Chapter 19 Human immunodeficiency virus (HIV) and acquired immunodeficiency syndrome (AIDS)-associated cutaneous diseases
Introduction
Worldwide, approximately 30–36 million people are currently infected with the human immunodeficiency virus (HIV). An estimated 1.8 to 2.3 million people succumbed to the acquired immunodeficiency syndrome (AIDS) in 2007 alone, including some 270 000 children.1,2
HIV is an enveloped RNA virus belonging to the genus Lentivirus within the family Retroviridae.3 The disease causing viruses are HIV-1 and HIV-2.4 The former is detected mainly in the United States and Europe and the latter, mainly in West Africa. Both HIV types cause a similar clinical disease profile and patients are commonly coinfected. HIV-2, however, is associated with a five- to eightfold less rate of transmissibility, rare vertical transmission, a longer latency period, and a more gradual CD4+ T-lymphocytic decline and clinical progression.4 The lack of natural eliminatory mechanisms of HIV following primary infection and perpetuation of viral replication throughout the course of the disease are pivotal to the initiation, establishment, and propagation of HIV infection. Chronic stimulation and destruction of lymphoid tissue are responsible for the spectrum of organ manifestations associated with HIV infection.
More than 90% of HIV-infected individuals will develop one or more dermatological disorders during the course of their illness, either as a result of AIDS or due to the effects of treatment. Furthermore, cutaneous disease is often the first manifestation of undiagnosed HIV infection or AIDS.5 A wide variety of skin diseases may arise in concert with or be modified by the progressively declining CD4 lymphocyte count. HIV/AIDS should always be suspected when the clinical history reveals that a common skin disorder has presented with atypical clinical features, followed an abnormal clinical course, displayed greater clinical severity than anticipated, or failed to exhibit a satisfactory clinical response to standard therapy for that particular condition.2,5 The spectrum of cutaneous HIV disease includes AIDS-defining opportunistic infections and neoplasms (these are summarized at the end of the chapter and discussed in depth elsewhere in the book), drug-induced cutaneous manifestations, and a range of noninfectious dermatoses that may occur in all stages of HIV progression. This latter group comprises dermatoses peculiar to HIV infection (e.g., acute HIV exanthem, pruritic papular eruption), those occurring with greater frequency or modified by HIV/AIDS (e.g., seborrheic dermatitis, psoriasis), and a variety of less common conditions in which an association with AIDS has been reported (e.g., Reiter disease, pityriasis rubra pilaris). HIV-positive patients may present with more than one skin disorder, a fact of which the practicing histopathologist should always remain cognizant when examining skin biopsies in this clinical context.5 Children, for example particularly in developing countries, may have cutaneous stigmata of a concomitant nutritional disorder in addition to features of a common HIV-associated condition such as seborrheic dermatitis.2
HIV-associated papulosquamous dermatoses
HIV-associated seborrheic dermatitis
Clinical features
