Pharmacotherapy of Congestive Heart Failure
Congestive heart failure (CHF) is responsible for more than half a million deaths annually in the U.S., carries a 1-year mortality rate of more than 50% in patients with advanced forms of the condition. Substantive advances in CHF pharmacotherapy have altered clinical practice by shifting the paradigm of its management from exclusively symptom palliation to modification of disease progression and prolonged survival.
DEFINING CONGESTIVE HEART FAILURE. The onset and progression of clinically evident CHF from left ventricular (LV) systolic dysfunction follows a pathophysiologic sequence in response to an initial insult to myocardial dysfunction. A reduction in forward cardiac output leads to expanded activation of the sympathetic nervous system and the renin–angiotensin–aldosterone axis that, together, maintain perfusion of vital organs by increasing LV preload, stimulating myocardial contractility, and increasing arterial tone. Acutely, these mechanisms sustain cardiac output by allowing the heart to operate at elevated end-diastolic volumes, while peripheral vasoconstriction promotes regional redistribution of the cardiac output to the CNS, coronary, and renal vascular beds.
Unfortunately, these compensatory mechanisms over time propagate disease progression. Intravascular volume expansion increases diastolic and systolic wall stress that disrupts myocardial energetics and causes pathologic LV hypertrophy. By increasing LV afterload, peripheral arterial vasoconstriction also adversely affects diastolic ventricular wall stress, thereby increasing myocardial O2 demand. Finally, neurohumoral effectors such as NE and AngII are associated with myocyte apoptosis, abnormal myocyte gene expression, and pathologic changes in the extracellular matrix that increase LV stiffness.
Clinically, the term CHF describes a final common pathway for the expression of myocardial dysfunction. While some emphasize the clinical distinction between systolic versus diastolic heart failure, many patients demonstrate dysfunction in both contractile performance and ventricular relaxation/filling. Indeed, these physiologic processes are interrelated; for example, the rate and duration of LV diastolic filling are directly influenced by impairment in systolic contractile performance. The following definitions are useful for establishing a conceptual framework to describe this clinical syndrome:
Congestive heart failure is the pathophysiologic state in which the heart is unable to pump blood at a rate commensurate with the requirements of metabolizing tissues, or can do so only from an elevated filling pressure.
Heart failure is a complex of symptoms—fatigue, shortness of breath, and congestion—that are related to the inadequate perfusion of tissue during exertion and often to the retention of fluid. Its primary cause is an impairment of the heart’s ability to fill or empty the left ventricle properly.
One may consider CHF as a condition in which failure of the heart to provide adequate forward output at normal end-diastolic filling pressures results in a clinical syndrome of decreased exercise tolerance with pulmonary and systemic venous congestion. Numerous cardiovascular comorbidities are associated with CHF, including coronary artery disease, MI, and sudden cardiac death.
PHARMACOLOGIC TREATMENT OF HEART FAILURE
The abnormalities of myocardial structure and function that characterize CHF are often irreversible. These changes narrow the end-diastolic volume range that is compatible with normal cardiac function. Although CHF is predominately a chronic disease, subtle changes to an individual’s hemodynamic status (e.g., increased circulating volume from high dietary sodium intake, increased systemic blood pressure from medication nonadherence) often provoke an acute clinical decompensation.
Not surprisingly, therefore, CHF therapy has for many years used diuretics to control volume overload and subsequent worsening in LV function. Other proven pharmacotherapies target ventricular wall stress, the renin–angiotensin–aldosterone axis, and the sympathetic nervous system to decrease pathologic ventricular remodeling, attenuate disease progression, and improve survival in selected patients with severe CHF and low LV ejection fraction. Figure 28–1 provides an overview of the sites of action for major drug classes commonly used to improve cardiac hemodynamics and function through preload reduction, afterload reduction, and enhancement of inotropy (i.e., myocardial contractility).
