Treatment of Myocardial Ischemia and Hypertension
PATHOPHYSIOLOGY OF ISCHEMIC HEART DISEASE
Angina pectoris, the primary symptom of ischemic heart disease, is caused by transient episodes of myocardial ischemia that are due to an imbalance in the myocardial oxygen supply–demand relationship that may be caused by an increase in myocardial oxygen demand or by a decrease in myocardial oxygen supply or sometimes by both (Figure 27–1). Typical angina is experienced as a heavy, pressing substernal discomfort (rarely described as a “pain”), often radiating to the left shoulder, flexor aspect of the left arm, jaw, or epigastrium. However, a significant minority of patients note discomfort in a different location or of a different character. Myocardial ischemia also may be silent, with electrocardiographic, echocardiographic, or radionuclide evidence of ischemia appearing in the absence of symptoms.
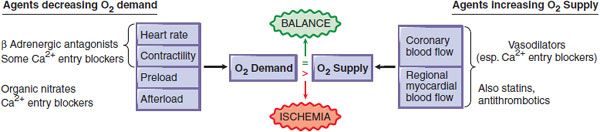
Figure 27–1 Pharmacological modification of the major determinants of myocardial O2 supply. When myocardial O2 requirements exceed O2 supply, an ischemic episode results. This figure shows the primary hemodynamic sites of actions of pharmacological agents that can reduce O2 demand (left side) or enhance O2 supply (right side). Some classes of agents have multiple effects. Stents, angioplasty, and coronary bypass surgery are mechanical interventions that increase O2 supply. Both pharmacotherapy and mechanotherapy attempt to restore a dynamic balance between O2 demand and O2 supply.
This section describes the principal pharmacological agents used in the treatment of angina: nitrovasodilators, β adrenergic receptor antagonists, and Ca2+ channel antagonists. These anti-anginal agents improve the balance of myocardial O2 supply and O2 demand, increasing supply by dilating the coronary vasculature and/or decreasing demand by reducing cardiac work (see Figure 27–1).
Drugs used in typical angina function principally by reducing myocardial O2 demand by decreasing heart rate, myocardial contractility, and/or ventricular wall stress. By contrast, the principal therapeutic goal in unstable angina is to increase myocardial blood flow; strategies include the use of antiplatelet agents and heparin to reduce intracoronary thrombosis, often accompanied by efforts to restore flow by mechanical means, including percutaneous coronary interventions using coronary stents, or (less commonly) emergency coronary bypass surgery. The principal therapeutic aim in variant or Prinzmetal angina is to prevent coronary vasospasm.
ORGANIC NITRATES
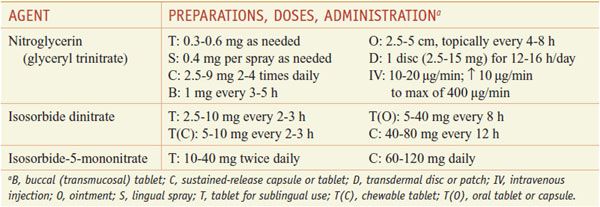
These agents are prodrugs that are sources of nitric oxide (NO) (Table 27–1).
Table 27–1
Organic Nitrates
Organic nitrates are polyol esters of nitric acid, whereas organic nitrites are esters of nitrous acid. Nitrate esters (—C—O—NO2) and nitrite esters (—C—O—NO) are characterized by the sequence of carbon–oxygen–nitrogen. Organic nitrates of low molecular mass (such as nitroglycerin) are moderately volatile, oily liquids, whereas the high-molecular-mass nitrate esters (e.g., erythrityl tetranitrate, isosorbide dinitrate, and isosorbide mononitrate) are solids. The organic nitrates and nitrites, collectively termed nitrovasodilators, must be metabolized (reduced) to produce gaseous NO, the active principle of this class of compounds. NO gas also may be administered by inhalation.
PHARMACOLOGICAL PROPERTIES
Mechanism of Action. Nitrites, organic nitrates, nitroso compounds, and a variety of other nitrogen oxide–containing substances (including nitroprusside) are basically exogenous sources of NO. NO can activate guanylyl cyclase and thereby increase the cellular level of cyclic GMP, which activates PKG and can modulate the activities of cyclic nucleotide phosphodiesterases (PDEs 2, 3, and 5) in a variety of cell types. In smooth muscle, the net result is phosphorylative activation of myosin light chain phosphatase, reduced phosphorylation of myosin light chain, reduced Ca2+ concentration in the cytosol, and relaxation. The pharmacological and biochemical effects of the nitrovasodilators appear to be identical to those of EDRF (endothelium-derived relaxing factor), now known to be NO. This NO-mediated pathway leads to relaxation of smooth muscle in the vasculature, bronchi, and GI tract, and inhibition of platelet aggregation. Chapter 3 presents details of NO biosynthesis and action.
