Tumors have also been described on the cheek, nipple, and fingertip.11,31,32 Clinical data are limited in the majority of published cases, but most tumors present as single or multiple, sometimes ulcerated, often slowly growing nodules or plaques covered by erythematous or purple skin. A presentation as carcinoma erysipeloides has also been documented.1–3,33 In some instances, tumors have been present for 30 years before diagnosis.12 Occasional tumors have arisen within a nevus sebaceous and invasive as well as in situ carcinoma has been documented to arise in association with apocrine adenoma in the perianal area.18,28,34–38 Patients with bilateral axillary apocrine carcinomas and associated apocrine hyperplasia have been reported in the Japanese literature.16,39 Telangiectatic and inflammatory cutaneous metastatic disease similar to that described with breast carcinoma has been described.24 Age at presentation is variable (18–91 years, mean 60 years) and the sex incidence is approximately equal.1,2,18 There is no racial predilection.2
Apocrine carcinoma is often characterized by a prolonged course and, although recurrences (28%) and nodal metastases (50%) are common, the overall mortality is low. Bone and lung secondary deposits or more disseminated disease and tumor-related deaths have, however, been described.1,2,5,11,12,18,22,24,40–43 Disease-related mortality was 24% in a recent study.18 In some patients, metastases are a very late development and therefore a very careful, prolonged follow-up is indicated.
Histological features
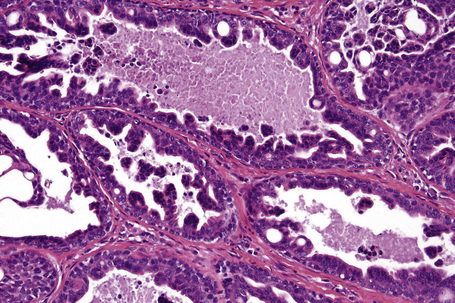
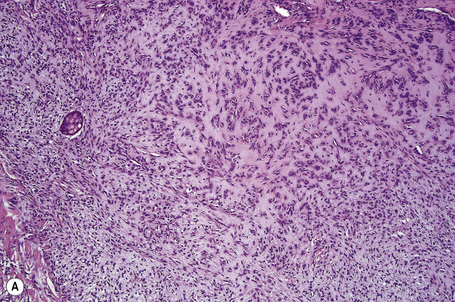
Apocrine carcinoma is characterized by a variable glandular, tubular, papillary, tubulopapillary or diffuse or solid growth pattern centered on the deeper dermis and frequently involving the subcutaneous fat (Figs 33.21–33.24).1–3,18,44 Occasional tumors are cystic, and foci of necrosis are sometimes evident.2 In contrast to apocrine adenoma, the tumor is usually poorly circumscribed and typically an infiltrating border is present. Epidermotropism is sometimes a feature and in some tumors frank Paget’s disease is present (Fig. 33.25).2,11,13,18,23,24,45–47
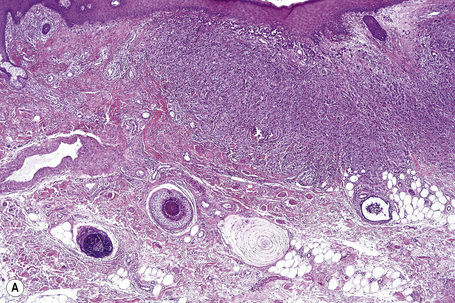
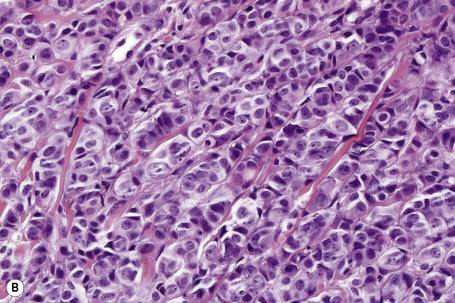
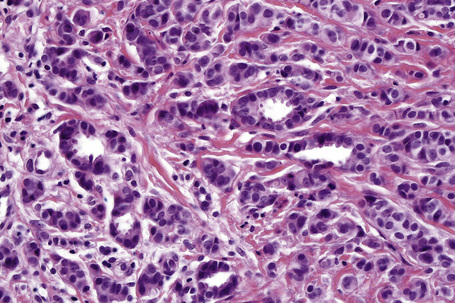
Fig. 33.22 Apocrine carcinoma: focal glandular differentiation and decapitation secretion is evident.