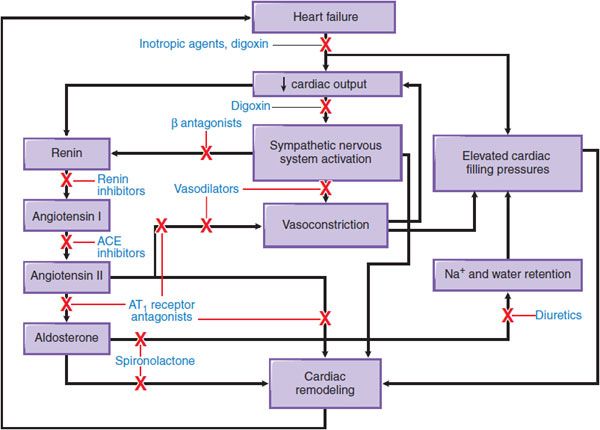
Figure 28–1 Pathophysiologic mechanisms of heart failure and major sites of drug action. Congestive heart failure is accompanied by compensatory neurohormonal responses, including activation of the sympathetic nervous and renin–angiotensin–aldosterone axis. Increased ventricular afterload, due to systemic vasoconstriction and chamber dilation, causes depression in systolic function. In addition, increased afterload and the direct effects of angiotensin and norepinephrine on the ventricular myocardium cause pathologic remodeling characterized by progressive chamber dilation and loss of contractile function. Key congestive heart failure medications and their targets of action are presented. ACE, angiotensin-converting enzyme; AT1 receptor, type 1 angiotensin receptor.
DIURETICS
Diuretics reduce extracellular fluid volume and ventricular filling pressure (or “preload”). Because CHF patients often operate on a “plateau” phase of the Frank-Starling curve (Figure 28–2), incremental preload reduction occurs under these conditions without a reduction in cardiac output. Sustained natriuresis and/or a rapid decline in intravascular volume, however, may “push” one’s profile leftward on the Frank-Starling curve, resulting in an unwanted decrease in cardiac output. In this way, excessive diuresis is counterproductive secondary to reciprocal neurohormonal overactivation from volume depletion. Thus, it is preferable to avoid diuretics in patients with asymptomatic LV dysfunction and to only administer the minimal dose required to maintain euvolemia in those patients symptomatic from hypervolemia. Despite the efficacy of loop or thiazide diuretics in controlling congestive symptoms and improving exercise capacity, their use is not associated with a reduction in CHF mortality.
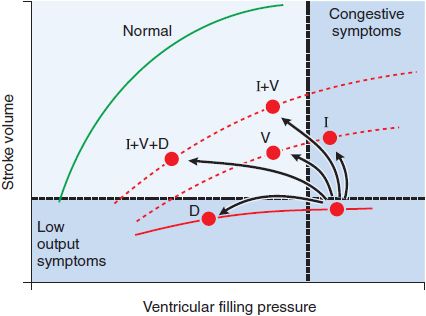
Figure 28–2 Hemodynamic responses to pharmacologic interventions in heart failure. The relationships between diastolic filling pressure (preload) and stroke volume (ventricular performance) are illustrated for a normal heart (green line; the Frank-Starling relationship) and for a patient with heart failure due to predominant systolic dysfunction (red line). Note that positive inotropic agents (I), such as cardiac glycosides or dobutamine, move patients to a higher ventricular function curve (lower dashed line), resulting in greater cardiac work for a given level of ventricular filling pressure. Vasodilators (V), such as angiotensin-converting enzyme (ACE) inhibitors or nitroprusside, also move patients to improved ventricular function curves while reducing cardiac filling pressures. Diuretics (D) improve symptoms of congestive heart failure by moving patients to lower cardiac filling pressures along the same ventricular function curve.
Dietary Na+ Restriction. All patients with clinically significant LV dysfunction, regardless of symptom status, should be advised to limit dietary sodium intake to 2-3 g/day. More stringent salt restriction is seldom necessary and may be counterproductive, as it can lead to hyponatremia, hypokalemia, and hypochloremic metabolic alkalosis when combined with loop diuretic.