Seborrheic dermatitis is the most common cutaneous disease to affect HIV-infected patients. It often occurs early on in HIV infection or it may reflect disease progression.1 Notwithstanding, it is seen in up to 85% of all HIV-infected individuals at some stage during the course of their disease.1,2 Although seborrheic dermatitis is characterized by erythema and greasy scaling of the nasolabial and postauricular areas, eyebrows, external ears, and scalp, as is seen in the general population, it can be more widespread, involving the chest, trunk, groin, and extremities with occasional progression to erythroderma in the AIDS setting.1–4 Other AIDS-associated clues of seborrheic dermatitis include a predominance of inflammatory and hyperkeratotic lesions, papular and scaly plaquelike lesions resembling psoriasis, hypo- and hyperpigmentation, a ‘cradle cap’ appearance of the scalp, and a sudden onset or acute worsening of seborrheic dermatitis.1,4,5–7
Pathogenesis and histological features
The exact pathogenesis of AIDS-associated seborrheic dermatitis is not known. A relationship with AIDS-associated dementia and central nervous system disease has nevertheless been documented in 20% of AIDS patients and in HIV-negative patients with neurologic diseases.1 HIV-1 possesses neurotropic characteristics. It has therefore been proposed that the same pathogenetic mechanism promoting seborrheic dermatitis in neurologic diseases occurs in AIDS.1 Seborrheic dermatitis is associated with Malassezia furfur infection in a minority of patients, but the exact pathogenetic role thereof is unconfirmed.8–11
The histological features include those seen in seborrheic dermatitis in HIV-negative patients, including hyperkeratosis, parakeratotic mounds localized particularly to the ostia of hair follicle infundibula, acanthosis with regular elongation of the rete ridges, and mild spongiosis with intraepidermal lymphocytes and neutrophils.1,2,12,13 Features specific for AIDS-related seborrheic dermatitis include spotty keratinocyte necrosis, hyperkeratosis, leukocytic exocytosis, a perivascular plasma cell infiltrate in the dermis, and focal leukocytoclasis.1
HIV-associated psoriasis
Clinical features
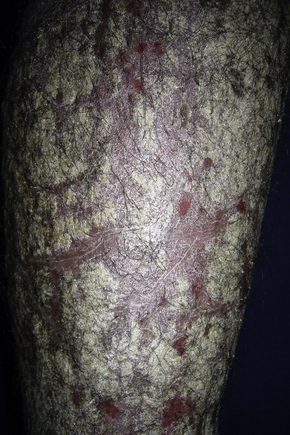
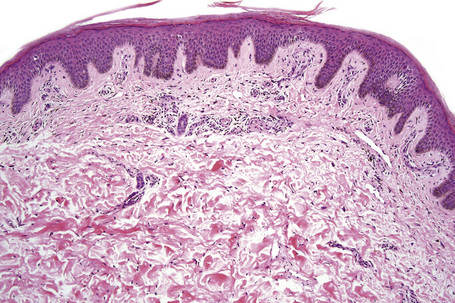
The overall incidence of HIV-associated psoriasis does not appear to exceed that of the general population.1 Occurring in 1–3% of HIV-infected individuals, the spectrum of clinical manifestations may be similar to psoriasis in the non-HIV-exposed group. Psoriasis may be the first clue to HIV infection and may undergo remission with advanced AIDS.2,3 Although all degrees of severity of psoriasis may occur at any stage of HIV infection, there may be deterioration with AIDS progression. Pre-existing psoriasis may undergo severe exacerbation with HIV exposure (Figs 19.1, 19.2).4 Two overlapping but clinically different groups of patients have been described.5 Group 1 is associated with preceding HIV infection, a family history of psoriasis, and chronic plaque or guttate psoriasis occurring in individuals aged 10 to 30 (mean = 19) years. Group 2, in contrast, occurs after HIV exposure in patients 23 to 58 (mean = 36) years of age, with a higher prevalence of psoriatic arthritis. Acral psoriasis and sebopsoriasis are the main cutaneous presentations.
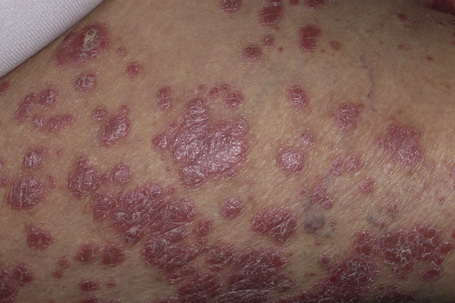
Fig. 19.1 HIV-associated psoriasis: there are numerous silver scaly plaques as seen in psoriasis vulgaris.