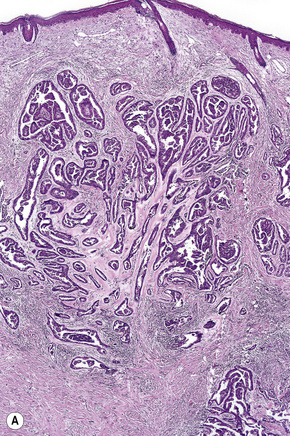
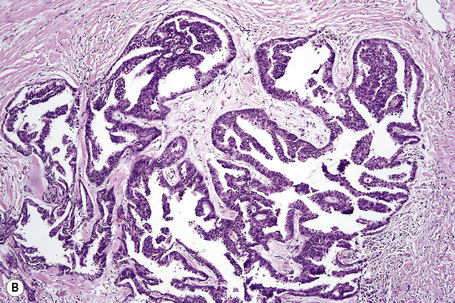
Fig. 33.23 (A, B) Apocrine carcinoma: this example from the face shows a striking papillary growth pattern.
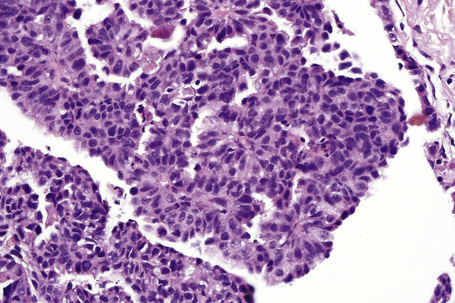
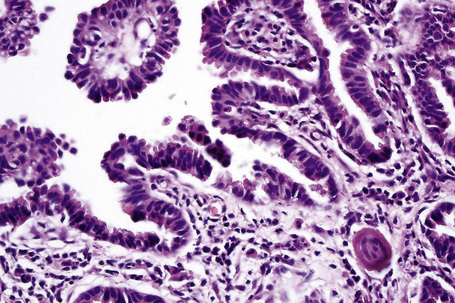
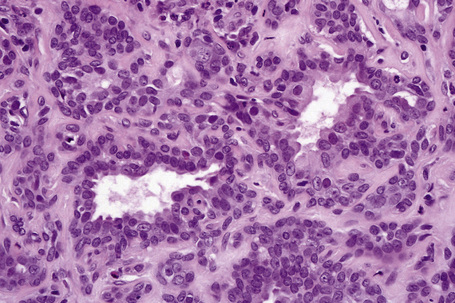
The epithelial cells have abundant eosinophilic cytoplasm, and decapitation secretion (albeit often focal) is invariably present (Fig. 33.26).2,6 Nuclei are round or oval and vesicular and commonly contain a solitary prominent nucleolus (Figs. 33.27, 33.28, 33.29). Focal squamous differentiation may occasionally be seen.2 Exceptionally, sebaceous differentiation has been described.26 Pleomorphism and mitotic activity are variable features, but become more prominent in poorly differentiated variants. The tumor is commonly accompanied by a dense hyaline stroma. A single filing growth pattern may be encountered in poorly differentiated tumors.8–10,18,24 Apocrine carcinoma is characterized by intracytoplasmic diastase-resistant, (PAS)-positive granules and intracytoplasmic iron is sometimes demonstrable.2,16,22 Alcian blue (pH 2.5) and mucicarmine may also be positive but glycogen is uniformly absent.9,13
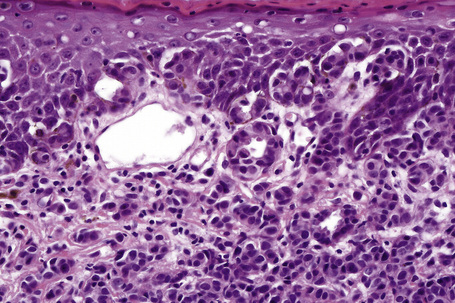
Fig. 33.26 Apocrine carcinoma: focal epidermotropism is evident (same case as Figures 33.21 and 33.22).