Loop Diuretics. Furosemide (LASIX, others), bumetanide (BUMEX, others), and torsemide (DEMADEX, others) are widely used in the treatment of CHF. Due to the increased risk of ototoxicity, ethacrynic acid (EDECRIN) is recommended only for patients with sulfonamides allergies or who are intolerant to alternative options. Loop diuretics inhibit a specific ion transport protein, the Na+-K+-2Cl– symporter on the apical membrane of renal epithelial cells in the ascending limb of the loop of Henle to increase Na+ and fluid delivery to distal nephron segments (see Chapter 25). These drugs also enhance K+ secretion, particularly in the presence of elevated aldosterone levels, as is typical in CHF.
The bioavailability of orally administered furosemide ranges from 40-70%. High drug concentrations often are required to initiate diuresis in patients with worsening symptoms or in those with impaired gastrointestinal absorption, as may occur in severely hypervolemic patients with CHF-induced gut edema. In contrast, the oral bioavailabilities of bumetanide and torsemide are >80%, and as a result, these agents are more consistently absorbed but are financially more costly. Furosemide and bumetanide are short-acting drugs, and rebound Na+ retention that occurs with sub-steady state drug levels make ≥2/day dosing an acceptable treatment strategy when using these agents, provided adequate monitoring of daily body weight and blood electrolyte level monitoring is possible.
Thiazide Diuretics. Monotherapy with thiazide diuretics (DIURIL, HYDRODIURIL, others) has a limited role in CHF. However, combination therapy with loop diuretics is often effective in those refractory to loop diuretics alone. Thiazide diuretics act on the Na+ Cl– co-transporter in the distal convoluted tubule (see Chapter 25) and are associated with a greater degree K+ wasting per fluid volume reduction when compared to loop diuretics.
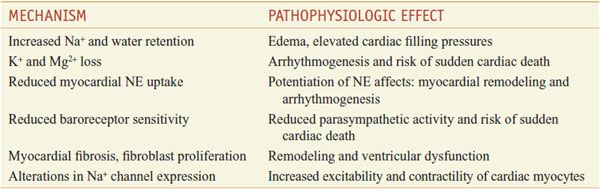
K+-Sparing Diuretics. K+-sparing diuretics (see Chapter 25) inhibit apical membrane Na+-conductance channels in renal epithelial cells (e.g., amiloride, triamterene) or are mineralocorticoid (e.g., aldosterone) receptor antagonists (e.g., canrenone [not commercially available in the U.S.], spironolactone, and eplerenone). Collectively, these agents are weak diuretics, but have been used to achieve volume reduction with limited K+ and Mg2+ wasting.
Diuretics in Clinical Practice. The majority of CHF patients will require chronic administration of a loop diuretic to maintain euvolemia. In patients with clinically evident fluid retention, furosemide typically is started at a dose of 40 mg once or twice daily, and the dosage is increased until an adequate diuresis is achieved. A larger initial dose may be necessary in patients with advanced CHF and azotemia. Serum electrolytes and renal function are monitored frequently. If present, hypokalemia from therapy may be corrected by oral or intravenous K+ supplementation or by the addition of a K+-sparing diuretic.
Diuretics in the Decompensated Patient. In patients with decompensated CHF warranting hospital admission, repetitive intravenously administered boluses or a constant infusion titrated to achieve a desired response may be needed to provide expeditious diuresis. A typical continuous furosemide infusion is initiated with a 40-mg bolus injection followed by a constant rate of 10 mg/h, with uptitration as necessary. If renal perfusion is reduced, drug efficacy may be enhanced by coadministration of drugs that increase cardiac output (e.g., dobutamine).
Diuretic Resistance. (Table 28–1). A compensatory increase in renal tubular Na+ reabsorption may prevent effective diuresis when dosed daily; as a result, reduction of diuretic dosing intervals may be warranted. In advanced CHF, invasive assessment of intracardiac filling pressures and cardiac output may be required to distinguish between low intravascular volume from aggressive diuresis versus low cardiac output states. Other factors can contribute to diuretic resistance (see Table 28–1).
Table 28–1
Causes of Diuretic Resistance in Heart Failure

Metabolic Consequences of Diuretic Therapy. With regard to diuretic use in CHF, the most important adverse sequelae of diuretics are electrolyte abnormalities, including hyponatremia, hypokalemia, and hypochloremic metabolic alkalosis.