Pathogenesis and histological features
The pathogenesis of HIV-induced psoriasis is multifaceted and complicated.1,4,5 Psoriasis is considered to be a T-helper-1 dominant disease and the cytokine response involves interleukin-2 and interferon-δ.6 Immune dysregulation is postulated to play a pivotal role in HIV-associated psoriasis.5 HLA-Cw6 is the most frequently described genetic factor in association with psoriasis. A significantly higher proportion of patients with HIV-associated psoriasis carry the HLA-Cw*0602 allele.4 It has been suggested that immune dysregulation may activate microorganisms to trigger psoriasis in predisposed patients carrying the Cw*0602 allele. This allele is postulated to be a putative target for CD8+ T lymphocytes responding to processed peptides from microorganisms, via molecular mimicry. One recent study showed a significant association between a CD4 count of < 200 × 106/l and the evolution of psoriasis in HIV-positive patients.7
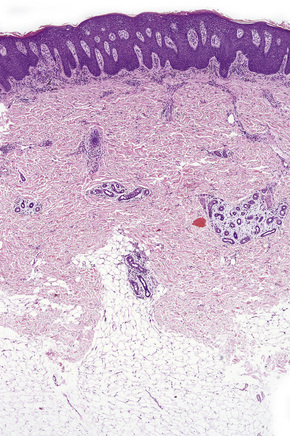
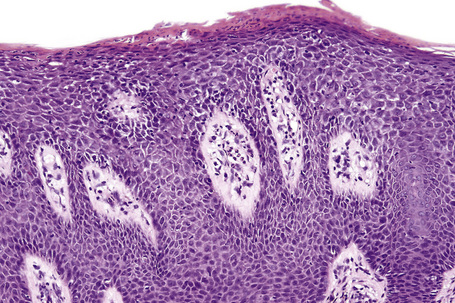
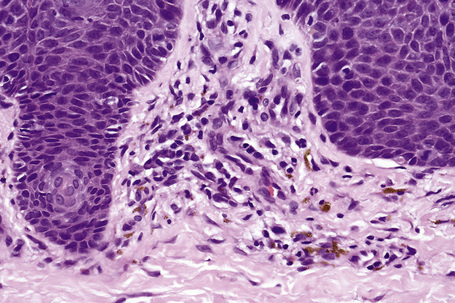
The histological spectrum of HIV-associated psoriasis is similar to that of psoriasis vulgaris, but the dermal infiltrate contains fewerT lymphocytes and significantly more plasma cells (Figs 19.3–19.5).1
HIV-associated pityriasis rubra pilaris
Clinical features
HIV-associated pityriasis rubra pilaris (PRP), also referred to as pityriasis rubra pilaris type VI (PRP-VI), is characterized by cutaneous lesions of PRP, and variable association with acne conglobata, hidradenitis suppurativa, and lichen spinulosus.1 Although PRP-VI is a disease usually encountered in young adults, children may be afflicted.2 It occurs predominantly in males and may be the sentinel of HIV infection. Clinically, PRP-VI is characterized by pruritic, symmetrical, erythematous, desquamative follicular papules that tend to become confluent, frequently resulting in erythroderma.3 The eruption occurs mainly over the extensor surfaces. There is variable palm and sole keratoderma, scalp scaling, and nail hyperkeratosis.1 The CD4 cell count is highly variable, ranging from values as low as 8 cells/mm3 to > 1000 cells/mm3. Patients with higher CD4 counts (> 500 cells/mm3) may suffer fatal PRP complications, even in the absence of opportunistic infections or neoplasms.1,4,5
Pathogenesis and histological features
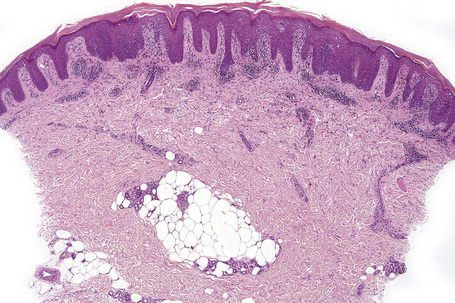
Although the association between HIV infection and PRP seems clear, the exact pathogenetic mechanisms involved are unknown. A direct role for HIV has been proposed because of the association with HIV and the response to zidovudine.1–6 Infection of the follicular hair bulge by HIV and secondary follicular inflammation have been suggested, as has precipitation of PRP in genetically predisposed individuals.5–13 The coexistence of PRP, acne conglobata and follicular plugging was a rare occurrence prior to the AIDS epidemic. The overlapping histological features have heralded the possibility that the spectrum of clinicopathological features may be due to a single disorder of follicular keratinization. PRP-VI may therefore be part of a unique and distinctive disease, more appropriately placed under the wider rubric of HIV-associated follicular syndrome.1,3,14–16
