- Bacteria can be classified according to their staining by the Gram stain (Gram positive, Gram negative, and mycobacteria) and their shape.
- Most bacterial (prokaryotic) cells differ from mammalian (eukaryotic) cells in that they have a cell wall and cell membrane, have no nucleus or organelles, and have different biochemistry.
- Bacteria can be identified by microscopy, or by using chromogenic (or fluorogenic) media or molecular diagnostic methods (e.g. real-time polymerase chain reaction (PCR)).
- Bacterial resistance to an antibacterial agent can occur as the result of alterations to a target enzyme or protein, alterations to the drug structure, and alterations to an efflux pump or porin.
- Antibiotic stewardship programmes are designed to optimise antimicrobial prescribing in order to improve individual patient care and slow the spread of antimicrobial resistance.
1.1.1 Classification
There are two basic cell types: prokaryotes and eukaryotes, with prokaryotes predating the more complex eukaryotes on earth by billions of years. Bacteria are prokaryotes, while plants, animals, and fungi (including yeasts) are eukaryotes. For our purposes in the remainder of this book, we will further subdivide bacteria into Gram positive, Gram negative, and mycobacteria (we will discuss prokaryotic cell shapes a little later).
As you are probably already aware, we can use the Gram stain to distinguish between groups of bacteria, with Gram positive being stained dark purple or violet when treated with Gentian violet then iodine/potassium iodide (Figures 1.1.1 and 1.1.2). Gram negative bacteria do not retain the dark purple stain, but can be visualised by a counterstain (usually eosin or fuschin, both of which are red), which does not affect the Gram positive cells. Mycobacteria do not retain either the Gram stain or the counterstain and so must be visualised using other staining methods. Hans Christian Joachim Gram developed this staining technique in 1884, while trying to develop a new method for the visualisation of bacteria in the sputum of patients with pneumonia, but the mechanism of staining, and how it is related to the nature of the cell envelopes in these different classes of bacteria, is still unclear.
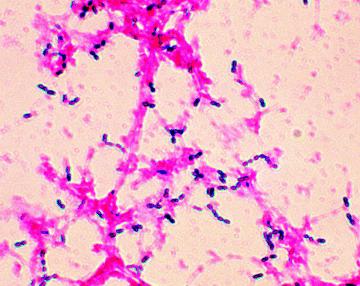
Figure 1.1.2 Example of a Gram stain showing Gram positive (Streptococcus pneumoniae) and Gram negative bacteria (Image courtesy of Public Health Image Library, Image ID 2896, Online, [http://phil.cdc.gov/phil/home.asp, last accessed 26th March 2012].)
Some of the Gram positive and Gram negative bacteria, as well as some mycobacteria, which we shall encounter throughout this book, are listed in Table 1.1.1.
Table 1.1.1 Examples of Gram positive and Gram negative bacteria, and mycobacteria.
| Gram positive | Gram negative | Mycobacteria |
| Bacillus subtilis | Burkholderia cenocepacia | Mycobacterium africanum |
| Enterococcus faecalis | Citrobacter freundii | Mycobacterium avium complex (MAC) |
| Enterococcus faecium | Enterobacter cloacae | Mycobacterium bovis |
| Staphylococcus epidermis | Escherichia coli | Mycobacterium leprae |
| Staphylococcus aureus | Morganella morganii | Mycobacterium tuberculosis |
| Meticillin-resistant Staphylococcus aureus (MRSA) | Pseudomonas aeruginosa | |
| Streptococcus pyogenes | Salmonella typhimurium | |
| Listeria monocytogenes | Yersinia enterocolitica |
1.1.2 Structure
The ultimate aim of all antibacterial drugs is selective toxicity – the killing of pathogenic1 bacteria (bactericidal agents) or the inhibition of their growth and multiplication (bacteriostatic agents), without affecting the cells of the host. In order to understand how antibacterial agents can achieve this desired selectivity, we must first understand the differences between bacterial (prokaryote) and mammalian (eukaryote) cells.