Cardiovascular Effects; Hemodynamic Effects. Low concentrations of nitroglycerin preferentially dilate veins more than arterioles. This venodilation decreases venous return, leading to a fall in left and right ventricular chamber size and end-diastolic pressures, but usually results in little change in systemic vascular resistance. Systemic arterial pressure may fall slightly; heart rate is unchanged or may increase slightly in response to a decrease in blood pressure. Pulmonary vascular resistance and cardiac output are slightly reduced. Doses of nitroglycerin that do not alter systemic arterial pressure may still produce arteriolar dilation in the face and neck, resulting in a facial flush, or dilation of meningeal arterial vessels, causing headache.
Higher doses of organic nitrates cause further venous pooling and may decrease arteriolar resistance as well, thereby decreasing systolic and diastolic blood pressure and cardiac output and causing pallor, weakness, dizziness, and activation of compensatory sympathetic reflexes. The reflex tachycardia and peripheral arteriolar vasoconstriction tend to restore systemic vascular resistance; this is superimposed on sustained venous pooling. Coronary blood flow may increase transiently as a result of coronary vasodilation but may decrease subsequently if cardiac output and blood pressure decrease sufficiently. In patients with autonomic dysfunction and an inability to increase sympathetic outflow, the physiological means to compensate for this fall in blood pressure are absent. In these clinical contexts, nitrates may reduce arterial pressure and coronary perfusion pressure significantly, producing potentially life-threatening hypotension and even aggravating angina. The appropriate therapy in patients with orthostatic angina and normal coronary arteries is to correct the orthostatic hypotension by expanding volume (fludrocortisone and a high-sodium diet), to prevent venous pooling with fitted support garments, and to carefully titrate use of oral vasopressors. Because patients with autonomic dysfunction occasionally may have coexisting coronary artery disease, the coronary anatomy should be defined before therapy is undertaken.
Effects on Coronary Blood Flow. Myocardial ischemia is a powerful stimulus to coronary vasodilation, and regional blood flow is adjusted by autoregulatory mechanisms. In the presence of atherosclerotic coronary artery narrowing, ischemia distal to the lesion stimulates vasodilation; if the stenosis is severe, much of the capacity to dilate is used to maintain resting blood flow and further dilation may not be possible when demand increases. Significant coronary stenoses disproportionately reduce blood flow to the subendocardial regions of the heart, which are subjected to the greatest extravascular compression during systole; organic nitrates tend to restore blood flow in these regions toward normal. The hemodynamic mechanisms responsible for these effects are likely the capacity of organic nitrates to cause dilation and prevent vasoconstriction of large epicardial vessels without impairing autoregulation in the small vessels. An important indirect mechanism for a preferential increase in subendocardial blood flow is the nitroglycerin-induced reduction in intracavitary systolic and diastolic pressures that oppose blood flow to the subendocardium (see below).
Effects on Myocardial O2 Requirements. By their effects on the systemic circulation, the organic nitrates can reduce myocardial O2 demand. The major determinants of myocardial O2 consumption include left ventricular wall tension, heart rate, and myocardial contractility. Ventricular wall tension is affected by a number of factors that may be considered under the categories of preload and afterload. Preload is determined by the diastolic pressure that distends the ventricle (ventricular end-diastolic pressure). Decreasing end-diastolic volume reduces ventricular wall tension (by the law of Laplace, tension is proportional to pressure times radius). Increasing venous capacitance with nitrates decreases venous return to the heart, decreases ventricular end-diastolic volume, and thereby decreases O2 consumption. An additional benefit of reducing preload is that it increases the pressure gradient for perfusion across the ventricular wall, which favors subendocardial perfusion. Afterload is the impedance against which the ventricle must eject. In the absence of aortic valvular disease, afterload is related to peripheral resistance. Decreasing peripheral arteriolar resistance reduces afterload and thus myocardial work and O2 consumption.
Organic nitrates decrease both preload and afterload as a result of respective dilation of venous capacitance and arteriolar resistance vessels. They do not directly alter the inotropic or chronotropic state of the heart. Since nitrates reduce the primary determinants of myocardial O2 demand, their net effect usually is to decrease myocardial O2 consumption. The effect of nitrovasodilators to inhibit platelet function may contribute to their anti-anginal efficacy, this effect appears to be modest.
Mechanism of Relief of Symptoms of Angina Pectoris. The capacity of nitrates to dilate epicardial coronary arteries, even in areas of atherosclerotic stenosis, is modest, and the preponderance of evidence continues to favor a reduction in myocardial work, and thus in myocardial O2 demand, as their primary effect in chronic stable angina. Paradoxically, high doses of organic nitrates may reduce blood pressure to such an extent that coronary flow is compromised; reflex tachycardia and adrenergic enhancement of contractility also occur. These effects may override the beneficial action of the drugs on myocardial O2 demand and can aggravate ischemia. Additionally, sublingual nitroglycerin administration may produce bradycardia and hypotension, probably owing to activation of the Bezold-Jarisch reflex.
