3.2.1 Discovery
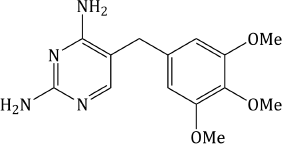
Trimethoprim (Figure 3.2.1) is an example of early rational drug design; it resulted from the work of George H. Hitchings and his group at Burroughs Wellcome in the USA during the 1940s, 1950s, and 1960s, when they were studying the cellular actions of purines and pyrimidines, with the rationale that interference in the processes associated with these biologically important heterocycles could result in therapeutic outcomes.
Using Lactobacillus casei as a representative organism, the group showed that metabolism of nucleic acids could be inhibited by selected purine and pyrimidine analogues and that this inhibition could be reversed by the addition of folic acid. In particular, 2,4-diaminopyrimidines were found to be competitive inhibitors of the folate cycle in L. casei. The synthesis of many substituted analogues of 2,4-diaminopyrimidine eventually led to the identification of trimethoprim as the optimal agent for the inhibition of this biochemical process in bacteria (Roth et al., 1962; Burchall and Hitchings, 1965). In addition, the extensive studies on L. casei provided much important information on biochemical cycles, which led to the identification of similarities and differences between species, and aided the subsequent development of selective therapeutic agents.
The Hitchings group was highly successful and developed many active agents, including: the prophylactic antimalarial agent pyrimethamine; the antileukaemic/immunosuppressant 6-mercaptopurine; the xanthine oxidase inhibitor allopurinol, used to treat gout; and, of course, the antibacterial agent trimethoprim (Hitchings, 1969; Elion, 1989). In fact, Hitchings, along with his colleague Gertrude B. Elion, shared the Nobel Prize for Physiology or Medicine in 1998 with a British pharmacologist, Sir James Black; their work which led to the awarding of the Nobel Prize has been recently reviewed (Raviña, 2011).
Strangely, although the in vitro and clinical results with trimethoprim were more than encouraging, it was several years after its discovery before it was adopted into regular clinical practice, despite further articles detailing its success across a range of infections (Cooper and Wald, 1964; Martin and Arnold, 1967; Darrell et al., 1968; Grüneberg and Kolbe, 1969).
3.2.2 Synthesis
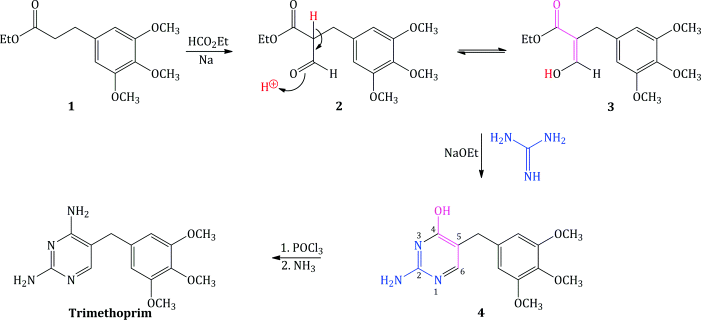
Trimethoprim was originally synthesised from the appropriate trimethoxy-substituted ethyl dihydrocinnamate 1 (Falco et al., 1951). Formylation of the dihydrocinnamate 1, with ethyl formate and sodium, gave the ethyl 3′,4′,5′-trimethoxybenzylmalonic semialdehyde 2 (Scheme 3.2.1), which tautomerises9 to its enol form 3. This mixture of tautomers was reacted with guanidine and sodium ethoxide to form 2-amino-4-hydroxy-5-(3′,4′,5′-trimethoxybenzyl)pyrimidine 4. Substitution of chlorine for the hydroxyl group was effected by phosphorus oxychloride (a reaction that is still commonly used for introducing the chloro group, which is a much better leaving group than hydroxyl) and, finally, amination of the 4-chloro compound using ammonia resulted in trimethoprim (Hitchings and Roth, 1959; Roth et al., 1962). The overall yield from the cinnamic acid precursor to the dihydrocinnamate 1 was poor (around 20%) and better syntheses were required for industrial-scale production.
Scheme 3.2.1 The original Hitchings synthesis of trimethoprim (Hitchings and Roth, 1959; Roth et al., 1962)
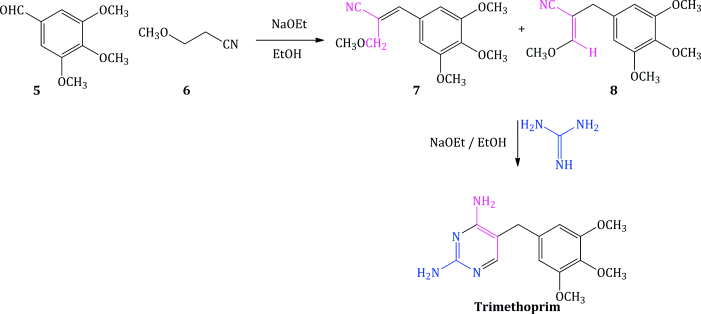
Since then, there have been several synthetic routes developed, some of which show significant yield improvements over the others, and the most common routes have been summarised (Vardanyan and Hruby, 2006). One route worth mentioning allows for the synthesis of a wide range of analogues, although again in moderate yield. It employs an appropriately substituted aldehyde 5, to which is added a 3-alkoxypropanenitrile 6, under strongly basic conditions, to provide the corresponding cinnamonitrile 7 (or its enol ether 8), followed by condensation with guanidine, as illustrated in Scheme 3.2.2 for trimethoprim (Stenbuck et al., 1963).