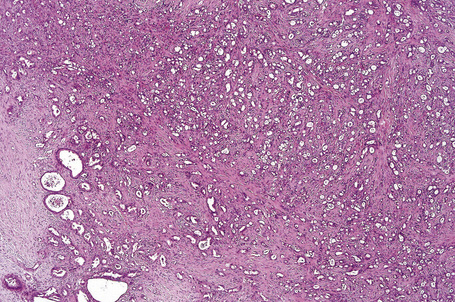
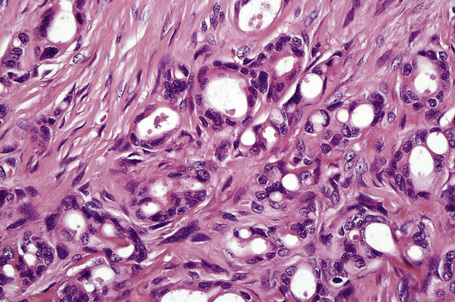
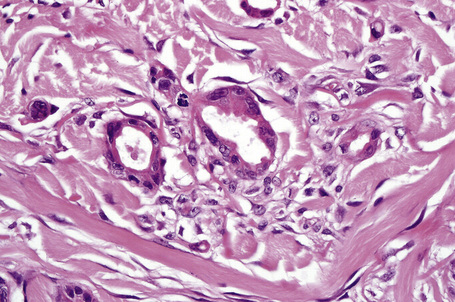
Normal apocrine glands are often found in close proximity to the tumor and occasionally longstanding pre-existent benign apocrine lesions (including hyperplasia, cystadenoma, cylindroma, syringocystadenoma papilliferum, and tubular adenoma) may be evident, raising the possibility of malignant transformation.2,3,6,14,20,48 The apocrine glands sometimes show tumor infiltration/in situ carcinoma.1,2,13,26 Perineural infiltration and lymphovascular invasion is occasionally seen.18,22,26
A small number of apocrine carcinomas showing signet ring cells reminiscent of invasive lobular carcinoma of breast have been reported.18,49,50 These show a striking predilection for elderly males (10:1).10 Although these have most frequently been described on the eyelids, they may also present in the axilla.8–10 Apocrine carcinoma with focal mucinous carcinoma-like features has been documented.23 An unusual tumor characterized by more circumscribed borders, epidermal connection, and atypical basaloid cells with duct formation and decapitation secretion has recently been reported.51
Immunocytochemically, the tumor shows low molecular weight keratin (CAM 5.2), AE1/AE3, EMA, CEA, CK15, and GCDFP-15 expression.2,3,11,19 Lysozyme, α1-antitrypsin, α1-antichymotrypsin, and in some tumors S-100 protein, are also present.2,10 Myoepithelial cells as demonstrated by SMA are usually lost.13,26 In a significant subset of tumors expression of estrogen receptor, progesterone receptor or androgen receptor is seen.18 If fresh tissue is available, assessment of apocrine enzymes, including acid phosphatase and non-specific esterase, may be of diagnostic value.6
Electron microscopic findings, including luminal microvilli, conspicuous mitochondria, and large electron dense granules, have supported apocrine differentiation.11,23
Ceruminous gland tumors
Clinical features
Ceruminous gland tumors (ceruminoma) are rare and present as an often pedunculated nodule or cystic lesion in the external auditory canal, often associated with deafness and less commonly with tinnitus or otorrhea.1–8 Pain is sometimes a feature as a result of otitis externa, ulceration or malignancy.7 Facial nerve palsy has occasionally been documented.2,9 The age distribution is wide but tumors are most commonly seen in adulthood.8 The sex incidence is equal.5,8
In the earlier literature, the term ceruminoma was often applied to all glandular tumors arising in the external auditory meatus. This resulted in considerable confusion since it lumped together both benign and malignant variants, the latter being the more common. Currently, ceruminous gland tumors are classified as benign, including apocrine adenoma and mixed tumor (pleomorphic adenoma), and malignant – ceruminous gland adenocarcinoma and adenoid cystic carcinoma.1,2,6 In addition, cylindroma, syringocystadenoma papilliferum, and mucoepidermoid carcinoma have exceptionally been documented.2,10
Histological features
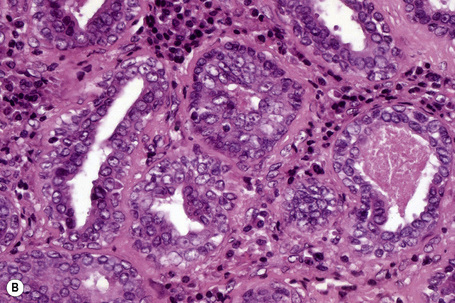
Ceruminous apocrine adenoma presents as a circumscribed nodule composed of glands lined by a double layer of epithelium (Fig. 33.30).4,8 The inner are cuboidal to columnar with eosinophilic cytoplasm often showing decapitation secretion. The outer are myoepithelial cells. Cystic change is sometimes present. Solid, acinar, and trabecular variants have been described.5,8 Pleomorphism is absent and mitoses are scarce. Abnormal forms, by definition, are not a feature.