The name ‘prokaryote’ means ‘pre-nucleus’, while eukaryote cells possess a true nucleus, so one of the major differences between bacterial (prokaryotic) and mammalian (eukaryotic) cells is the presence of a defined nucleus (containing the genetic information) in mammalian cells, and the absence of such a nucleus in bacterial cells. Except for ribosomes, prokaryotic cells also lack the other cytoplasmic organelles which are present in eukaryotic cells, with the function of these organelles usually being performed at the bacterial cell membrane.
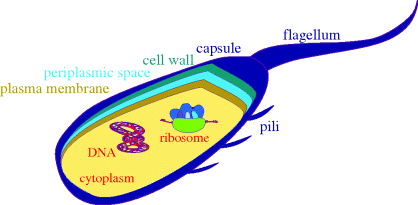
A schematic diagram of a bacterial cell is given in Figure 1.1.3, showing the main features of the cells and the main targets for antibacterial agents. As eukaryotic cells are much more complex, we will not include a schematic diagram for them here, and will simply list the major differences between the two basic cell types:
- Bacteria have a cell wall and plasma membrane (the cell wall protects the bacteria from differences in osmotic pressure and prevents swelling and bursting due to the flow of water into the cell, which would occur as a result of the high intracellular salt concentration). The plasma membrane surrounds the cytoplasm and between it and the cell wall is the periplasmic space. Surrounding the cell wall, there is often a capsule (there is also an outer membrane layer in Gram negative bacteria). Mammalian eukaryotic cells only have a cell membrane, whereas the eukaryotic cells of plants and fungi also have cell walls.
- Bacterial cells do not have defined nuclei (in bacteria the DNA is present as a circular double-stranded coil in a region called the ‘nucleoid’, as well as in circular DNA plasmids), are relatively simple, and do not contain organelles, whereas eukaryotic cells have nuclei containing the genetic information, are complex, and contain organelles,2 such as lysosomes.
- The biochemistry of bacterial cells is very different to that of eukaryotic cells. For example, bacteria synthesise their own folic acid (vitamin B9), which is used in the generation of the enzyme co-factors required in the biosynthesis of the DNA bases, while mammalian cells are incapable of folic acid synthesis and mammals must acquire this vitamin from their diet.
Figure 1.1.3 Simplified representation of a prokaryotic cell, showing a cross-section through the layers surrounding the cytoplasm and some of the potential targets for antibacterial agents
Whenever we discuss the mode of action of a drug, we will be focussing on the basis of any selectivity. As you will see from the section headings, we have classified antibacterial agents into those which target DNA (Section 2), metabolic processes (Section 3), protein synthesis (Section 4), and cell-wall synthesis (Section 5). In some cases, the reasons for antibacterial selectivity are obvious, for example mammalian eukaryotic cells do not have a peptidoglycan-based cell wall, so the agents we will discuss in Section 5 (which target bacterial cell-wall synthesis) should have no effect on mammalian cells. In other cases, however, the basis for selectivity is not as obvious, for example agents targeting protein synthesis act upon a process which is common to both prokaryotic and eukaryotic cells, so that in these cases selective toxicity towards the bacterial cells must be the result of a more subtle difference between the ribosomal processes in these cells.
We will now look at these antibacterial targets in detail, in preparation for our in-depth study of the modes of action of antibacterial agents and bacterial resistance in the remaining sections.
1.1.3 Antibacterial targets
1.1.3.1 DNA Replication
DNA replication is a complex process, during which the two strands of the double helix separate and each strand acts as a template for the synthesis of complementary DNA strands. This process occurs at multiple, specific locations (origins) along the DNA strand, with each region of new DNA synthesis involving many proteins (shown in italics below), which catalyse the individual steps involved in this process (Figure 1.1.4):
- The separation of the two strands at the origin to give a replication fork (DNA helicase).
- The synthesis and binding of a short primer DNA strand (DNA primase).
- DNA synthesis, in which the base (A, T, C, or G) that is complementary to that in the primer sequence is added to the growing chain, as its triphosphate; this process is continued along the template strand, with the new base always being added to the 3′-end of the growing chain (DNA polymerase) in the leading strand.
- The meeting and termination of replication forks.
- The proofreading and error-checking process to ensure the new DNA strand’s fidelity; that is, that this strand (red) is exactly complementary to the template (black) strand (DNA polymerase and endonucleases).
Figure 1.1.4 DNA replication fork (adapted from http://commons.wikimedia.org/wiki/File:DNA_replication_en.svg, last accessed 7 March 2012.)