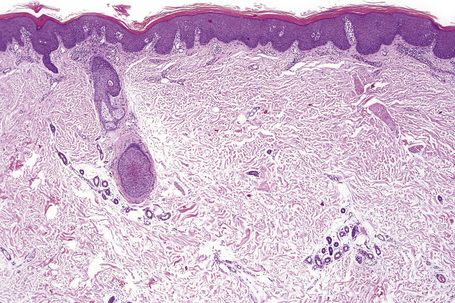
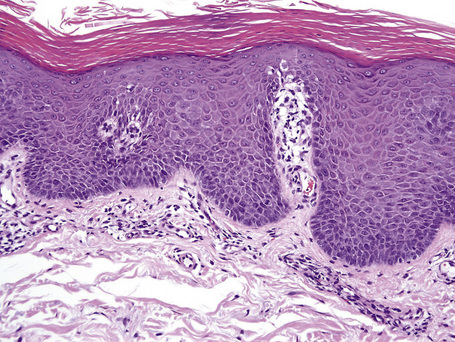
Histologically, there is variable epidermal psoriasiform hyperplasia and lamellar hyperkeratosis, with alternating zones of vertical and horizontal parakeratosis and an intact granular cell layer (Figs 19.6, 19.7). There is follicular hyperkeratosis and variable infundibular dilation with orthokeratotic and hyperkeratotic keratin.17 A sparse mononuclear infiltrate is present around the superficial vascular plexus and hair follicles. Perifollicular mucinosis may be present.11
Xerosis
Clinical features
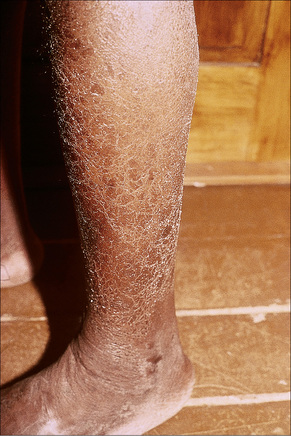
Xerosis is a relatively common scaling dermatosis in HIV infection, occurring at any time throughout disease progression.1,2 Labeled as “generalized dry skin syndrome” xerosis is usually severely pruritic, may be refractory to H1 antihistaminic therapy and may be the first and most common clinical manifestation of AIDS.1,3 Xerotic dermatitis is encountered in 4.5% to 80% of the AIDS population.1,4,5 Severe xerosis occurs in 20% of AIDS cases. It is characterized by diffuse dryness of the skin with hyperpigmented scales and focal crusting (Fig. 19.8).1,2,6,7 It is often prominent on the extremities and is worse in the winter months.3 There may be fissuring of lesional skin, leading to eczema craquelé and secondary infection in some patients.4 Occasionally, discrete thickened patches occur.1
Pathogenesis and histopathology
The pathogenesis of xerosis, although obscure, may be related to a range of factors, including cutaneous microcirculation and nutritional alterations, altered sweat or sebaceous gland activity, alterations in the composition of sweat, and changes in the cutaneous mast cell population.3,6–8 Recently, decreased calcitonin gene-related peptide and substance P levels have been documented in HIV-associated xerosis.2 These factors may result in decreased epidermal integrity, with a resultant reduction in the effectiveness of the epidermal barrier.2,3
Skin biopsies often demonstrate minimal superficial epidermal hyperkeratosis with parakeratosis, mild acanthosis, and focal spongiosis in the absence of microvesiculation (Fig. 19.9). The dermis demonstrates a minimal perivascular lymphocytic infiltrate. An inconsistent finding is the presence of early alterations of acquired ichthyosis, characterized by dense orthokeratosis with a diminished granular cell layer and minimal inflammation. Other pruritic scaling dermatoses, such as scabies, dermatophytosis, and the ‘flaky skin’ appearance of kwashiorkor are clinicopathological mimickers, as all of these conditions may occur in individuals with advanced AIDS.1,4
HIV-associated photodistributed eruptions
Photosensitivity refers to an abnormal response to nonionizing radiation.1 The spectrum of photodistributed eruptions in HIV-infected patients includes porphyria cutanea tarda, granuloma annulare, hypertrophic lichen planus, erythroderma, chronic actinic dermatitis, photodistributed hyperpigmentation, and lichenoid photoeruptions.1–8 Some of the aforementioned conditions are discussed in further detail below.