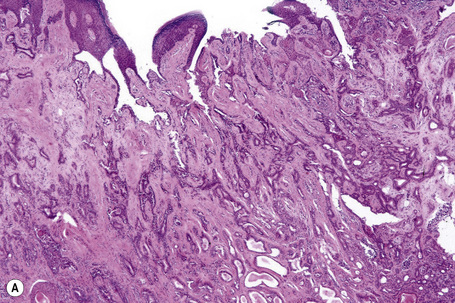
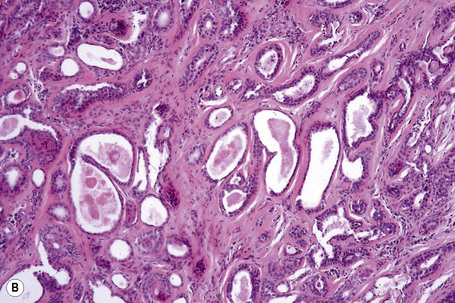
Ceruminous gland adenocarcinoma is characterized by an infiltrating growth pattern. Pure glandular and papillary variants are recognized. The cytology varies from deceptive, well-differentiated forms to high-grade tumors with marked pleomorphism and conspicuous mitotic activity (Fig. 33.31). Perineural infiltration is sometimes a feature and rarely extramammary Paget’s disease is seen.11
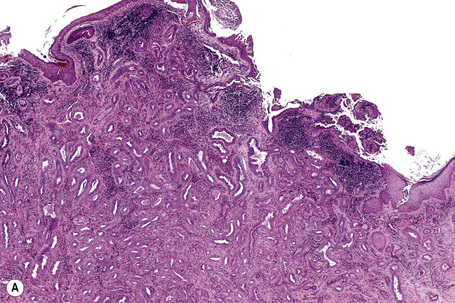
Differential diagnosis
Well-differentiated ceruminous gland adenocarcinoma may be difficult to distinguish from adenoma if the infiltrative border is not visible, as may be the case in small biopsy specimens. If there is any doubt, it is recommended that tumors be reported as having uncertain malignant potential with the final diagnosis deferred until the full excision specimen is available for study.5
Mixed tumor of the skin
Clinical features
Mixed tumor of the skin (chondroid syringoma) is not uncommon and presents as a slowly growing, firm, circumscribed, lobulated nodule within the dermis or subcutaneous fat. It is usually solitary and asymptomatic and most often affects the head and neck, particularly the nose, cheek, upper lip, scalp, forehead, chin, eyelids, and ear including the external auditory meatus, in descending order of frequency, although on occasions it may involve the axillae, trunk or extremities (Figs 33.32, 33.33).1–9 Scrotal involvement has rarely been described.10,11 Exceptional giant variants affecting the cheek and axilla have been documented.8,12–15 Males are more commonly affected than females and the tumor typically presents in the middle aged.9 Recurrences are rare.
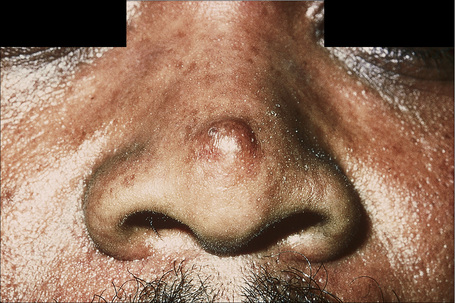
Fig. 33.32 Mixed tumor: this erythematous nodule is present at a characteristic site.
By courtesy of R.A. Marsden, MD, St George’s Hospital, London, UK.