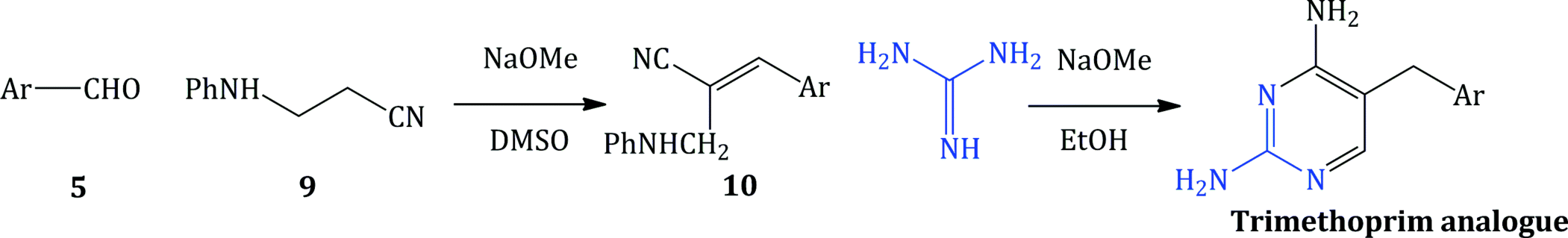
Many newer substituted 2,4-diaminopyrimidines, such as iclaprim (which you will meet in Subsection 3.2.9) (Jaeger et al., 2011), are accessed by a modification of this synthesis, which replaces the 3-alkoxypropionitrile 6 with an N-substituted 3-aminopropionitrile 9 and proceeds through the analogous β-cyano-N-substituted cinnamylamine intermediate 10, followed by reaction with guanidine to give the appropriately substituted 5-benzyl-2,4-diaminopyrimidines (analogues of trimethoprim), in moderate to good yields (Scheme 3.2.3) (Poe and Ruyle, 1981).
Using this method, numerous analogues of trimethoprim have been prepared and used to explore the requirements of binding in the enzyme active site, with particular emphasis on understanding the selectivity (see, for example, Roth and Aig, 1987; Roth et al., 1987; Davis et al., 1989; Selassie et al., 1998). We will consider some of these derivatives and their contribution to the knowledge of the mode of action in Subsection 3.2.4.
3.2.3 Bioavailability
The bioavailability of trimethoprim is almost 100%, with rapid absorption following oral administration and no impact of food upon the rate. It has a half-life of 10 hours, allowing twice-daily administration for acute infections and once-daily for its prophylactic action, although differences in the half-life were observed between adults and children when treated orally with trimethoprim (Hoppu and Arjomaa, 1984; Rylance et al., 1985). The volume of distribution is considerable at 71–136 L/kg, so that it is widely distributed throughout tissues, organs, and fluids. For example, it is well distributed into lymph tissue, cerebrospinal fluid, prostatic fluid, saliva, sinus secretions, and breast milk, readily achieving therapeutic concentrations (Brogden et al., 1982). Although trimethoprim is partly metabolised by demethylation and oxidation at the N1 and N3 positions (Sigel et al., 1973), the majority is excreted unchanged in the urine – one reason why it is commonly used to treat urinary infections (Kasanen et al., 1978; Libecco and Powell, 2004). As for the sulfonamides, the urinary excretion of trimethoprim is influenced by pH; if you look at the structure of trimethoprim, you will not be surprised to find that it is weakly basic, with a pKa of about 7.3. In blood (pH 7.2–7.4) then, you would expect it to be approximately 50% ionised and 50% unionised.10 A more acidic pH will increase ionisation (greater formation of N1H+) and so solubility, leading to greater excretion, while a higher pH will decrease ionisation and thus lead to lower excretion (Brogden et al., 1982). The clinical pharmacokinetics of trimethoprim and trimethoprim in combination with a sulfonamide have been reviewed (Patel and Welling, 1980; Friesen et al., 1981; Watson et al., 1988).
3.2.3.1 Bacterial Uptake of Trimethoprim
Similarly to the sulfonamides, trimethoprim is sufficiently small and lipophilic that it can enter bacterial cells by passive diffusion. Poor concentrations achieved in Gram negative bacteria have been linked to an active efflux pump.