HIV-associated lichenoid photoeruptions and chronic actinic dermatitis
Clinical features
Lichenoid photoeruptions are frequently seen as the CD4 cell count decreases and may be related to potentially photosensitizing drugs.1,2 The cutaneous disease may be persistent and extend to involve nonexposed areas and become generalized.1 Lower lip involvement may be present but oral involvement is typically absent.1 Chronic actinic dermatitis (CAD) is associated with advanced HIV disease.3 On photobiologic testing, reproducible sensitivity in the UVB range (290 to 320 nm) occurs.1,3 Vitiligo-like depigmentation may be encountered in some cases.4–6
Pathogenesis and histological features
The pathogenesis of CAD is unclear but a delayed-type immune mechanism is postulated. Regardless of the cause, the immune response in CAD is regulated by CD8-positive T lymphocytes reacting to a photoinduced self-antigen. The release of control from such antiself response is lost in HIV seropositive individuals by a decrease in the CD4/CD8 cell ratio.1,3,7,8
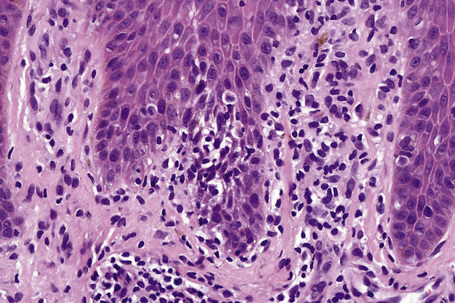
Histologically, lichenoid photoeruptions are characterized by marked hyperkeratosis, focal parakeratosis, acanthosis, papillomatosis, hydropic degeneration, and pigmentary incontinence (Figs 19.10, 19.11). There is variable keratinocyte necrosis with involvement of the upper half of the epidermis, a superficial perivascular infiltrate with a lichenoid pattern and a mid to deep dermal infiltrate of mononuclear cells (including plasma cells) and eosinophils. Occasional features include a diffuse dermal infiltrate, wedge-shaped hypergranulosis, and a microscopic subepidermal blister. CAD is characterized by subacute dermatitis with variable psoriasiform dermatitis and an atypical lymphocytic infiltrate. It has been postulated that the vitiligo-like depigmentation observed in some cases is potentiated by cytotoxic destruction of basal melanocytes by CD8+ lymphocytes.6
HIV-associated granuloma annulare
Clinical features
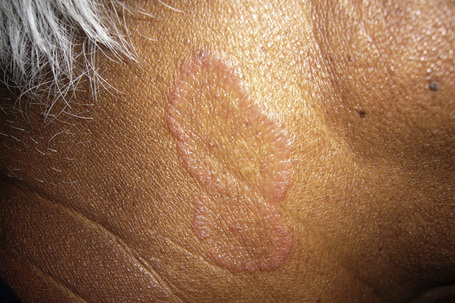
HIV infection is associated with atypical forms of granuloma annulare (GA), including oral, perforating, and generalized forms (Fig. 19.12).1–5 The generalized form occurs most frequently in HIV-infected patients.6 HIV-associated GA (HAGA) occurs predominantly in males, usually homo/bisexual, with an age range of 25 to 58 years.3 HAGA is characterized by a transient or chronic course and a more frequent occurrence on the extremities. A photodistributed generalized form of HAGA may occur.1
Pathogenesis and histological features
The exact pathogenesis of HAGA is uncertain. Polymerase chain reaction investigation and in situ hybridization have failed to confirm Epstein-Barr virus as a causative agent. A type IV cell-mediated hypersensitivity reaction to an unknown antigen causing degenerative change has been proposed. The release of cytokines from activated fibroblasts and macrophages in GA has been proposed as the cause of degeneration of elastic tissue and collagen.3,4,7 Although immunophenotypic analyses have demonstrated a CD4-positive T-helper cell response in most cases, similar to that in GA in HIV-negative patients, a predominant CD8-positive lymphocytic infiltrate may be present.2,4
The histopathological picture is one of a necrobiotic granulomatous pattern that may be interstitial, palisaded or mixed, similar to that in HIV-negative patients.8 Increased amounts of interstitial mucin can be demonstrated by Alcian blue or colloidal iron staining. The histological changes in the incomplete (or interstitial) form of GA are often subtle.9
HIV-associated porphyria cutanea tarda
Clinical features
Porphyria cutanea tarda (PCT) has been reported with increased frequency in HIV-infected patients.1 The clinical signs of type 1 PCT are confined to the skin and include pruritic, fluid-filled vesicles and bullae that develop in sun-exposed sites, including the face, dorsa of hands, and forearms. Additional findings include hyperpigmentation, hypertrichosis, and scarring.2–6
Pathogenesis and histological features