OTHER EFFECTS. The nitrovasodilators act on almost all smooth muscle tissues. Bronchial smooth muscle is relaxed irrespective of the preexisting tone. The muscles of the biliary tract, including those of the gallbladder, biliary ducts, and sphincter of Oddi, are effectively relaxed. Smooth muscle of the GI tract, including that of the esophagus, can be relaxed and its spontaneous motility decreased by nitrates both in vivo and in vitro. The effect may be transient and incomplete in vivo, but abnormal “spasm” frequently is reduced. Indeed, many incidences of atypical chest pain and “angina” are due to biliary or esophageal spasm, and these too can be relieved by nitrates. Similarly, nitrates can relax ureteral and uterine smooth muscle, but these responses are of uncertain clinical significance.
ADME AND PREPARATIONS
Nitroglycerin. In humans, peak concentrations of nitroglycerin are found in plasma within 4 min of sublingual administration; the drug has a t1/2 of 1-3 min. The onset of action of nitroglycerin may be even more rapid if it is delivered as a sublingual spray rather than as a sublingual tablet. Glyceryl dinitrate metabolites, which have about one-tenth the vasodilator potency, appear to have half-lives of ~40 min.
Isosorbide Dinitrate. The major route of metabolism of isosorbide dinitrate in humans appears to be by enzymatic denitration followed by glucuronide conjugation. Sublingual administration produces maximal plasma concentrations of the drug by 6 min, and the fall in concentration is rapid (t1/2 of ~45 min). The primary initial metabolites, isosorbide-2-mononitrate and isosorbide-5-mononitrate, have longer half-lives (3-6 h) and presumably contribute to the therapeutic efficacy of the drug.
Isosorbide-5-Mononitrate. This agent is available in tablet form. It does not undergo significant first-pass metabolism and so has excellent bioavailability after oral administration. The mononitrate has a significantly longer t1/2 than does isosorbide dinitrate and has been formulated as a plain tablet and as a sustained-release preparation; both preparations have longer durations of action than the corresponding dosage forms of isosorbide dinitrate.
Inhaled NO. Nitric oxide gas administered by inhalation appears to exert most of its therapeutic effects on the pulmonary vasculature; systemically, it rapidly interacts with heme groups in hemoglobin in the blood. Inhaled NO is used to treat pulmonary hypertension in hypoxemic neonates, where inhaled NO reduces morbidity and mortality.
TOLERANCE
Sublingual organic nitrates should be taken at the time of an anginal attack or in anticipation of exercise or stress. Such intermittent treatment provides reproducible cardiovascular effects. However, frequently repeated or continuous exposure to high doses of organic nitrates leads to a marked attenuation in the magnitude of most of their pharmacological effects. The magnitude of tolerance is a function of dosage and frequency of use. Tolerance may result from a reduced capacity of the vascular smooth muscle to convert nitroglycerin to NO, true vascular tolerance, or to the activation of mechanisms extraneous to the vessel wall, pseudotolerance. Multiple mechanisms have been proposed to account for nitrate tolerance, including volume expansion, neurohumoral activation, cellular depletion of sulfhydryl groups, and the generation of free radicals. Inactivation of mitochondrial aldehyde dehydrogenase, an enzyme implicated in biotransformation of nitroglycerin, is seen in models of nitrate tolerance, potentially associated with oxidative stress. A reactive intermediate formed during the generation of NO from organic nitrates may itself damage and inactivate the enzymes of the activation pathway; tolerance could involve endothelium-derived reactive oxygen species.
An effective approach to restoring responsiveness is to interrupt therapy for 8-12 h each day, which allows the return of efficacy. It is usually most convenient to omit dosing at night in patients with exertional angina either by adjusting dosing intervals of oral or buccal preparations or by removing cutaneous nitroglycerin. However, patients whose anginal pattern suggests its precipitation by increased left ventricular filling pressures (i.e., occurring in association with orthopnea or paroxysmal nocturnal dyspnea) may benefit from continuing nitrates at night and omitting them during a quiet period of the day. Tolerance also has been seen with isosorbide-5-mononitrate; an eccentric twice-daily dosing schedule appears to maintain efficacy. Some patients develop an increased frequency of nocturnal angina when a nitrate-free interval is employed using nitroglycerin patches; such patients may require another class of anti-anginal agent during this period. Tolerance is not universal, and some patients develop only partial tolerance. The problem of anginal rebound during nitrate-free intervals is especially problematic in the treatment of unstable angina with intravenous nitroglycerin. As tolerance develops, increasing doses are required to achieve the same therapeutic effects; eventually, despite dose escalation, the drug loses efficacy.