3.2.4 Mode of Action and Selectivity
Through their many experiments, as discussed at the beginning of this chapter, Hitchings and coworkers learned that trimethoprim was interfering with the folic acid cycle, but the actual bacterial enzyme target was not isolated and characterised until 1958. Like the sulfonamides, such as sulfamethoxazole, trimethoprim acts upon the folic acid cycle (Scheme 3.2.4).
Scheme 3.2.4 Targets for inhibition of the folic acid cycle by trimethoprim and the sulfonamides (such as sulfamethoxazole)
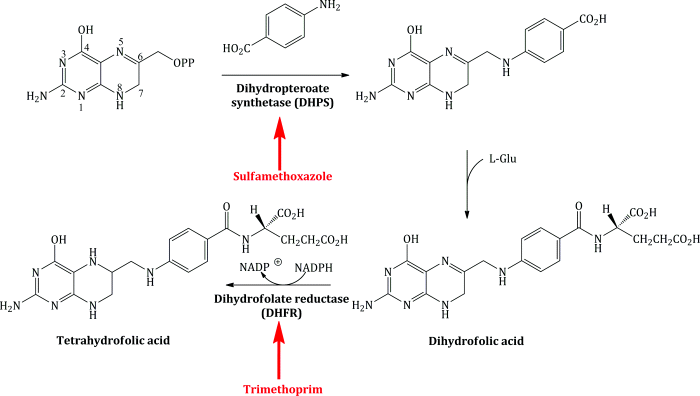
As Scheme 3.2.4 shows, trimethoprim acts upon the enzyme dihydrofolate reductase (DHFR), EC 1.5.1.3. Details about the enzyme itself can be accessed from one of the excellent Internet resource sites, such as BRENDA (www.brenda-enzymes.info) or the Kyoto Encyclopedia of Genes and Genomes (KEGG; www.genome.jp/kegg), from which the folic acid pathway is also available.11
Trimethoprim is a DHFR inhibitor and its bacteriostatic effect occurs through the disruption of the folate cycle and depletion of tetrahydrofolate (Quinlivan et al., 2000), which, as we saw in Subsection 1.1.3.2, is required as a co-factor in the biosynthesis of thymidine (Kasanen et al., 1978).
More than 200 crystal structures of dihydrofolate reductase are available on the RCSB protein crystal structure database (www.pdb.org), almost 100 of which are of DHFR from a range of bacterial species, either alone or in complex with an inhibitor.
Perhaps surprisingly for an enzyme that catalyses the same reaction across a wide range of species, there is only about 30% homology between the amino acid sequences of the DHFR from humans, chickens, mice, Escherichia coli, L. casei, and Plasmodium falciparum. When examined more closely using X-ray crystallography, however, it becomes apparent that the overall tertiary structure and 3D shape of the enzymes show much greater similarity, with relatively high conservation of the amino acids in and around the active sites of the mammalian and avian DHFR enzyme. The bacterial enzymes show similarity to one other but have key differences to the mammalian and avian forms (Selassie et al., 1998; Schweitzer et al., 1990).
If you look again at Scheme 3.2.4, you will see that dihydrofolate is reduced to tetrahydrofolate in the usual biochemical reaction catalysed by DHFR, using NADPH as a co-factor, which transfers a hydride (H−) to C6 (where the side chain is located) of dihydrofolate (Matthews et al., 1986). The pteridine ring of the substrate binds close to the NADPH in the active site, presenting the imine (N=C6) reduction site to the co-factor in order for hydride transfer to occur (Bolin et al., 1982). Protonation of N5, from a conserved water molecule located in the active site, follows, and this is assisted by Asp-27 in bacterial DHFR. Folic acid is also a substrate of DHFR (being reduced first to dihydrofolic acid, then tetrahydrofolic acid,12 although it is reduced at around a 10–1000 times slower rate than the reduction of dihydrofolic acid and requires two transfers of hydride from the co-factor) (Oefner et al., 1988). In order for a reversible competitive inhibitor to be successful without the formation of a covalent bond to the enzyme or co-factor, it needs to bind more strongly to the enzyme than the natural substrate, which is effectively blocked from the active site. How does trimethoprim do this in bacterial DHFR?
Like the natural substrate, trimethoprim and other bacterial DHFR inhibitors bind to the enzyme along with NADPH in a ternary complex, in which the reducing end of the co-factor is sited near to the pyrimidine ring of trimethoprim. Much work has gone into studying the binding of trimethoprim to bacterial DHFR and identifying the key interactions that facilitate tight binding to the active site. NMR spectroscopy and X-ray crystallography indicate that N1 is protonated in the bound state (Cocco et al., 1983; Matthews et al., 1985a), and X-ray crystallography also shows that the pyrimidine ring of trimethoprim locates in a deep cleft of the protein, with favourable non-covalent interactions helping to maintain its position. This binding mode enables ionic interactions between the protonated N1 and the carboxylate group of the conserved Asp-27 residue, and hydrogen-bonding interactions (Figure 3.2.2) between:
- N1 and Asp-27;
- 2-NH2 and Asp-27 and a conserved water molecule;
- the same conserved water molecule and Thr-113;
- 4-NH2 and Ile-5 and Ile-94.