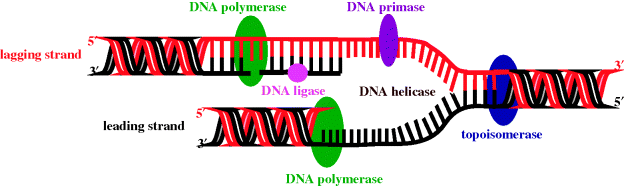
Due to the antiparallel nature of DNA, synthesis of the strand that is complementary (black) to the lagging strand (red) must occur in the opposite direction, and this is more complex than the process which takes place in the leading strand.
DNA helicase is the enzyme which separates the DNA strands and in so doing, as a result of the right-handed helical nature of DNA, produces positive supercoils (knots) ahead of the replication site. In order for DNA replication to proceed, these supercoils must be removed by enzymes (known as topoisomerases) relaxing the chain. By catalysing the formation of negative supercoils, through the cutting of the DNA chain(s) and the passing of one strand through the other, these enzymes remove the positive supercoils and give a tension-free DNA double helix so that the replication process can continue. Type I topoisomerases relax DNA by cutting one of the DNA strands, while, you’ve guessed it, type II cut both strands (Champoux, 2001). In Section 2.1 we will look at a class of drugs which target the topoisomerases: the quinolone antibacterials, which, as DNA replication is obviously common to both prokaryotes and eukaryotes, must act on some difference in the DNA relaxation process between these cells.
1.1.3.2 Metabolic Processes (Folic Acid Synthesis)
As mentioned above, metabolic processes represent a key difference between prokaryotic and eukaryotic cells and an example of this is illustrated by the fact that bacteria require para-aminobenzoic acid (PABA), an essential metabolite, for the synthesis of folic acid. Bacteria lack the protein required for folate uptake from their environment, whereas folic acid is an essential metabolite for mammals (as it cannot be synthesised by mammalian cells and must therefore be obtained from the mammalian diet). Folic acid is indirectly involved in DNA synthesis, as the enzyme co-factors which are required for the synthesis of the purine and pyrimidine bases of DNA are derivatives of folic acid. If the synthesis of folic acid is inhibited, the cellular supply of these co-factors will be diminished and DNA synthesis will be prevented.
Bacterial synthesis of folic acid (actually dihydrofolic acid3) involves a number of steps, with the key steps shown in Schemes 1.1.1 and 1.1.2. A nucleophilic substitution is initially involved, in which the free amino group of PABA substitutes for the pyrophosphate group (OPP) introduced on to 6-hydroxymethylpterin by the enzyme 6-hydroxymethylpterinpyrophosphokinase (PPPK). In the next step, amide formation takes place between the free amino group of L-glutamic acid and the carboxylic acid group derived from PABA (Achari et al., 1997).
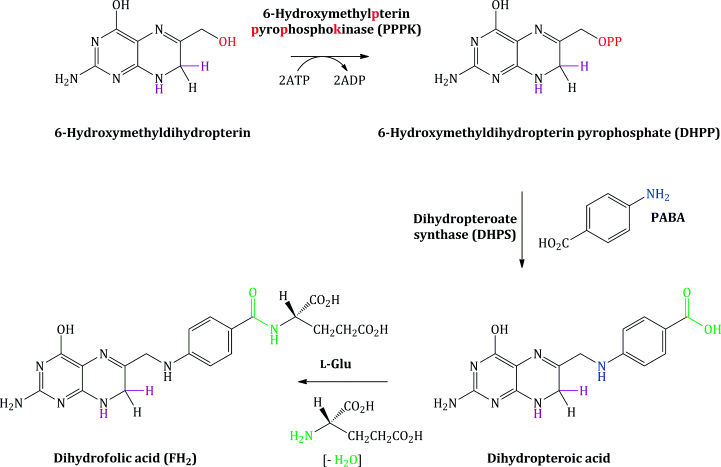
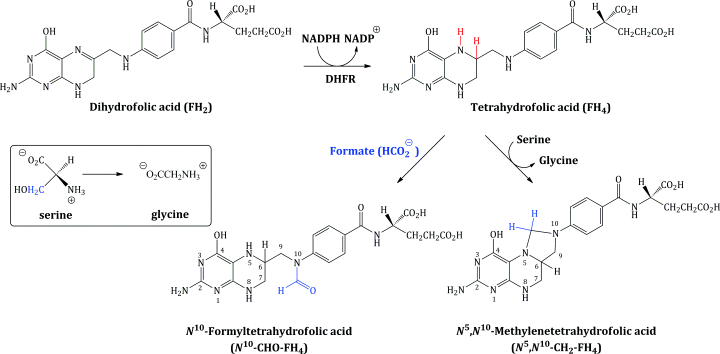
Dihydrofolic acid (FH2) is further reduced to tetrahydrofolic acid (FH4), a step which is catalysed by the enzyme dihydrofolate reductase (DHFR), and FH4 is then converted into the enzyme co-factors N5,N10-methylenetetrahydrofolic acid (N5,N10-CH2-FH4) and N10-formyltetrahydrofolic acid (N10-CHO-FH4) (Scheme 1.1.2).