A special form of nitroglycerin tolerance is observed in individuals exposed to nitroglycerin in the manufacture of explosives. If protection is inadequate, workers may experience severe headaches, dizziness, and postural weakness during the first several days of employment. Tolerance then develops, but headache and other symptoms may reappear after a few days away from the job—the “Monday disease.” The most serious effect of chronic exposure is a form of organic nitrate dependence. Workers without demonstrable organic vascular disease have been reported to have an increase in the incidence of acute coronary syndromes during the 24-72-h periods away from the work environment. Because of the potential problem of nitrate dependence, it seems prudent not to withdraw nitrates abruptly from a patient who has received such therapy chronically.
TOXICITY AND UNTOWARD RESPONSES. Untoward responses to the therapeutic use of organic nitrates are almost all secondary to actions on the cardiovascular system. Headache is common and can be severe. It usually decreases over a few days if treatment is continued and often can be controlled by decreasing the dose. Transient episodes of dizziness, weakness, and other manifestations associated with postural hypotension may develop, particularly if the patient is standing immobile, and may progress occasionally to loss of consciousness, a reaction that appears to be accentuated by alcohol. It also may be seen with very low doses of nitrates in patients with autonomic dysfunction. Even in severe nitrate syncope, positioning and other measures that facilitate venous return are the only therapeutic measures required. All the organic nitrates occasionally can produce drug rash.
INTERACTION OF NITRATES WITH PDE5 INHIBITORS. Erectile dysfunction is a frequently encountered problem whose risk factors parallel those of coronary artery disease. Thus, many men desiring therapy for erectile dysfunction already may be receiving (or may require, especially if they increase physical activity) anti-anginal therapy. The combination of sildenafil and other phosphodiesterase 5 (PDE5) inhibitors with organic nitrate vasodilators can cause extreme hypotension.
Cells in the corpus cavernosum produce NO during sexual arousal in response to nonadrenergic, noncholinergic neurotransmission. NO stimulates the formation of cyclic GMP, which leads to relaxation of smooth muscle of the corpus cavernosum and penile arteries, engorgement of the corpus cavernosum, and erection. The accumulation of cyclic GMP can be enhanced by inhibition of the cyclic GMP–specific PDE5 family. Sildenafil (VIAGRA, REVATIO) and congeners inhibit PDE5 and have been demonstrated to improve erectile function in patients with erectile dysfunction. Not surprisingly, PDE5 inhibitors have assumed the status of widely used recreational drugs. Since the introduction of sildenafil, 3 additional PDE5 inhibitors have been developed for use in therapy of erectile dysfunction. Tadalafil (CIALIS, ADCIRCA), vardenafil (LEVITRA), and avanafil (STENDRA) share similar therapeutic efficacy and side-effect profiles with sildenafil; tadalafil has a longer time to onset of action and a longer therapeutic t1/2 than the other PDE5 inhibitors. Sildenafil has been the most thoroughly characterized of these compounds, but all 3 PDE5 inhibitors are contraindicated for patients taking organic nitrate vasodilators, and the PDE5 inhibitors should be used with caution in patients taking α or β adrenergic receptor antagonists (see Chapter 12).
The side effects of sildenafil and other PDE5 inhibitors are largely predictable on the basis of their effects on PDE5. Headache, flushing, and rhinitis may be observed, as well as dyspepsia owing to relaxation of the lower esophageal sphincter. Sildenafil and vardenafil also weakly inhibit PDE6, the enzyme involved in photoreceptor signal transduction (see Chapters 3 and 64), and can produce visual disturbances, most notably changes in the perception of color hue or brightness. In addition to visual disturbances, sudden one-sided hearing loss has also been reported. Tadalafil inhibits PDE11, a widely distributed PDE isoform, but the clinical importance of this effect is not clear. The most important toxicity of all these PDE5 inhibitors is hemodynamic. When given alone to men with severe coronary artery disease, these drugs have modest effects on blood pressure, producing >10% fall in systolic, diastolic, and mean systemic pressures and in pulmonary artery systolic and mean pressures. However, sildenafil, tadalafil, and vardenafil all have a significant and potentially dangerous interaction with organic nitrates, which act therapeutically via enhancing cyclic GMP production in smooth muscle. In the presence of a PDE5 inhibitor, nitrates cause profound increases in cyclic GMP and can produce dramatic reductions in blood pressure. This drug class toxicity is the basis for the warning that PDE5 inhibitors should not be prescribed to patients receiving any form of nitrate and dictates that patients should be warned about the prior use of PDE5 inhibitors within 24 h of administration of nitrates. A period of longer than 24 h may be needed following administration of a PDE5 inhibitor for safe use of nitrates, especially with tadalafil because of its prolonged t1/2. In the event that patients develop significant hypotension following combined administration of sildenafil and a nitrate, fluids and α adrenergic receptor agonists, if needed, should be used for support. These same hemodynamic responses to PDE5 inhibition also may underlie the efficacy of sildenafil in the treatment of patients with primary pulmonary hypertension, in whom chronic treatment with the drug appears to result in enhanced exercise capacity associated with a decrease in pulmonary vascular resistance. PDE5 inhibitors also are being studied in patients with congestive heart failure and cardiac hypertrophy (see Chapter 28).