Figure 3.2.2 Simplified illustration of hydrogen bonding between trimethoprim and the active site of E. coli DHFR. The DHFR residues are shown in blue, the conserved water molecule in pink, and the hydrogen bonds in red (adapted from Matthews et al., 1985a, 1985b; Champness et al., 1986.)
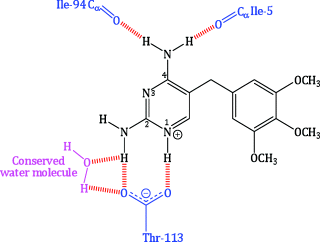
Several favourable van der Waals interactions between the pyrimidine and hydrophobic groups in the active site, involving the side chains of Ile-5, Ala-7, Leu-28, Phe-31, Ile-94, and the backbone of Ala-6, Ala-7, and Ile-94, contribute to the strong binding of trimethoprim to the DHFR from E. coli and other bacteria. The trimethoxybenzyl group is orientated at an angle to the pyrimidine ring, extended towards the cleft opening, with van der Waals interactions to Leu-28, Phe-31, and Met-20 enhancing the binding (Matthews et al., 1985a, 1985b; Champness et al., 1986). Further interactions of the three methoxy groups are possible with the proximal amino acid residues and with the hydrophilic exterior, into which the 4-OMe group extends (Selassie et al., 1998). You can imagine how this complex balance of intermolecular interactions, holding the structures tightly together, along with complementarity of size and shape, results in the potent inhibitory effect of trimethoprim upon bacterial DHFR.
The analysis of the structures of potential inhibitors and their activity against bacterial DHFR enzymes (in comparison to DHFR from other species) has been used to aid the design of improved inhibitors (Roth et al., 1988; Johnson et al., 1989; Rauckman et al., 1989; Chan and Roth, 1991; Ohemeng and Roth, 1991; Selassie et al., 1998), while the structures of various DHFR enzymes have been compared, and their binding to trimethoprim analysed (Schweitzer et al., 1990).
The selectivity of trimethoprim for the bacterial enzyme over the mammalian enzyme has been appreciated for over 50 years (Hitchings, 1969, 1988; Elion, 1989). The mammalian and avian DHFR enzymes were isolated in 1958 (Osborn and Huennekens, 1958; Peters and Greenberg, 1958), followed by a bacterial form (Blakley and McDougall, 1961), but it was some years before it was confirmed that trimethoprim, pyrimethamine, and methotrexate all act as inhibitors of the same enzyme, with the selective inhibition of a particular DHFR enzyme dictating their pharmaceutical use (Burchall and Hitchings, 1965). The structures of these three classic DHFR inhibitors, all of which are still used clinically, are shown in Figure 3.2.3, alongside the natural substrate, dihydrofolic acid, with which they compete for the DHFR active site. You can see that the 2,4-aminopyrimidine group of these inhibitors resembles part of the pteridine nucleus of dihydrofolate, while the differently substituted benzene rings make a major contribution to the selective binding to the various DHFR enzymes.
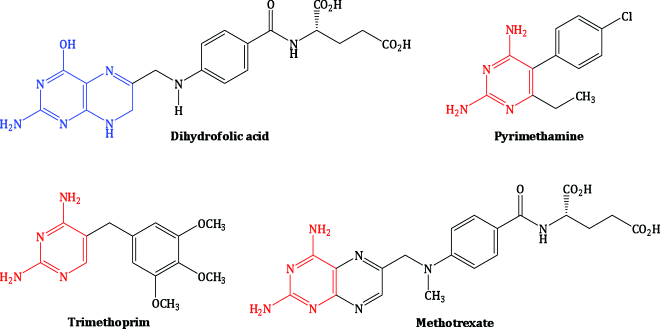
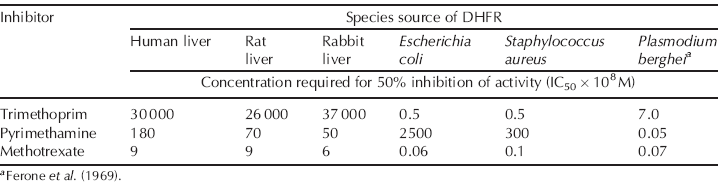
If you consider the data in Table 3.2.1 (Burchall and Hitchings, 1965; Ferone et al., 1969), you can see that trimethoprim inhibits the bacterial enzyme with great selectivity over mammalian enzymes, while pyrimethamine is selective for the protozoal enzyme. Although methotrexate has good inhibitory activity against the bacterial, protozoal, and mammalian DHFR enzymes, and you would be forgiven for thinking that it could have wide antimicrobial application, it has poor uptake into bacterial cells, which lack the membrane-bound folate transporter used by methotrexate to enter mammalian cells, so it has very little antibacterial activity in vivo (Allegra et al., 1987).