The tetrahydrofolate enzyme co-factors are the donors of one-carbon fragments in the biosynthesis of the DNA bases. Crucially, each time these co-factors donate a C-1 fragment, they are converted back to dihydrofolic acid, which, in an efficient cell cycle, is reduced to FH4, from which the co-factors are regenerated. For example, in the biosynthesis of deoxythymidine monophosphate (from deoxyuridine monophosphate), the enzyme thymidylate synthetase utilises N5,N10-CH2-FH4 as the source of the methyl group introduced on to the pyrimidine ring (Scheme 1.1.3).
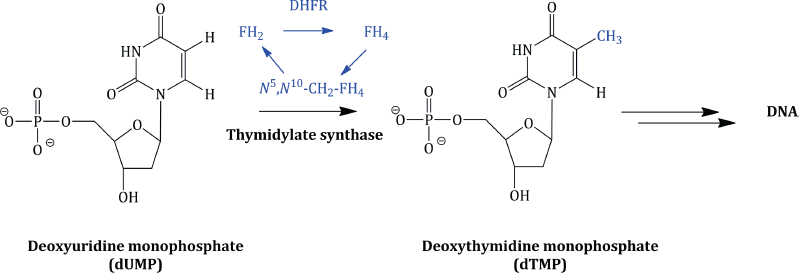
Similarly, N10-CHO-FH4 serves as the source of a formyl group in the biosynthesis of the purines and, onceagain, is converted to dihydrofolic acid (which must be converted to tetrahydrofolic acid and then N10-CHO-FH4, again in a cyclic process).
Cells which are proliferating thus need to continually regenerate these enzyme co-factors due to their increased requirement for the DNA bases. If a drug interferes with any step in the formation of these co-factors then their cellular levels will be depleted and DNA replication, and so cell proliferation, will be halted. In Section 3 we will look more closely at drugs which target these processes: the sulfonamides (which interfere with dihydrofolic acid synthesis) and trimethoprim (a DHFR inhibitor).
1.1.3.3 Protein Synthesis
Protein synthesis, like DNA replication, is a truly awe-inspiring process, involving:
- Transcription – the transfer of the genetic information from DNA to messenger RNA (mRNA).
- Translation – mRNA carries the genetic code to the cytoplasm, where it acts as the template for protein synthesis on a ribosome, with the bases complementary to those on the mRNA being carried by transfer RNA (tRNA).
- Post-translational modification – chemical modification of amino acid residues.
- Protein folding – formation of the functional 3D structure.
Throughout this process, any error in transcription or translation may result in the inclusion of an incorrect amino acid in the protein (and thus a possible loss of activity), so it is essential that all of the enzymes involved in this process carry out their roles accurately. (For further information on protein synthesis, see Laursen et al., 2005; Steitz, 2008.)