The involvement of HIV in the pathogenesis of PCT is unclear because porphyrinogenic factors other than HIV such as alcohol use, drug therapy, and hepatitis C virus infection have been identified in the majority of reported cases.7–14 The deposition of iron in the liver in patients consuming alcohol may be responsible for triggering PCT through direct inhibition of uroporphyrinogen decarboxylase activity.2 HIV may alter porphyrin metabolism through interference with the hepatic oxidase system or by creating ineffective erythropoiesis, which results in increased iron deposition. The altered steroid metabolism in patients with HIV infection could increase endogenous estrogen production, causing disruption of heme synthesis.6 Concomitant HIV and hepatitis C virus infection increases hepatitis C virus RNA levels, promoting clinical manifestations of PCT.10–12 The coexistent use of drugs may trigger PCT in the setting of HIV infection.15
HIV-associated pruritic papular eruptions
HIV-associated pruritic papular eruption
Clinical features
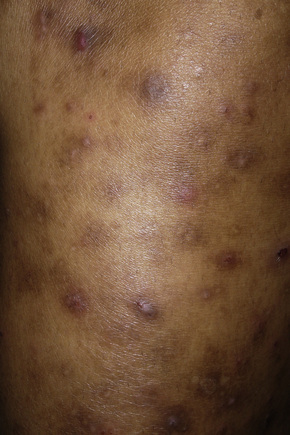
Pruritic papular eruption (PPE), sometimes referred to as papulopruritic eruption, is a unique manifestation of HIV disease characterized by chronic, pruritic, symmetrical, 3–5-mm diameter, firm, discrete, erythematous urticarial papules (Fig. 19.13).1–3 Clinically similar cutaneous lesions, however, may be encountered in eosinophilic folliculitis and suppurative folliculitis in HIV-positive patients.3 The condition is said to occur in 10–50% of HIV-infected patients, and is encountered mainly in Africa, Haiti, and Thailand. It has only rarely been documented in Europe and North America.1–5 It occurs in adults and children.1 Some studies confirm a roughly equal sex distribution, while others have shown a striking female predominance.1,4 The distribution of the disease may be localized or generalized, occurring mainly on the extremities. The lesions tend to heal with hyperpigmentation, and prurigo nodularis-like lesions may develop. An increase in the absolute eosinophil count and relative peripheral eosinophilia are common. While blood CD4-positive T-lymphocyte levels may be normal or high, the CD4 count is more commonly reduced, serving as a marker of failing immunity.1,4
Pathogenesis and histological features
Whether PPE is a distinct entity or whether it is part of the spectrum of eosinophilic folliculitis remains controversial. Unproven pathogenetic theories include the role of infections (Staphylococcus aureus, Demodex folliculorum, and Sarcoptes scabiei), drug eruptions, a direct HIV effect, and a primary abnormality of the pilosebaceous unit. The relationship between mosquito bites and PPE, documented for two decades, is more complex. Proposed mechanisms include the hypothesis that the individual lesions in PPE represent mosquito bites, or that the eruption represents a generalized hypersensitivity reaction to mosquito saliva, similar to papular urticaria.1,4,6,7 A recent study has highlighted the role of arthropod bites, mainly those of the mosquito, in the pathogenesis of PPE, emphasizing that PPE represents an exaggerated immune response to arthropod antigens in predisposed HIV-infected patients.4 Increased concentrations of interleukin-2, interleukin-12, δ-interferon and interleukin-5, in association with a decreased CD4-positive T-lymphocyte count in the blood and increased CD8-positive T lymphocytes in lesional skin, are hypothesized to explain the occurrence of a mixed Th1/Th2 or Th0 pattern, leading to cytokine production and an influx of eosinophils in the cutaneous infiltrate.8,9
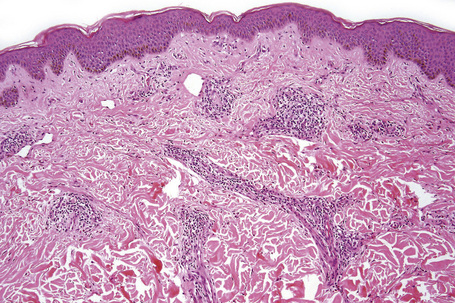
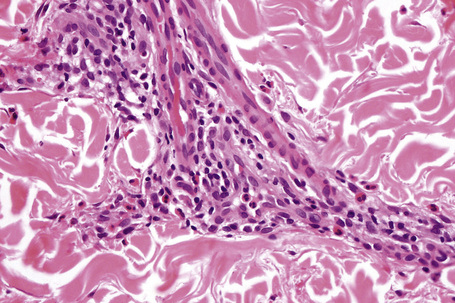
The histological features vary, depending on whether older, scarred, and excoriated nodules or new lesions are biopsied. The early lesions demonstrate a moderately dense to dense, superficial, and deep, perivascular, perifollicular, perieccrine, and interstitial infiltrate of lymphocytes and eosinophils, with variable extension into the subcutis, and epidermal hyperplasia (Figs 19.14, 19.15).2,4 There is a predominance of CD8-positive T lymphocytes. Healed lesions demonstrate variable scarring and postinflammatory hyperpigmentation.1,2,4,10 Longstanding nodular lesions that are the result of chronic pruritus and excoriation show histological features of prurigo nodularis.