Table 3.2.1 Selective inhibition of dihydrofolate reductase from varying sources by trimethoprim, pyrimethamine, and methotrexate (Burchall and Hitchings, 1965).
A detailed analysis of the structure–activity relationship for DHFR inhibitors was made by Hansch13 and coworkers in the 1980s (Blaney et al., 1984), when more than 1500 DHFR inhibitors were analysed for their structural and electronic properties, to arrive at a number of conclusions:
- Substitution of the 4-hydroxy group in dihydrofolate (corresponding to the 4-oxo group in folate) by the 4-NH2 group in 2,4-diaminopteridines and 2,4-diaminopyrimidines is the most important feature for the switch from agonist to antagonist activity.
- Binding of NADPH enhances inhibitor binding to the DHFR active site, and increased hydrophobicity in the inhibitor increases binding to the bacterial enzyme.
- Bacterial DHFR enzymes are the most inhibited by 5-benzyl-2,4-diaminopyrimidines and the greatest selectivity is achieved by the 3′,4′,5′-trisubstituted derivatives.
- In contrast to the mammalian enzymes, the size of the substituents on the benzyl ring is crucial for bacterial DHFR inhibition (Blaney et al., 1984).
Since then, there has been a lot of effort directed at delineating the requirements for potent bacterial inhibition, while retaining the excellent selectivity displayed by trimethoprim. You would expect that the similarity of the active sites of DHFR across the species would make it difficult to design selectivity for one over another, but X-ray crystallography has shown that there are two possible binding sites for the trimethoxybenzyl in the active-site cleft: a lower, deeper cleft, and another sited higher. In non-bacterial DHFR, both of these active-site clefts are slightly wider, by a mere 1.5–2.0 Å, than that of the bacterial enzyme, possibly due to a three-amino-acid insertion found in all vertebrate DHFR enzymes near the active site (Matthews et al., 1985b). These differences mean that trimethoprim cannot bind into both lower and upper binding sites, occupying the lower site alone, and lead to a conformational change in trimethoprim when it binds to mammalian DHFR, causing rotation of the trimethoxybenzyl group into a less favourable conformation for trimethoprim, as well as a close contact with part of the active site, disturbing one of the pyrimidine hydrogen bonds (4-NH2 to Ile-94) (Matthews et al., 1985b; Schweitzer et al., 1990). In combination, these differences in the binding of trimethoprim to bacterial and mammalian DHFR lead to the selectivity observed.
Considering all of the information above, isn’t it remarkable that Hitchings and his coworkers achieved such impressive selectivity for the DHFR of different species with little detailed knowledge of the structural requirements? Indeed, much effort since then (reviewed in Then, 2004; da Cunha et al., 2005; Kompis et al., 2005; Chan and Anderson, 2006; Hawser et al., 2006) has focussed on the design of new selective DHFR inhibitors, with only a few notable successes, despite the addition of X-ray crystal structure information (Baccanari and Kuyper, 1993; Beierlein et al., 2008), molecular modelling (Roth, 1986; Ohemeng and Roth, 1991), and the use of NMR to probe the mechanism and structural requirements of binding (Cocco et al., 1983; Roberts 1991; Kovalevskaya et al., 2005, 2007). A considerable amount of research continues into improving DHFR activity and selectivity across species, such as Bacillus anthrax (Beierlein et al., 2008; Bourne et al., 2009), Pneumocytis carinii, Toxoplasma gondii, and Mycobacterium avium (these last three are of particular clinical relevance as opportunistic infections in AIDS patients) (Forsch et al., 2004), and in overcoming resistance (Dale et al., 1997), which is discussed in more detail in Subsection 3.2.5.
Another discovery of great clinical relevance was made in 1947, when the synergy between a pterin (based on the structure of folic acid) and sulfonamides was observed (Daniel and Norris, 1947); others followed with investigations into the use of early diaminopyrimidines with sulfonamides (Elion et al., 1954; Hitchings and Burchall, 1965), and eventually the antibacterial effects of trimethoprim were combined with a range of different sulfonamides in order to try to identify the optimal combination and doses (Bushby and Hitchings, 1968). Although the synergy is generally believed to be due to sulfonamide and trimethoprim inhibition of the same pathway at two different points, it has also been suggested that both sulfonamide and trimethoprim can bind together to the active site of DHFR when present in sub-therapeutic concentrations, raising the possibility that at least some sulfonamides can act at two stages of the folate pathway (Lacey, 1982). The combined use of trimethoprim and sulfonamides has been under discussion almost since it was first used, as there is some doubt that the synergy is achieved routinely in clinical cases (Watson et al., 1988) and concern that the side effects may outweigh the benefits (Brumfitt and Hamilton-Miller, 1994; Howe and Spencer, 1996).
