Chapter 28 Treatment of Parkinson’s and Alzheimer’s Diseases
| Abbreviations | |
|---|---|
| ACh | Acetylcholine |
| AChE | Acetylcholinesterase |
| BBB | Blood-brain barrier |
| ChE | Cholinesterase |
| COMT | Catechol-O-methyl transferase |
| DA | Dopamine |
| l-DOPA | 3,4-Dihydroxy-phenylalanine |
| MAO | Monoamine oxidase |
| NMDA | N-methyl-d-aspartate |
Therapeutic Overview
While several neurotransmitter systems deteriorate in Alzheimer’s disease, one of the first and most pronounced reductions occurs within the acetylcholine (ACh) containing projections from the nucleus basalis in the basal forebrain to the cerebral cortex. The septo-hippocampal cholinergic pathway is similarly affected, whereas the
| Therapeutic Overview |
|---|
| Parkinson’s Disease |
| Pathology |
| Degeneration of nigrostriatal DA neurons |
| Presence of Lewy bodies in surviving neurons |
| Treatment |
| l-DOPA |
| MAO inhibitors |
| COMT inhibitors |
| DA agonists |
| Muscarinic receptor antagonists |
| NMDA receptor antagonists |
| Alzheimer’s Disease |
| Pathology |
| Degeneration of basal forebrain cholinergic neurons |
| Presence of amyloid plaques and neurofibrillary tangles |
| Neuron and synapse loss in cerebral cortex and hippocampus |
| Treatment |
| AChE inhibitors |
| NMDA receptor antagonists |
intrinsic striatal cholinergic system remains largely intact. Muscarinic cholinergic receptors in the cerebral cortex and hippocampus remain more or less intact, but nicotinic cholinergic receptors decline.
Patients with both Parkinson’s and Alzheimer’s diseases may manifest neuropsychiatric disturbances as a result of their primary disease, including psychoses, depression, anxiety, and agitation. These can be treated with the atypical antipsychotics, antidepressants, and anxiolytic compounds discussed in Chapters 29 to Chapters 31. A summary of the treatment of Parkinson’s and Alzheimer’s diseases is provided in the Therapeutic Overview Box.
Mechanisms of Action
Current strategies for the treatment of Parkinson’s disease are directed at increasing dopaminergic activity in the striatum to compensate for the loss of nigrostriatal DA neurons (see Fig. 27-8). The major drugs used include compounds that increase the synthesis and decrease the catabolism of DA or directly stimulate DA receptors; secondary compounds block muscarinic cholinergic receptors, enhance DA release, and perhaps antagonize NMDA receptors.
l-DOPA was introduced for the treatment of Parkinson’s disease in 1970. It is the precursor of DA and crosses the BBB, whereas DA does not. l-DOPA increases DA synthesis but does not stop progression of the disease. When given alone, only 1% to 3% of an administered dose of l-DOPA reaches the brain; the rest is metabolized peripherally as shown in Figure 28-1. To prevent its peripheral metabolism and increase its availability to the brain, l-DOPA is administered with carbidopa, an aromatic l-amino acid decarboxylase inhibitor that does not cross the BBB. Carbidopa does not have any therapeutic benefit when used alone but increases the amount of l-DOPA available to the brain. However, as a consequence of peripheral inhibition of aromatic l-amino acid decarboxylase, more precursor is metabolized by plasma catechol-O-methyltransferase (COMT) producing 3-o-methyldopa. To overcome this, the COMT inhibitors tolcapone or entacapone are used in combination with l-DOPA/carbidopa. These compounds prolong the plasma t1/2 of l-DOPA, increasing the time the drug is available to cross the BBB. They also prevent the buildup of 3-o-methyldopa, which competitively inhibits l-DOPA transport across the BBB. Entacapone does not cross the BBB, and its actions are limited to the periphery. However, tolcapone does cross the BBB and also prevents the formation of 3-o-methyldopa in brain and the catabolism of DA (Fig. 28-1).
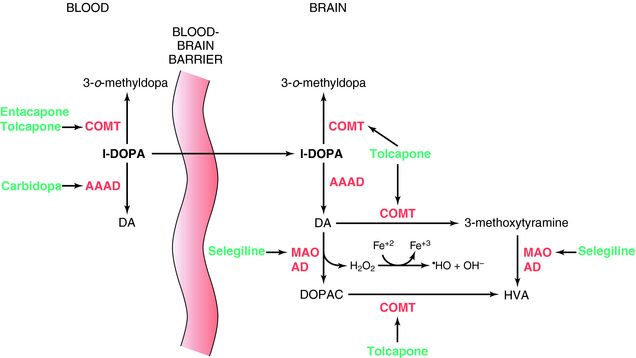
Another approach to increase brain DA levels involves the use of the monoamine oxidase type B (MAO-B) irreversible inhibitors selegiline or rasagiline to inhibit the catabolism of DA in the brain (see Fig. 28-1). In addition, by inhibiting the catabolism of DA to DOPAC, these compounds decrease production of the byproduct hydrogen peroxide, limiting the possible formation of free radicals that form when the peroxide reacts with ferrous iron.