Differential diagnosis
The histological differential diagnoses include folliculitis (suppurative or eosinophilic), scabies, secondary syphilis, and a drug eruption. Against folliculitis and scabies are the absence of a dominant folliculocentric localization of the pathology and the absence of mite parts of S. scabiei or mite’s feces, respectively. Examination of multiple serial sections is required to exclude the aforementioned conditions.3,11 A drug eruption may be a close mimic but the inflammatory infiltrate in this setting usually does not extend into the subcutis, and is not as dense as that seen in PPE.4 A lichenoid interface reaction pattern and the presence of a dominant infiltrate of histiocytes and plasma cells in the cellular infiltrate distinguishes secondary syphilis from PPE.
HIV-associated eosinophilic folliculitis
Clinical features
HIV-associated eosinophilic folliculitis (HIV-EF) is a distinctive, chronic pruritic skin eruption characterized by discrete, urticarial follicular papules scattered on the trunk, the head and neck, and the proximal extremities.1–5 Excoriation and crusting are frequent. Less commonly, small pustules are noted atop the papules.1 There may be significant clinical overlap with other HIV-related papular and/or follicular eruptions such as HIV-associated pruritic papular eruption (PPE) or suppurative folliculitis.3 Increased absolute or relative peripheral eosinophil counts and elevated serum IgE levels have been documented. HIV-EF is associated with advanced HIV disease and may be a manifestation of the immune restoration syndrome. Although the disease is encountered almost exclusively in adults, rare cases have been documented in infancy and childhood.6
Pathogenesis and histological features
The exact etiology and pathogenesis of HIV-EF remain unestablished.2,5,7 Fungal (Pityrosporum ovale), parasitic (Demodex folliculorum), and bacterial (Leptotrichia, Mycobacterium, Staphylococcus) organisms have been the focus of several theories, but these infective agents have been reported scarcely and inconsistently in association with HIV-EF, evoking a possible bystander role.1–5,7–13 A lipid-soluble chemotactic factor for eosinophils and neutrophils has been proposed as a causative factor. Follicular disruption and exposure of the sebaceous antigen are implicated in the stimulation of an inflammatory process involving lymphocytes, cytokines, and eosinophils.2–5,7–13 The presence of mast cells and evidence of their degranulation has implicated a role for mast cells in the evolution of HIV-EF.1
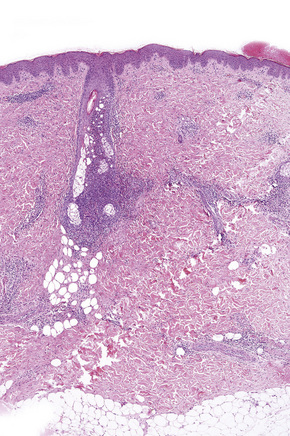
The histopathological features are those of follicular disruption and infiltration by eosinophils, with the surrounding dermis showing perivascular and interstitial infiltration by lymphocytes, eosinophils, and mast cells (Figs 19.16, 19.17). Occasionally, elaborated Demodex folliculorum mites are observed in the perifollicular dermis when intradermal rupture of an involved, distended follicle has occurred.3
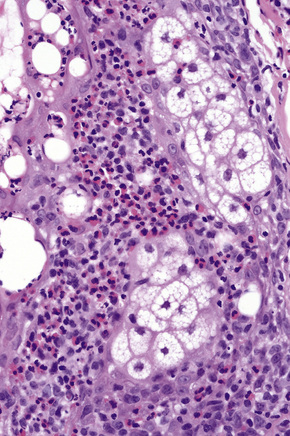
Differential diagnosis
The main histological differential diagnoses are eosinophilic pustular folliculitis (Ofuji disease) and PPE. Follicular eosinophil accumulation is common to both HIV-EF and Ofuji disease. The latter condition is more prevalent in the East and occurs in healthy individuals, with an increased peripheral leukocyte count, pruritus in < 50% of patients and coalescence of papulopustular plaques, with central clearing and postinflammatory hyperpigmentation. A similar histologic reaction pattern may sometimes be triggered by fungal infections of the hair shaft.14 PPE shares many of the clinical features of HIV-associated PPE. The follicular involvement by eosinophils, however, serves as the main distinguishing feature.1,3,15 To this end, examination of multiple serial sections is advised to assess the presence of folliculitis.3,16,17