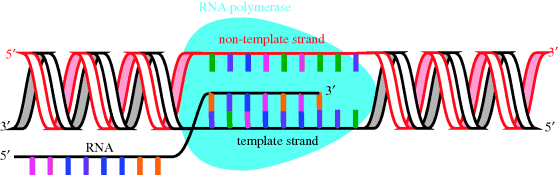
During transcription, DNA acts as a template for the synthesis of mRNA (Figure 1.1.5), a process which is catalysed by DNA-dependent RNA polymerase (RNAP), a nucleotidyl transferase enzyme (Floss and Yu, 2005; Mariani and Maffioli, 2009). In bacteria, the transcription process can be divided into a number of distinct steps in which the RNAP holoenzyme4 binds to duplex promoter DNA to form the RNAP-promoter complex, then a series of conformational changes leads to local unwinding of DNA to expose the transcription start site. RNAP can then initiate transcription, directing the synthesis of short RNA products, with synthesis of the RNA taking place in the 5′ → 3′ direction (with the DNA template strand being read in the 3′ → 5′ direction).
RNAP is a complex system, comprising five subunits (α2ββ′ω), each of which has a different function. The α subunits assemble the enzyme and bind regulatory factors, the β subunit contains the polymerase, the β′ subunit binds non-specifically to DNA, and the ω subunit promotes the assembly of the subunits and constrains the β′unit. The core structure of RNAP is thought to resemble a crab’s claw, with the active centre on the floor of the cleft between the two ‘pincers’, the β and β′ subunits, and also contains a secondary channel, by which the nucleotide triphosphates access the active centre, and an RNA-exit channel (for a really good interactive tutorial showing the structure of RNAP, see http://www.pingrysmartteam.com/RPo/RPo.htm, last accessed 26 March 2012). Bacterial RNAP contains only these conserved subunits, while eukaryotic RNAP contains these and seven to nine other units (Ebright, 2000).
In bacteria, the transcription of a particular gene requires the binding of a further subunit, a σ factor (a transcription initiation factor), which increases the specificity of RNAP binding to a particular promoter region and is involved in promoter melting, and so results in the transcription of a particular DNA sequence. Once the assembly process is complete, the holoenzyme (the active form containing all the subunits: α2ββ′ωσ) catalyses the synthesis of RNA, which is complementary to the DNA sequence characterised by the σ factor (Figure 1.1.5) (eukaryotic RNAP also requires the binding of transcription factors, as do some bacterial RNAP). Proofreading of the transcription process is less effective than that involved in the copying of DNA, so this is the point in the transfer of genetic information which is most susceptible to errors. As we will see in Subsection 2.2.4, DNA-dependent RNA polymerase is the target of the rifamycin antibiotics.
Ochoa and Kornberg were awarded the Nobel Prize for Physiology or Medicine in 1959 ‘for their discovery of the mechanisms in the biological synthesis of ribonucleic acid and deoxyribonucleic acid’ (http://nobelprize.org/nobel_prizes/medicine/laureates/1959/#, last accessed 26 March 2012).
Once the mRNA has been synthesised, it moves to the cytoplasm, where it binds to the ribosome, a giant ribonucleoprotein which catalyses protein synthesis from an mRNA template (translation). In 2009, Ramakrishnan, Steitz, and Yonath were awarded the Nobel Prize in Chemistry for their ‘studies of the structure and function of the ribosome’ (http://nobelprize.org/nobel_prizes/chemistry/laureates/2009/press.html, last accessed 26 March 2012).
The ribosome (Steitz, 2008), a large assembly consisting of RNA and proteins (ribonucleoproteins), has two subunits (30S and 50S in bacteria (complete ribosome 70S), 40S and 60S in eukaryotic cells (complete ribosome 80S)), and the large ribosome subunit has three binding sites, peptidyl-tRNA (P), aminoacyl-tRNA (A), and the exit (E) site, in the peptidyl transferase centre (PTC). Protein synthesis is initiated by the binding of a tRNA charged with methionine5 to its AUG codon on the mRNA. tRNAs (or charged tRNAs) then carry amino acids to the ribosome site where mRNA binds. tRNA has three nucleotides which code for a specific amino acid (a triplet) and bind to the complementary sequence on the mRNA. The ribosome moves along the mRNA from the 5′- to the 3′-end and, once the peptide bond has formed, the non-acylated tRNA leaves the P site and the peptide-tRNA moves from the A to the P site. A new tRNA-amino acid (as specified by the mRNA codon) then enters the A site and the peptide chain grows as amino acids are added, until a stop codon is reached, when it leaves the ribosome through the nascent protein exit tunnel (Figure 1.1.6). One thing which has probably already occurred to you is that every protein does not have a methionine residue at its amino terminus; this is a result of modifications once the protein has been synthesised. In bacteria, the formyl group is removed by peptide deformylase and the methionine is then removed by a methionine aminopeptidase. Although you might not agree, this is actually a simplification of protein synthesis, which also involves other processes and species, including initiation factors, elongation factors, and release factors.