How did clinicians know which of the many sulfonamides to combine with trimethoprim? A great deal of effort went into evaluating the right combination, as the components must have similar clinical pharmacokinetics in order to achieve the required therapeutic concentrations simultaneously. Several factors were investigated, including the penetration of the drugs into the active site, their volumes of distribution, and their elimination half-lives (Ortengren et al., 1979; Bernstein, 1982). The two sulfonamides with the closest clinical pharmacokinetics to those of trimethoprim were found to be sulfadiazine and sulfamethoxazole (Vree et al., 1978; Patel and Welling, 1980; Watson et al., 1988). Both have been used in combination therapy with trimethoprim, and the combinations studied for their clinical pharmacokinetics and outcomes across a range of infections and complications (Bergan et al., 1986; Chin et al., 1995; Klepser et al., 1996; Pokrajac et al., 1998; Nyleen, et al., 1999; Mistri et al., 2010). Although both possible combinations are still available in some parts of the world, as co-trimazine (trimethoprim with sulfadiazine) and co-trimoxazole (trimethoprim with sulfamethoxazole), the former is not licensed in all countries and the latter is the more commonly used. Despite the fact that considerable effort was directed at matching the pharmacokinetics of the two components, it is necessary to administer sulfamethoxazole and trimethoprim in the ratio 5 : 1 in order to achieve appropriate concentrations in vivo. The differences in the rates of absorption and their abilities to distribute throughout various body compartments have also led to controversy about the combined use of sulfamethoxazole and trimethoprim (O’Grady, 1975; Watson et al., 1988).
The observation of synergy was of great clinical importance, because it allowed both agents to be used at lower doses than is required in monotherapy, and the combined effect is that of a bactericidal agent against many different pathogenic bacteria, rather than the bacteriostatic effect of each agent separately. Another benefit is the lower risk of resistance developing due to a mutation in the enzyme targets, as the probability of a resistance-causing mutation arising in both enzymes (DHPS and DHFR) concurrently is very small (Bushby, 1975; Lacey, 1982).
3.2.5 Bacterial Resistance
Within a few years of the clinical use of trimethoprim, resistance was reported across a range of bacteria (Datta and Hedges, 1972; Fleming et al., 1972; May and Davies, 1972), although for many years it was restricted to Gram negative species. The incidence of resistance has continued to increase but has not yet resulted in the redundancy of trimethoprim as an antibacterial agent (Amyes and Towner, 1990; Huovinen, 2001; Roberts, 2002).
Low-level resistance to trimethoprim has been observed, and is linked to spontaneous chromosomal mutations, leading to decreased uptake or alterations to the target enzyme, DHFR (Goldstein, 1977). It is the high-level resistance, however, that causes clinical concerns, leading to increased resistance in the majority of regions and a disturbingly high incidence of resistant bacteria in some countries (Murray et al., 1985; Mathai et al., 2004; Guneysel et al., 2009). In Gram negative bacteria, high-level resistance to trimethoprim is primarily due to the transmission of plasmids encoding a resistant form of DHFR (Amyes and Towner, 1990). In 1987, nine different plasmid-encoded trimethoprim-resistant DHFR enzymes and/or associated genes were known (Huovinen, 1987); by 2010, 30 different trimethoprim-resistant forms of the gene encoding DHFR (named dfr) had been described, most of which were associated with integrons (Brolund et al., 2010). In Gram positive bacteria, resistance is largely due to chromosomal mutations, which lead to point mutations of amino acids in the DHFR enzyme; plasmid-borne dfr genes can also be observed in these species and generally give rise to similar resistant enzymes with point mutations (Huovinen, 1987; Bourne et al., 2009).
The effect of mutation in a dfr gene can be seen, for example, in Staphylococcus aureus, in which Phe-98 is replaced by Tyr in the enzyme active site of S1 DHFR, a trimethoprim-resistant enzyme encoded by plasmid dfrA, located on transposon Tn4003, leading to high-level resistance to trimethoprim. The same mutation is seen in the chromosomally encoded DHFR and leads to intermediate-level resistance (Dale et al., 1997). This same mutation was observed across a range of trimethoprim-resistant S. aureus clinical isolates, sometimes alongside other mutations in which His-30 was replaced by Asn (or His-149 was replaced by Arg), and may cause the loss of a hydrogen bond between trimethoprim and a conserved water molecule in the active site (Frey et al., 2010). Mutations due to a variety of active-site amino acids have been observed in trimethoprim-resistant bacteria (DeGroot et al., 1991; Adrian and Klugman, 1997; Coque et al., 1999; Woodford, 2005).