HIV-associated vasculitic disorders
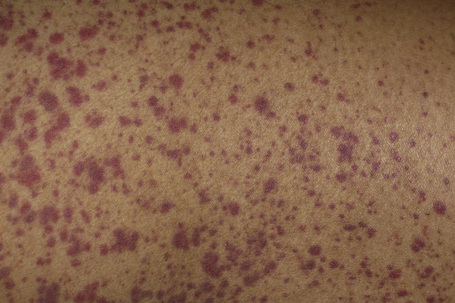
Excluding drug-related hypersensitivity reactions, vasculitic syndromes are not common in HIV-positive patients.1–4 These have been divided into four basic categories (Table 19.1).3 Category 1 diseases do not currently have a convincing association with HIV. Category 2 conditions are common because hypersensitivity reactions to drugs are common in HIV-positive patients since they consume a large number and range of drugs.1,5,6 Hypersensitivity reactions have been reported with almost all the HIV antiretroviral medications, typically involving small vessels, producing a lymphocytic or leukocytoclastic vasculitis (Fig. 19.18). Category 3 diseases involve a direct or indirect role for infectious agents, especially in advanced HIV disease. Category 4 vasculitides are largely inferential, based on the absence of a defined etiology, a disproportionate number of rare illnesses among HIV-positive patients, and unusual presentations that do not fit prior defined clinical diseases.1
Table 19.1 Vasculitides in HIV-infected patients1
| Category 1 | Probable incidental vasculitis syndromes Behçet’s syndrome Churg-Strauss syndrome Essential mixed cryoglobulinemia (hepatitis C) Henoch-Schönlein purpura Takayasu’s arteritis Temporal arteritis Wegener’s granulomatosis |
| Category 2 | Adverse drug reactions |
| Category 3 | Vasculitides with known infectious etiologies Cytomegalovirus (gastrointestinal tract, pulmonary, skin) Toxoplasma gondii (central nervous system) Pneumocystis jiroveci (pulmonary) Hepatitis B (polyarteritis nodosa) |
| Category 4 | Vasculitis syndromes probably facilitated by HIV pathogenesis Erythema elevatum diutinum Microscopic polyangiitis Nonhepatitis B (polyarteritis nodosa) Kawasaki-like syndromes Primary angiitis of the central nervous system Acute occlusion syndromes |
Erythema elevatum diutinum
Clinical features
Erythema elevatum diutinum (EED), a rare chronic disease of unknown etiology, is part of the spectrum of cutaneous leukocytoclastic vasculitis.1 The disease is characterized by symmetrical, persistent, and raised erythematous plaques and nodules over the extensor surfaces of the extremities. There is a predilection for involvement of skin overlying the elbow, knee, ankle, and interphalangeal joints.1–7 Bulla formation has been reported. Early lesions are soft and yellow and may contain petechiae or exhibit purpura. The duration of the disease is typified by a chronic course, varying between 1 and 39 years. The lesions of EED are usually asymptomatic, but may be pruritic or painful. Later lesions are doughy to firm and of red to purple color.
Pathogenesis and histological features
The leukocytoclastic vasculitis is the result of ongoing immune complex-mediated, neutrophil-induced, dermal small vessel damage. Antigen–antibody complexes from hepatitis B, C, and D viruses and HIV have been implicated.1
Histopathologically, the early lesions demonstrate leukocytoclastic vasculitis and capillary proliferation.5 This is followed by a fascicular proliferation of spindle cells and macrophages in the background of leukocytoclastic vasculitis.7 Late lesions demonstrate homogenization and fibrosis of the dermis. Serial sectioning may be required to identify foci of leukocytoclastic vasculitis. The fibrosis may assume an acellular, laminated appearance, with a parallel, wavy orientation of collagen with respect to the overlying epidermis. Capillaries are orientated vertically. Skin biopsy is essential to differentiate EED from a range of clinically similar HIV-associated lesions, including Kaposi’s sarcoma and bacillary angiomatosis. Stains to exclude infective causes for the fibrovascular and cellular alterations may be required.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