Sildenafil, tadalafil, vardenafil, and avanafil are metabolized via CYP3A4, and their toxicity may be enhanced in patients who receive other substrates of this enzyme, including macrolide and imidazole antibiotics, some statins, and antiretroviral agents (see individual chapters and Chapter 6). PDE5 inhibitors also may prolong cardiac repolarization by blocking the IKr. In patients with coronary artery disease whose exercise capacity indicates that sexual activity is unlikely to precipitate angina and who are not currently taking nitrates, the use of PDE5 inhibitors can be considered. Such therapy needs to be individualized, and appropriate warnings must be given about the risk of toxicity if nitrates are taken subsequently for angina. Alternative non-nitrate anti-anginal therapy, such as β adrenergic receptor antagonists, should be used during these time periods.
THERAPEUTIC USES
ANGINA. Diseases that predispose to angina should be treated as part of a comprehensive therapeutic program with the primary goal being to prolong life. Conditions such as hypertension, anemia, thyrotoxicosis, obesity, heart failure, cardiac arrhythmias, and acute anxiety can precipitate anginal symptoms in many patients. Patients should be counseled to stop smoking, lose weight, and maintain a low-fat, high-fiber diet; hypertension and hyperlipidemia should be corrected; and daily aspirin (or clopidogrel if aspirin is not tolerated) (see Chapter 30) should be prescribed. Exposure to sympathomimetic agents (e.g., those in nasal decongestants and other sources) probably should be avoided. The use of drugs that modify the perception of pain is a poor approach to the treatment of angina because the underlying myocardial ischemia is not relieved. Table 27–1 lists the preparations and dosages of the nitrites and organic nitrates.
Sublingual Administration. Because of its rapid action, long-established efficacy, and low cost, nitroglycerin is the most useful drug of the organic nitrates given sublingually. The onset of action is within 1-2 min, but the effects are undetectable by 1 h after administration.
Oral Administration. Oral nitrates often are used to provide prophylaxis against anginal episodes. Higher doses of either isosorbide dinitrate (e.g., 20 mg or more orally every 4 h) or sustained-release preparations of nitroglycerin decrease the frequency of anginal attacks and improve exercise tolerance. Effects peak at 60-90 min and last for 3-6 h. Administration of isosorbide mononitrate (typically starting at 20 mg) once or twice daily (in the latter case, with the doses administered 7 h apart) is efficacious in the treatment of chronic angina, and once-daily dosing or an eccentric twice-daily dosing schedule can minimize the development of tolerance.
Cutaneous Administration. Nitroglycerin ointment (2%) is applied to the skin (2.5-5 cm); the dosage must be adjusted for each patient. Effects are apparent within 30-60 min (although absorption is variable) and last for 4-6 h. The ointment is particularly useful for controlling nocturnal angina, which commonly develops within 3 h after the patient goes to sleep. To avoid tolerance, therapy should be interrupted for at least 8 h each day.
Transmucosal or Buccal Nitroglycerin. This formulation is inserted under the upper lip above the incisors, where it adheres to the gingiva and dissolves gradually in a uniform manner. Hemodynamic effects are seen within 2-5 min, and it is therefore useful for short-term prophylaxis of angina.
CONGESTIVE HEART FAILURE. The utility of nitrovasodilators to relieve pulmonary congestion and to increase cardiac output in congestive heart failure is addressed in Chapter 28.
UNSTABLE ANGINA PECTORIS AND NON-ST-SEGMENT–ELEVATION MYOCARDIAL INFARCTION. The term unstable angina pectoris has been used to describe a broad spectrum of clinical entities characterized by an acute or subacute worsening in a patient’s anginal symptoms. The variable prognosis of unstable angina reflects the broad range of clinical entities subsumed by the term. Efforts have been directed toward identifying patients with unstable angina on the basis of their risks for subsequent adverse outcomes such as MI or death. The term acute coronary syndrome has been useful in this context: Common to most clinical presentations of acute coronary syndrome is disruption of a coronary plaque, leading to local platelet aggregation and thrombosis at the arterial wall, with subsequent partial or total occlusion of the vessel. There is some variability in the pathogenesis of unstable angina, with gradually progressive atherosclerosis accounting for some cases of new-onset exertional angina. Less commonly, vasospasm in minimally atherosclerotic coronary vessels may account for some cases where rest angina has not been preceded by symptoms of exertional angina. For the most part, the pathophysiological principles that underlie therapy for exertional angina—which are directed at decreasing myocardial oxygen demand—have limited efficacy in the treatment of acute coronary syndromes characterized by an insufficiency of myocardial oxygen (blood) supply.