3.2.6 Clinical Applications
3.2.6.1 Spectrum of Activity
Trimethoprim has a range of activity against UTI-causing pathogens, such as Gram negative bacteria (including E. coli, Proteus mirabilis, and Klebsiella pneumoniae) and some Gram positive bacteria, including Staphylococcus saprophyticus. Unfortunately, trimethoprim shows poor activity against Pseudomonas aeruginosa and anaerobic bacteria (Rosenblatt and Stewart, 1974; Then and Angehrn, 1979; Köhler et al., 1996).
3.2.6.2 Urinary Tract Infections (UTIs)
The most common therapeutic use of trimethoprim is in the treatment of lower urinary tract infections (LUTIs) in women. LUTIs are very common, with symptoms including dysuria (pain when urinating) and polyuria (passage of large volumes of urine). In the case of upper urinary tract infections (UUTIs), symptoms are often suggestive of pyelonephritis (bacterial infection of the kidney) and can include fever and back pain. UUTIs are considered more serious than LUTIs, as they can be accompanied by bacteraemia, which is a potentially life-threatening condition.
E. coli is estimated to cause around 80% of UTIs, with other organisms such as S. saprophyticus and P. mirabilis also playing a role in some cases. Generally, empirical14 antibacterial treatment is used to treat uncomplicated UTIs; for example, to treat a LUTI in a non-pregnant woman, a 3-day course of trimethoprim is recommended. In cases where the UTI is resistant to trimethoprim (or the symptoms suggest an UUTI), a quinolone (such as ciprofloxacin) is sometimes used.
Three-day courses of antibiotic therapy are recommended because the evidence presented in a Cochrane systematic review suggests that this duration of antibiotic therapy is similar to 5–10 days in achieving symptomatic cures for uncomplicated UTIs in women (Milo et al., 2005). In addition, the 3-day course of antibiotic therapy offers the advantage of causing fewer adverse effects and decreases antibiotic exposure, thereby minimising the risk of developing resistance.
If a woman presents with symptoms of a LUTI and pregnancy is suspected, a pregnancy test should be done, as trimethoprim is not recommended due to its possible teratogenic risk (because of its antifolate effects). Although several studies have failed to demonstrate the increased risk of foetal abnormalities associated with trimethoprim use (Brumfitt and Pursell, 1972, 1973), others have shown that trimethoprim can cause birth defects in animals (Briggs et al., 2008). Trimethoprim should thus be avoided in pregnancy, unless the potential therapeutic benefit outweighs the possible risks.15 If pregnancy is confirmed, and the patient has a LUTI, an alternative agent, such as amoxicillin, should be prescribed.
The use of antibacterial agents in the treatment of UTIs highlights an interesting point concerning the prescribing of antibacterial drugs: if a patient presents with signs of an acute UTI, some doctors will prescribe trimethoprim without taking a urine sample. Some doctors, however, will take a urine sample for bacterial culture and sensitivity testing prior to starting trimethoprim therapy. If the culture from the urine sample later confirms the presence of a pathogen that is resistant to trimethoprim, the patient will be quickly contacted and their treatment will be altered accordingly. This latter method is preferred, as the threat of antibacterial resistance will be decreased by minimising the inappropriate prescribing of antibacterial drugs.
In cases where a patient is suffering from recurrent UTIs (typically more than three episodes in 12 months), the use of prophylactic trimethoprim should be considered to reduce the number of recurrences. When used for UTI prophylaxis, trimethoprim is typically given as a single dose at night and is taken at a lower dose than that used to treat acute infections.
3.2.6.3 Miscellaneous
Trimethoprim can also be used to treat acne vulgaris. Generally, systemic antibiotics are used in the management of acne when topical treatment has either been ineffective, not tolerated, or the acne is predominately inflammatory. Trimethoprim, at a dose of 300 mg twice daily (which is higher than that used for treatment of acute UTIs), is effective in the management of acne vulgaris (Bottomley and Cunliffe, 1993). However, as the long-term use of trimethoprim can be associated with severe adverse effects (see Subsection 3.2.7), it is only regarded as a third-line treatment. Consequently, for this indication, trimethoprim should only be initiated by specialists for the management of acne that is resistant to other antibacterial agents.
Trimethoprim also has a role in the treatment of acute and chronic prostatitis (inflammation of the prostate gland). Acute prostatitis is an uncommon complication of UTIs; it is considered a medical emergency and is managed using empirical antibiotic therapy. In contrast, however, chronic prostatitis often presents with a history of recurrent UTIs and, interestingly, it is thought that the bacteria that cause the UTIs (such as E. coli) also cause the chronic prostatitis. The quinolones are generally used first-line to treat both of these disorders, but if this is not suitable (for example, if the patient is taking an interacting medication, such as theophylline), trimethoprim may be used. The quinolones are preferred over trimethoprim as they have a broader spectrum of activity against the common urinary pathogens, but if trimethoprim is indicated for either acute or chronic prostatitis it should be taken twice daily for 28 days.