Figure 1.1.6 The sequence of events leading to protein synthesis on the ribosome: (a) the small ribosomal subunit binds to the mRNA product of transcription; (b) the initiation complex is formed as the initiator tRNA(formyl-methionine) binds; (c) the large ribosomal subunit binds – tRNA(formyl-methionine) bound in the P (peptidyl-tRNA) site of the peptidyl transfer centre (the small subunit is transparent to allow a view of molecular events within the ribosome); (d) the mRNA codon (CCG) dictates that tRNA(proline), with an anticodon of GGC, binds to the A (aminoacyl-tRNA) site of the peptidyl transfer centre; (e) a peptide bond forms between methionine (M) and proline(P), the ribosome moves along mRNA in the 5′ → 3′ direction, tRNA bearing M-P binds to the P site, leaving the A site free to bind the tRNA encoded by the next three bases of the mRNA. The exit (E site) binds the free tRNA before it exits the ribosome; (f) as the amino acids are added, the new protein exits the ribosome into the cytoplasm via the nascent protein exit tunnel
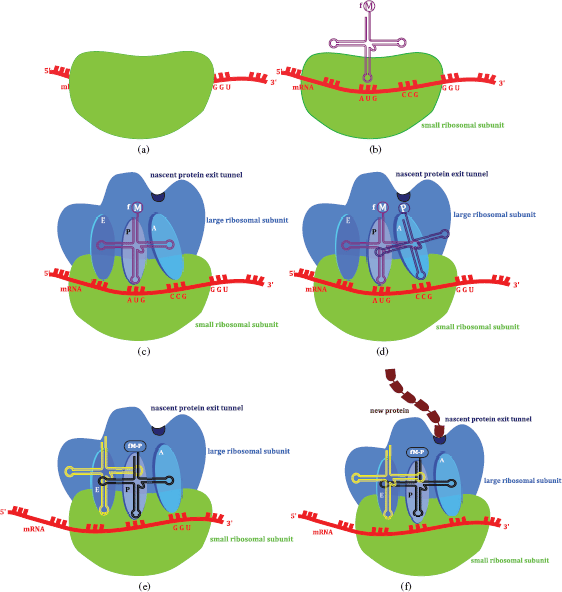
In Section 4 we will look at several drug classes which target protein synthesis by interfering with different aspects of the ribosomal translation process highlighted above. As with DNA replication, these antibiotics target processes common to both prokaryotes and eukaryotes and so any selectivity will be based on subtle differences in the structures of the ribosomes in the different cell types.
1.1.3.4 Bacterial Cell-Wall Synthesis
As mentioned earlier, bacteria have a cell wall and a cell (plasma) membrane, while mammalian eukaryotic cells only have a cell membrane. The prokaryotic cell wall is composed of peptidoglycan (a polymer consisting of sugar and peptide units) and other components, depending upon the type of bacterium.
Gram positive bacteria (which are stained dark purple/violet by Gentian violet-iodine complex) are surrounded by a plasma membrane and cell wall containing peptidoglycan (Figure 1.1.7) linked to lipoteichoic acids (which consist of an acylglycerol linked via a carbohydrate (sugar) to a poly(glycerophosphate) backbone, Figure 1.1.8).