Adenosine A1 Receptor Antagonists. Adenosine A1 receptor antagonists may provide a renal protective therapeutic strategy for enhanced volume loss in decompensated CHF. Adenosine is secreted from the macula densa in the renal arteriole in response to diuretic-induced increases in Na+ and Cl– tubular flow concentrations. This results in increased Na+ resorption, a volume-loss counterregulatory mechanism (see Chapter 26). Na+ reabsorption, in addition to adenosine-induced renal arteriole vasoconstriction, appears responsible (in part) for the development of complications common to the use of diuretics in decompensated CHF patients, particularly prerenal azotemia. The role of adenosine in the macula densa and juxtaglomerular (granular) cells suggests other effects of A1 antagonists on the renin-angiotensin system (see Figure 26–3). Administration of A1 antagonists KW-3902 (ROLOFYLLINE) or BG9179 (NAXIFYLLINE), to patients with decompensated CHF already treated with loop diuretics, has been associated with increased volume reduction, improved renal function, and lower diuretic dosing, however, a clinical trial failed to show significant benefits of rolofylline in patients with CHF and clinical development of the drug was stopped in 2009. No A1 antagonists are currently marketed in the U.S.
ALDOSTERONE ANTAGONISTS AND CLINICAL OUTCOME
LV systolic dysfunction decreases renal blood flow and results in overactivation of the renin–angiotensin–aldosterone axis and may increase circulating plasma aldosterone levels in CHF to 20-fold above normal. The pathophysiologic effects of hyperaldosteronemia are diverse (Table 28–2) and extend beyond Na+ and fluid retention; however, the precise mechanism by which aldosterone receptor blockade improves outcome in CHF remains unresolved.
Table 28–2
Potential Roles of Aldosterone in the Pathophysiology of Heart Failure
Aldosterone-receptor antagonists in combination with ACE inhibitor therapy have provided beneficial effects in clinical trials. In CHF patients with low LV ejection fraction, spironolactone (25 mg/day) decreased mortality by ~30% (from progressive heart failure or sudden cardiac death), and patients had fewer CHF-related hospitalizations compared with the placebo group. Treatment was well tolerated; however, 10% of men reported gynecomastia and 2% of all patients developed severe hyperkalemia (>6.0 mEq/L).
ORAL VASODILATORS
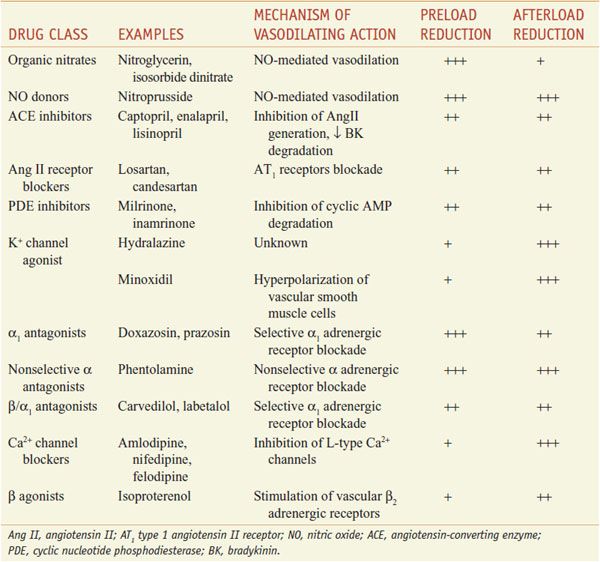
Although numerous vasodilators have been developed that improve CHF symptoms, only the hydralazine–isosorbide dinitrate combination, ACE inhibitors, and AT1 receptor blockers (ARBs) demonstrably improve survival. The therapeutic use of vasodilators in the treatment of hypertension and myocardial ischemia is considered in detail in Chapter 27. This chapter focuses on the uses for some of these same vasodilator drugs in the treatment of CHF, mainly through their capacity to reduce preload and afterload (Table 28–3).