OTHER AGENTS; DRUG COMBINATIONS. Drugs that reduce myocardial O2 consumption by reducing ventricular preload (nitrates) or by reducing heart rate and ventricular contractility (β adrenergic receptor antagonists) are efficacious, but additional therapies are directed at an atherosclerotic plaque and the consequences (or prevention) of its rupture. These therapies include combinations of:
• Antiplatelet agents, including aspirin and thioenopyridines such as clopidogrel or prasugrel
• Antithrombin agents such as heparin and the thrombolytics anti-integrin therapies that directly inhibit platelet aggregation mediated by glycoprotein (GP)IIb/IIIa mechano-pharmacological approaches with percutaneously deployed intracoronary stents
• Coronary bypass surgery for selected patients
Along with nitrates and β adrenergic receptor antagonists, antiplatelet agents represent the cornerstone of therapy for acute coronary syndrome. Aspirin inhibits platelet aggregation and improves survival. Heparin (either unfractionated or low-molecular-weight) also appears to reduce angina and prevent infarction. These and related agents are discussed in detail in Chapters 30 and 34. Anti-integrin agents directed against the platelet integrin GPIIb/IIIa (including abciximab, tirofiban, and eptifibatide) are effective in combination with heparin, as discussed later. Nitrates are useful both in reducing vasospasm and in reducing myocardial O2 consumption by decreasing ventricular wall stress. Intravenous administration of nitroglycerin allows high concentrations of drug to be attained rapidly. Because nitroglycerin is degraded rapidly, the dose can be titrated quickly and safely using intravenous administration. If coronary vasospasm is present, intravenous nitroglycerin is likely to be effective, although the addition of a Ca2+ channel blocker may be required to achieve complete control.
ACUTE MYOCARDIAL INFARCTION. Therapeutic maneuvers in MI are directed at reducing the size of the infarct, preserving or retrieving viable tissue by reducing the O2 demand of the myocardium, and preventing ventricular remodeling that could lead to heart failure.
Nitroglycerin is commonly administered to relieve ischemic pain in patients presenting with MI, but evidence that nitrates improve mortality in MI is sparse. Because they reduce ventricular preload through vasodilation, nitrates are effective in relief of pulmonary congestion. A decreased ventricular preload should be avoided in patients with right ventricular infarction because higher right-sided heart filling pressures are needed in this clinical context. Nitrates are relatively contraindicated in patients with systemic hypotension. According to the American Heart Association/American College of Cardiology (AHA/ACC) guidelines, “nitrates should not be used if hypotension limits the administration of β blockers, which have more powerful salutary effects.” Because the proximate cause of MI is intracoronary thrombosis, reperfusion therapies are critically important, employing, when possible, direct percutaneous coronary interventions (PCIs) for acute MI, usually using drug-eluting intracoronary stents. Thrombolytic agents are administered at hospitals where emergency PCI is not performed, but outcomes are better with direct PCI than with thrombolytic therapy.
VARIANT (PRINZMETAL) ANGINA. The large coronary arteries normally contribute little to coronary resistance. However, in variant angina, coronary constriction results in reduced blood flow and ischemic pain. Whereas long-acting nitrates alone are occasionally efficacious in abolishing episodes of variant angina, additional therapy with Ca2+ channel blockers usually is required.
CA2+ CHANNEL ANTAGONISTS
Voltage-sensitive Ca2+ channels (L-type or slow channels) mediate the entry of extracellular Ca2+ (~1.25 mM) into smooth muscle and cardiac myocytes and sinoatrial (SA) and atrioventricular (AV) nodal cells (cytosolic concentration in resting cell, ~100 nM) in response to electrical depolarization. In both smooth muscle and cardiac myocytes, Ca2+ is a trigger for contraction, albeit by different mechanisms. Ca2+ channel antagonists, also called Ca2+ entry blockers, inhibit Ca2+ channel function. In vascular smooth muscle, this leads to relaxation, especially in arterial beds. In the heart, these drugs can produce negative inotropic and chronotropic effects.
Clinically used Ca2+ channel antagonists include the phenylalkylamine compound verapamil, the benzothiazepine diltiazem, and numerous dihydropyridines, including nifedipine, amlodipine, felodipine, isradipine, nicardipine, nisoldipine, and nimodipine. The specificities of several of these drugs are shown in Table 27–2. Although these drugs are commonly grouped together as “Ca2+ channel blockers,” there are fundamental differences among verapamil, diltiazem, and the dihydropyridines, especially with respect to pharmacological characteristics, drug interactions, and toxicities.
Table 27–2
Comparative CV Effectsa of Ca2+ Channel Blockers
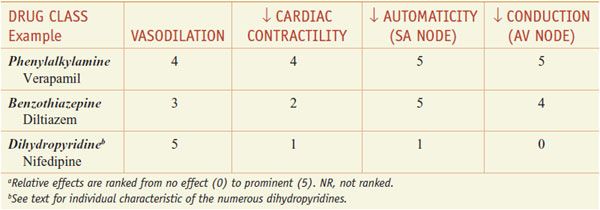
Mechanisms of Action. An increased concentration of cytosolic Ca2+ causes increased contraction in both cardiac and vascular smooth muscle cells. The entry of extracellular Ca2+ initiates contraction of cardiac myocytes (Ca2+-induced Ca2+ release, where the bulk of contractile Ca2+ is from the sarcoplasmic reticulum) and provides a major source of contractile Ca2+ in smooth muscle (in which the release of Ca2+ from IP3-sensitive intracellular storage vesicles and Ca2+ entry via receptor-operated channels can also contribute to contraction, particularly in some vascular beds). The Ca2+ channel antagonists produce their effects by binding to the α1 subunit of the L-type Ca2+ channels and reducing Ca2+ flux through the channel. The vascular and cardiac effects of some of the Ca2+ channel blockers are summarized in the next section and in Table 27–2.