3.2.7 Adverse Drug Reactions
Until recently trimethoprim was considered to be a relatively safe drug – with a similar structure to the potassium-sparing diuretic, amiloride, it is rapidly excreted and has been reported to cause hyperkalaemia in some patients (this can be more significant if a patient is already taking a drug which elevates potassium levels, such as, say, an ACE inhibitor). Relatively recent publications have, however, demonstrated that trimethoprim is associated with skin reactions, toxic epidermal necrolysis, and neutropenic reactions (Das et al., 1988; Hawkins et al., 1993).
3.2.8 Drug Interactions
Trimethoprim is primarily excreted unchanged in the renal tubule; however, approximately 20% of the drug is subject to drug metabolism by the CYP 450 system (Gleckman et al., 1981), so the half-life of trimethoprim can be extended by up to 50% in patients with a degree of hepatic failure (Rieder and Schwartz, 1975).
Trimethoprim has also been shown to inhibit CYP 2C8 (Wen et al., 2002), a recently identified isoenzyme, which it is expected has both arachidonic acid and retinoic acid as natural substrates. Other drug substrates known to be metabolised, wholly or in part, by CYP 2C8 are zopiclone, the ‘glitazones’, repaglinide, amodiaquine, amiodarone, chloroquine, dapsone, loperamide, retinoic acid derivatives, and paclitaxel (Lebovitz, 2000; Ohyama et al., 2000; Cresteil et al., 2002; Niemi et al., 2003a; Gil and Gil Berglund, 2007; Scheen, 2007). As trimethoprim inhibits CYP 2C8, one would expect to see a significant number of drug interactions, but so far, clinically at least, this is not the case.
3.2.8.1 Pioglitazone
An in vitro study predicted the possible interaction of trimethoprim with pioglitazone via inhibition of CYP 2C8*3 (Tornio et al., 2008), while the plasma levels of the antidiabetic drug repaglinide were raised by the concomitant use of trimethoprim (Niemi et al., 2003b).
3.2.8.2 Methotrexate
The concomitant use of either co-trimoxazole or trimethoprim with methotrexate has been associated with pancytopaenia (Groenendal and Rampen, 1990; Steur and Gumpel, 1998).
3.2.8.3 Lamivudine
The co-administration of trimethoprim increases the bioavailability of lamivudine by 40%, raising the possibility that trimethoprim could be exploited in HIV treatment regimens (Hudson and Nash, 1996).
3.2.8.4 Memantine
A clinical case report indicated that the co-administration of memantine (used in the treatment of Alzheimer’s disease) and trimethoprim caused myoclonus and exacerbated delirium in a 74-year-old patient (Moellentin et al., 2008). The structure of memantine resembles that of amantidine, which has also been shown to interact with trimethoprim (Speeg et al., 1989).
3.2.8.5 Digoxin
One report has demonstrated an up to 22% increase in serum digoxin levels when co-administered with trimethoprim (Petersen et al., 1985), and it has been suggested that serum digoxin levels are raised due to the decreased renal excretion of the drug and not due to extrarenal drug clearance.
3.2.9 Recent Developments
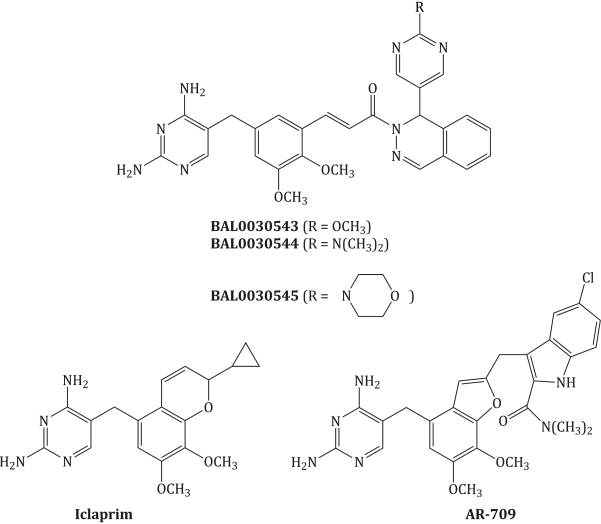
The exploitation of selectivity towards bacterial DHFR enzymes is still a target for rational drug design (Hawser et al., 2006) and it is hoped that new DHFR inhibitors, such as those shown in Figure 3.2.4 and Table 3.2.2, will exhibit improved bactericidal activity, extend the spectrum of antibacterial activity, and overcome the problems associated with trimethoprim resistance.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