Histological features
Although the earlier literature argued variably for an eccrine or apocrine derivation, more recently variants differentiating towards both are recognized.16,17
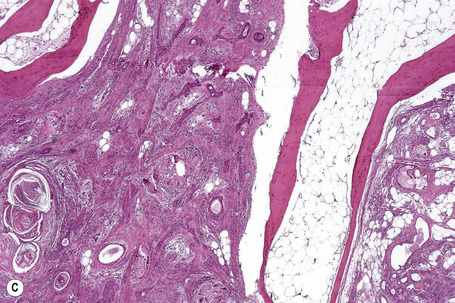
Most tumors are currently classified as apocrine type (apocrine chondroid syringoma). This is usually multilobulated and situated within the deep dermis and/or subcutaneous fat (Fig. 33.34). It forms a well-circumscribed mass in which a dominant component has a chondroid appearance (Figs. 33.35, 33.36). The lobules are separated by fibrous septa. The epithelial component is composed of nests and cords of cuboidal or polygonal cells with copious eosinophilic cytoplasm and basophilic nuclei.1–3,12,17 They are distributed singly, in cords and nests or as irregular tubuloalveolar and ductal structures.
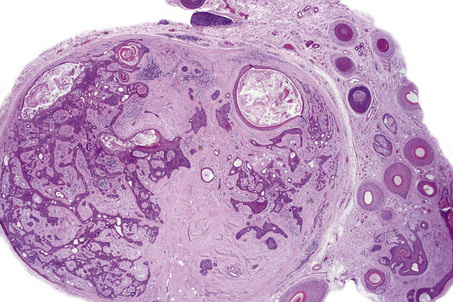
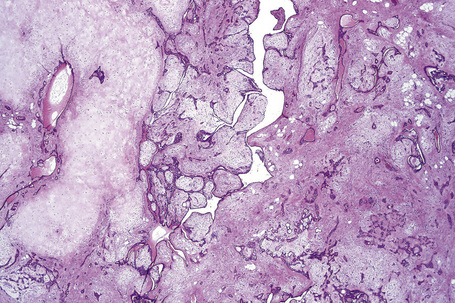
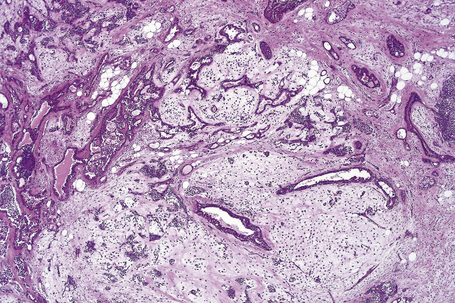
The tubuloalveolar foci, which are believed to represent differentiation towards the secretory coil, are lined by two or more rows of epithelial cells, the outer layer being somewhat flattened and of myoepithelial derivation (Fig. 33.37). Apocrine decapitation secretion is sometimes evident and occasionally the epithelium has a lacelike pattern (Fig. 33.38). PAS-positive, diastase sensitive, glycogen-rich clear cells may be present (Fig. 33.39).17 One or occasionally two layers of cells line the ducts, which represent differentiation towards the dermal sweat gland duct (Fig. 33.40). Cystic dilatation is common (Fig. 33.41). Occasionally, keratinous cysts and foci of squamous differentiation are present (Fig. 33.42).1,17 In addition, clear cell change as well as mucinous, columnar, oxyphilic, and hobnail metaplasia of the epithelial component may be evident.9 There is, however, no pleomorphism, mitoses are sparse, and necrosis is absent.