PHARMACOLOGICAL PROPERTIES
CARDIOVASCULAR EFFECTS
Vascular Tissue. At least 3 distinct mechanisms may be responsible for contraction of vascular smooth muscle cells. First, voltage-sensitive Ca2+ channels open in response to depolarization of the membrane, and extracellular Ca2+ moves down its electrochemical gradient into the cell. Second, agonist-induced contractions that occur without depolarization of the membrane result from stimulation of the Gq–PLC–IP3 pathway, resulting in the release of intracellular Ca2+ from the sarcoplasmic reticulum (see Chapter 3). Third, receptor-operated Ca2+ channels allow the entry of extracellular Ca2+ in response to receptor occupancy.
Ca2+ channel antagonists inhibit the voltage-dependent Ca2+ channels in vascular smooth muscle at significantly lower concentrations than are required to interfere with the release of intracellular Ca2+ or to block receptor-operated Ca2+ channels. All Ca2+ channel blockers relax arterial smooth muscle, but they have a less pronounced effect on most venous beds and hence do not affect cardiac preload significantly.
Cardiac Cells. In the SA and AV nodes, depolarization largely depends on the movement of Ca2+ through the slow channel. The effect of a Ca2+ channel blocker on AV conduction and on the rate of the sinus node pacemaker depends on whether or not the agent delays the recovery of the slow channel. Although nifedipine reduces the slow inward current in a dose-dependent manner, it does not affect the rate of recovery of the slow Ca2+ channel. The channel blockade caused by nifedipine and related dihydropyridines also shows little dependence on the frequency of stimulation. At doses used clinically, nifedipine does not affect conduction through the AV node. In contrast, verapamil not only reduces the magnitude of the Ca2+ current through the slow channel but also decreases the rate of recovery of the channel. In addition, channel blockade caused by verapamil (and to a lesser extent by diltiazem) is enhanced as the frequency of stimulation increases, a phenomenon known as frequency dependence or use dependence. Verapamil and diltiazem depress the rate of the sinus node pacemaker and slow AV conduction; the latter effect is the basis for their use in the treatment of supraventricular tachyarrhythmias (see Chapter 29). Bepridil, like verapamil, inhibits both slow inward Ca2+ current and fast inward Na+ current. It has a direct negative inotropic effect. Its electrophysiological properties lead to slowing of the heart rate, prolongation of the AV nodal effective refractory period, and importantly, prolongation of the QTc interval. Particularly in the setting of hypokalemia, the last effect can be associated with torsade de pointes, a potentially lethal ventricular arrhythmia (see Chapter 29).
In cardiac myocytes, initiation of Ca2+-induced Ca2+ release relies on entry Ca2+ of through the L-type channel in response to depolarization. By reducing this entry, Ca2+ channel blockers can produce a negative inotropic effect.
Hemodynamic Effects. All the Ca2+ channel blockers approved for clinical use decrease coronary vascular resistance and can lead to an increase in coronary blood flow. The dihydropyridines are more potent vasodilator than verapamil, which is more potent than diltiazem. The hemodynamic effects of these agents vary depending on the route of administration and the extent of left ventricular dysfunction. Drugs that significantly lower mean pressure will elicit a baroreceptor reflex response.
Nifedipine, the prototypical dihydropyridine, selectively dilates arterial resistance vessels. The decrease in arterial blood pressure elicits sympathetic reflexes, with resulting tachycardia and positive inotropy. Nifedipine has direct negative inotropic effects in vitro but relaxes vascular smooth muscle at significantly lower concentrations than those required for prominent direct effects on the heart. Thus, arteriolar resistance and blood pressure are lowered, contractility and segmental ventricular function are improved, and heart rate and cardiac output are increased modestly. After oral administration of nifedipine, arterial dilation increases peripheral blood flow; venous tone does not change. The other dihydropyridines—amlodipine, felodipine, isradipine, nicardipine, nisoldipine, nimodipine, and clevidipine—share many of the cardiovascular effects of nifedipine.