Figure 1.1.7 Schematic representation of the plasma membrane and cell wall of Gram positive bacteria
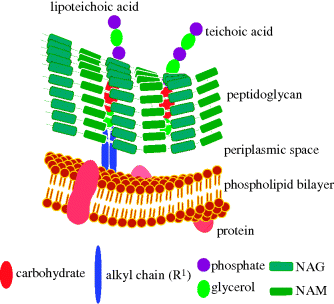
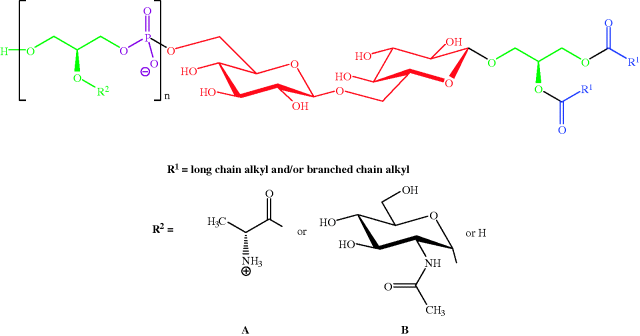
Figure 1.1.8 General structure of the lipoteichoic acid from Staphylococcus aureus (n = 40–50; ratio of R2 side-chains is D-Ala A (≈ 70%): N-acetylglucosamine B (≈ 15%): H (≈ 15%) (Reprinted from A. Stadelmaier, S. Morath, T. Hartung, and R. R. Schmidt, Angew. Chem. Int. Ed., 42, 916–920, 2003, with permission of John Wiley & Sons.)
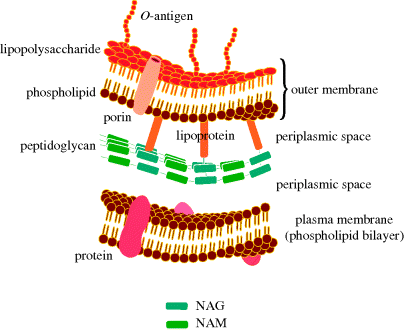
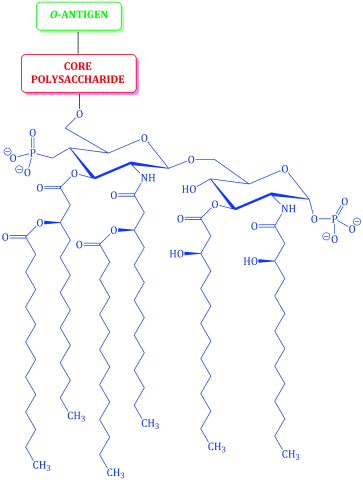
The cell wall of Gram negative bacteria is more complex. They have a plasma membrane and a thinner cell wall (peptidoglycan and associated proteins) surrounded by an outer membrane of phospholipid and lipopolysaccharide and proteins called porins (Figure 1.1.9). The outer membrane is thus the feature of the Gram negative cell wall which represents the greatest difference to that of Gram positive bacteria. The lipopolysaccharide (LPS) consists of: a phospholipid containing glucosamine rather than glycerol (lipid A6), a core polysaccharide (often containing some rather unusual sugars), and an O-antigen polysaccharide side chain (Figure 1.1.10). As this outer membrane poses a significant barrier for the uptake of any non-hydrophobic molecules, the outer membrane contains porins: protein pores which allow hydrophilic molecules to diffuse through the membrane. As a result of their more complex cell wall and membranes, Gram negative bacteria are not stained dark blue/violet by the Gram stain, but can be visualised with a counterstain (usually the pink dye fuschin).
Figure 1.1.9 Schematic representation of the plasma membrane, cell wall, and outer membrane of Gram negative bacteria
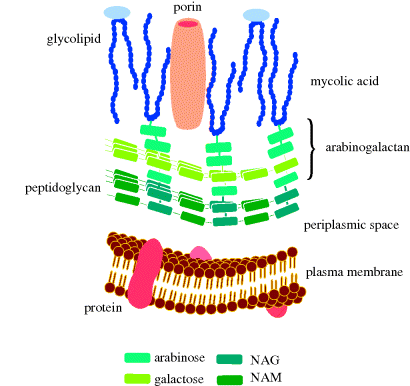
Finally, mycobacteria have a structure which includes a cell wall (Figure 1.1.11), composed of peptidoglycan and arabinogalactan, to which are anchored mycolic acids (long-chain α-alkyl-substituted β-hydroxyacids which can contain cyclopropyl or alkenyl groups, as well as a range of oxygenated functional groups); see Figure 1.1.12. Mycobacteria are resistant to antibacterial agents that target cell-wall synthesis (such as the β-lactams).
Figure 1.1.11 Schematic representation of the plasma membrane, cell wall, and mycomembrane (mycolic acid layer) of mycobacteria
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree



{kind=link}