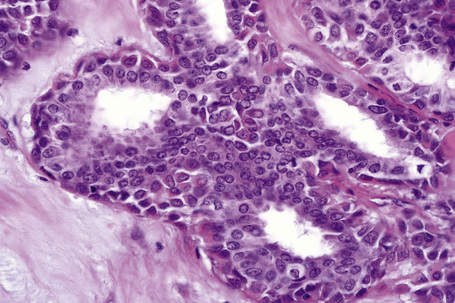
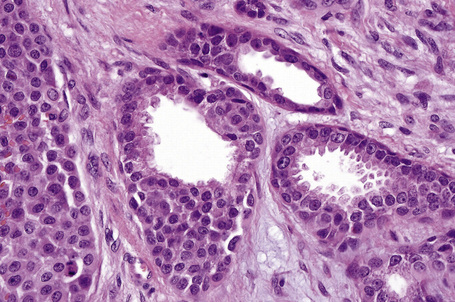
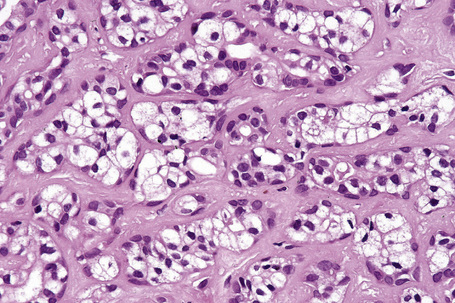
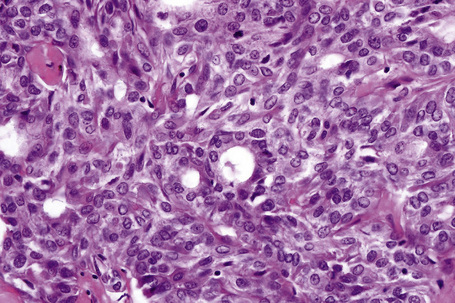
Fig. 33.40 Mixed tumor: the ducts are lined by cuboidal epithelium and show a well-developed cuticle.
Not uncommonly, mixed tumor shows additional foci of follicular and sebaceous differentiation.1,6,17–21 The former is characterized by the presence of infundibulocystic, isthmic as well as tricholemmal differentiation, hair bulbs and papillary mesenchyme in addition to foci of ghost cells reminiscent of pilomatrixoma.6,9,20,21 Sebaceous differentiation is present as foci of cells showing granular or bubbly cytoplasm and scalloped nuclei or as sebaceous ductlike structures with an eosinophilic, wavy, laminated, keratinized lining.6,9,18 Rarely, pigmented dendritic melanocytes are a feature.
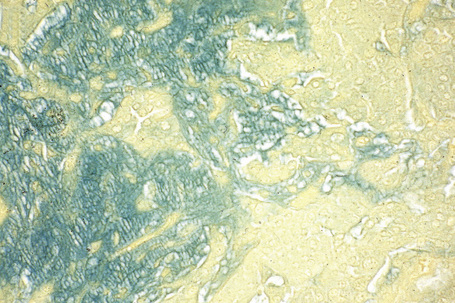
The myoepithelial component may show hyaline (plasmacytoid), spindled cell or clear cell differentiation.9,22–26 The hyaline (plasmacytoid) cells are characterized by abundant ground-glass eosinophilic cytoplasm and an eccentric nucleus. They are present singly within the stroma or as distinct noncohesive aggregates (Fig. 33.43).17,22–27,28 Spindled cell myoepithelial cells are also present within the chondroid matrix and are responsible for its production.29–31 Clear cell change of the myoepithelial component may be an additional finding. Transition between polyhedral cells and spindled cells or foci of squamous differentiation may be seen.27 The stroma is of variable appearance. Characteristically, it is composed of homogeneous bluish chondroid (Fig. 33.44). In other areas it is myxoid or densely collagenous, eosinophilic, and hyalinized (Fig. 33.45). Positive Alcian blue or green staining at pH 2.5 indicates the presence of acid mucopolysaccharides (Fig. 33.46).17 Sometimes, abundant mature fat is present (‘lipomatous mixed tumor’) (Fig. 33.47).8,9,32–36 Some tumors may show foci of calcification, and on rare occasions osteoid with marrow spaces has been described.6,16,18,37–42