Amlodipine has a slow absorption and a prolonged effect. Amlodipine produces both peripheral arterial vasodilation and coronary dilation. There is less reflex tachycardia with amlodipine than with nifedipine, possibly because the long t1/2 (35-50 h) produces minimal peaks and troughs in plasma concentrations. Felodipine may have even greater vascular specificity than does nifedipine or amlodipine. At concentrations producing vasodilation, there is no negative inotropic effect. Nicardipine has anti-anginal properties similar to those of nifedipine and may have selectivity for coronary vessels. Isradipine also produces the typical peripheral vasodilation seen with other dihydropyridines, but because of its inhibitory effect on the SA node, little or no rise in heart rate is seen. Despite the negative chronotropic effect, isradipine appears to have little effect on the AV node, so it may be used in patients with AV block or combined with a β adrenergic receptor antagonist. In general, because of their lack of myocardial depression and, to a greater or lesser extent, lack of negative chronotropic effect, dihydropyridines are less effective as monotherapy in stable angina than are verapamil, diltiazem, or a β adrenergic receptor antagonist. Nisoldipine is more than 1000 times more potent in preventing contraction of human vascular smooth muscle than in preventing contraction of human cardiac muscle in vitro, suggesting a high degree of vascular selectivity. Although nisoldipine has a short elimination t1/2, a sustained-release preparation that is efficacious as an anti-anginal agent has been developed. Nimodipine has high lipid solubility and is effective in inhibiting cerebral vasospasm and has been used primarily to treat patients with neurological defects associated with cerebral vasospasm after subarachnoid hemorrhage.
Clevidipine is a novel dihydropyridine L-type Ca2+ channel blocker—available for intravenous administration—that has a very rapid (t1/2 ~2 min) onset and offset of action. It is metabolized by esterases in blood, similar to the fate of esmolol. Clevidipine preferentially affects arterial smooth muscle compared to targeting veins or the heart. Infusions are typically started at a rate of 1-2 μg/kg/min and titrated to the desired effect on blood pressure.
Verapamil is a less potent vasodilator the dihydropyridines. Like dihydropyridines, verapamil causes little effect on venous resistance vessels at concentrations that produce arteriolar dilation. With doses of verapamil sufficient to produce peripheral arterial vasodilation, there are more direct negative chronotropic, dromotropic, and inotropic effects than with the dihydropyridines. Intravenous verapamil causes a decrease in arterial blood pressure owing to a decrease in vascular resistance, but the reflex tachycardia is blunted or abolished by the direct negative chronotropic effect of the drug. This intrinsic negative inotropic effect is partially offset by both a decrease in afterload and the reflex increase in adrenergic tone. Thus, in patients without congestive heart failure, ventricular performance is not impaired and actually may improve, especially if ischemia limits performance. In contrast, in patients with congestive heart failure, intravenous verapamil can cause a marked decrease in contractility and left ventricular function. Oral administration of verapamil reduces peripheral vascular resistance and blood pressure, often with minimal changes in heart rate. The relief of pacing-induced angina seen with verapamil is due primarily to a reduction in myocardial oxygen demand.
Intravenous administration of diltiazem can result initially in a marked decrease in peripheral vascular resistance and arterial blood pressure, which elicits a reflex increase in heart rate and cardiac output. Heart rate then falls below initial levels because of the direct negative chronotropic effect of the agent. Oral administration of diltiazem decreases both heart rate and mean arterial blood pressure. While diltiazem and verapamil produce similar effects on the SA and AV nodes, the negative inotropic effect of diltiazem is more modest.
ADME. Although the absorption of these agents is nearly complete after oral administration, their bioavailability is reduced, in some cases markedly, by first-pass hepatic metabolism. The effects of these drugs are evident within 30-60 min of an oral dose, with the exception of the more slowly absorbed and longer-acting agents amlodipine, isradipine, and felodipine. These agents all are bound extensively to plasma proteins (70-98%); their elimination half-lives vary widely, from 1.3 to 64 h. During repeated oral administration, bioavailability and t1/2 may increase because of saturation of hepatic metabolism. The bioavailability of some of these drugs may be increased by grapefruit juice, likely through inhibition of enzyme CYP3A4. A major metabolite of diltiazem is desacetyldiltiazem, which has about one-half of diltiazem’s potency as a vasodilator. N-Demethylation of verapamil results in production of norverapamil, which is biologically active but much less potent than the parent compound. The t1/2 of norverapamil is ~10 h. The metabolites of the dihydropyridines are inactive or weakly active. In patients with hepatic cirrhosis, the bioavailabilities and half-lives of the Ca2+ channel blockers may be increased, and dosage should be adjusted. The half-lives of these agents also may be longer in older patients. Except for diltiazem and nifedipine, all the Ca2+ channel blockers are administered as racemic mixtures.
Toxicity and Untoward Responses. The profile of adverse reactions to the Ca2+ channel blockers varies among the drugs in this class. Patients receiving immediate-release capsules of nifedipine develop headache, flushing, dizziness, and peripheral edema. Dizziness and flushing are much less of a problem with the sustained-release formulations and with the dihydropyridines having a long t1/2 and relatively constant concentrations of drug in plasma. Peripheral edema may occur in some patients with Ca2+
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