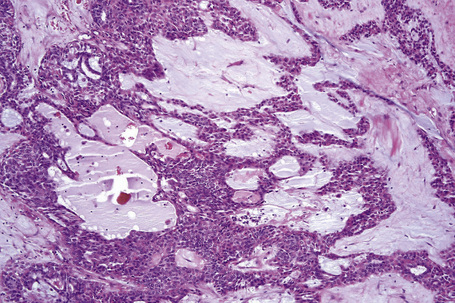
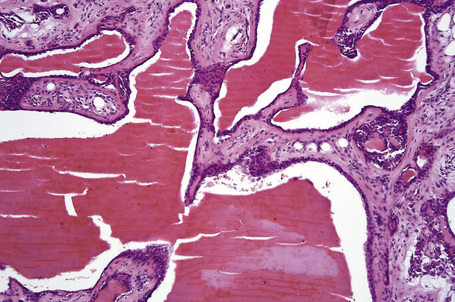
Less often, the tumor is composed of small gland and ductlike structures lined by a single layer of cuboidal epithelium dispersed in a mucinous and/or chondroid stroma (Figs 33.48, 33.49).16,17 This is sometimes referred to as the eccrine variant (eccrine chondroid syringoma).4,16
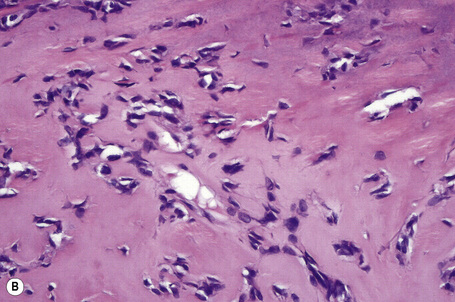
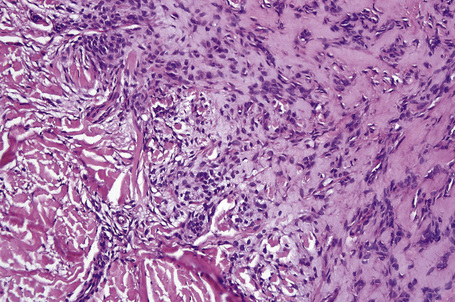
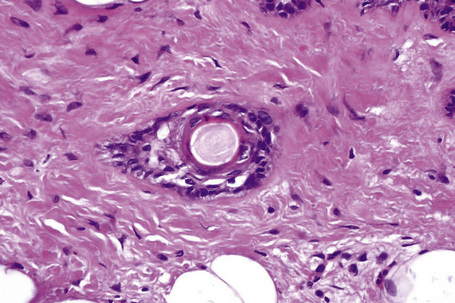
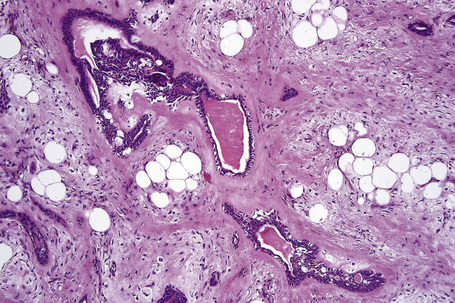
Exceptionally, hidrocystoma-like features have been described.43
Cutaneous mixed tumors with atypical histological features but benign clinical behavior have recently been described.44 Atypical architectural features include lesional asymmetry and slightly infiltrative tumor edges without capsular invasion.44 More often, the atypical findings relate to the cytological features. Mild cytological atypia of epithelial cells in ductular structures may be present but the most frequent finding is the presence of scattered multinucleated pleomorphic and bizarre-appearing cells within the myoepithelial component of the tumor. By immunohistochemistry, these cells are characterized by a myoepithelial phenotype.44
There are occasional reports of diagnosis of mixed tumor by fine needle aspiration cytology.45–47 In view of the difficulty sometimes encountered in distinguishing between benign and malignant variants, a complete excision with histological study should always be performed.
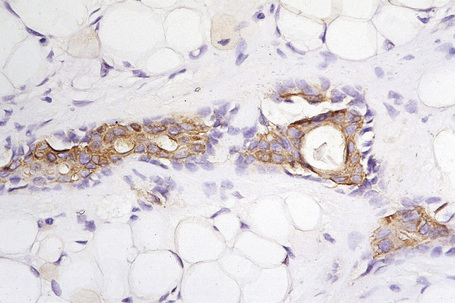
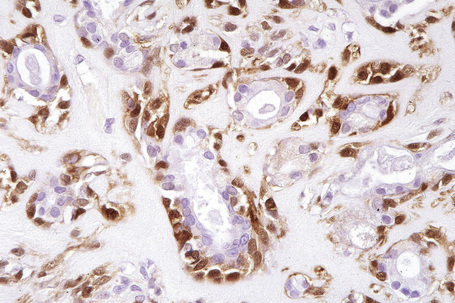
Immunocytochemically, the inner epithelial cell layer is characterized by high and low molecular weight keratin (AE1/AE3), EMA, CEA, and GCDFP-15 expression (Fig. 33.50).17,48–51 The outer cells express vimentin, S-100 protein, and sometimes SMA and muscle-specific actin (MSA) (Fig. 33.51).39,40,50–53 Stromal cells are vimentin and S-100 protein positive.39,40
< div class='tao-gold-member'>
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