Table 28–3
Vasodilator Drugs Used to Treat Heart Failure
NITROVASODILATORS. Nitrovasodilators are nitric oxide (NO) donors that activate soluble guanylate cyclase in vascular smooth muscle cells, leading to vasodilation. Unlike nitroprusside, which is converted to NO• by cellular reducing agents such as glutathione, nitroglycerin and other organic nitrates undergo a more complex enzymatic biotransformation to NO• or bioactive S-nitrosothiols. The activities of specific enzyme(s) and cofactor(s) required for this biotransformation appear to differ by target organ and even by different vasculature beds within a particular organ.
Organic Nitrates. Organic nitrates are available in a number of formulations that include rapid-acting nitroglycerin tablets or spray for sublingual administration, short-acting oral agents such as isosorbide dinitrate (ISORDIL, others), long-acting oral agents such as isosorbide mononitrate (ISMO, others), topical preparations such as nitroglycerin ointment and transdermal patches, and intravenous nitroglycerin. The principal action of these preparations in CHF is reducing LV filling pressure. This occurs, in part, by augmentation of peripheral venous capacitance that results in preload reduction. Additional effects of organic nitrates include pulmonary and systemic vascular resistance reduction, and coronary artery vasodilation for which systolic and diastolic ventricular function is enhanced by increased coronary blood flow. These beneficial physiologic effects translate into improved exercise capacity and CHF-symptom reduction. However, these drugs do not substantially influence systemic vascular resistance, and pharmacologic tolerance limits their utility over time. Organic nitrates are commonly used with other vasodilators (e.g., hydralazine) to increase the clinical effectiveness.
Nitrate Tolerance. Nitrate tolerance may limit the long-term effectiveness of these drugs in the treatment of CHF. Blood nitrate levels may be permitted to fall to negligible levels for at least 6-8 h each day (see Chapter 27). Patients with recurrent orthopnea or paroxysmal nocturnal dyspnea, e.g., might benefit from nighttime nitrate use. Likewise, co-treatment with hydralazine may decrease nitrate tolerance by an antioxidant effect that attenuates superoxide formation, thereby increasing the bioavailable NO levels.
PARENTERAL VASODILATORS
Sodium Nitroprusside. Sodium nitroprusside (NITRO-PRESS, others) is a direct NO donor and potent vasodilator that is effective in reducing both ventricular filling pressure and systemic vascular resistance (Figure 28–3). Onset to action is rapid (2-5 min) as the drug is quickly metabolized to NO. Nitroprusside is effective in treating critically ill patients with CHF who have elevated systemic vascular resistance or mechanical complications that follow acute MI (e.g., mitral regurgitation or ventricular septal defect-induced left-to-right shunts). It increases cardiac output and renal blood flow, improving both glomerular filtration and diuretic effectiveness. The most common adverse side effect of nitroprusside is hypotension. Excessive reduction of systemic arterial pressure may limit or prevent an increase in renal blood flow in patients with more severe LV contractile dysfunction.
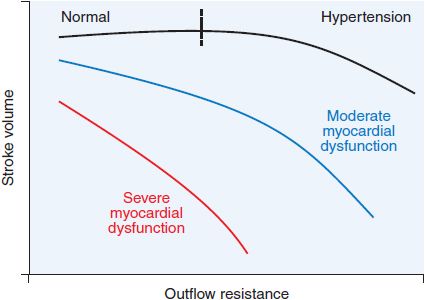
Figure 28–3 Relationship between ventricular outflow resistance and stroke volume in patients with systolic ventricular dysfunction. An increase in ventricular outflow resistance, a principal determinant of afterload, has little effect on stroke volume in normal hearts, as illustrated by the relatively flat curve. In contrast, in patients with systolic ventricular dysfunction, an increase in outflow resistance often is accompanied by a sharp decline in stroke volume. With more severe ventricular dysfunction, this curve becomes steeper. Because of this relationship, a reduction in systemic vascular resistance (1 component of outflow resistance) in response to an arterial vasodilator may markedly increase stroke volume in patients with severe myocardial dysfunction. The resultant increase in stroke volume may be sufficient to offset the decrease in systemic vascular resistance, thereby preventing a fall in systemic arterial pressure. (Adapted from Cohn and Franciosa, 1977.)
Cyanide produced during the biotransformation of nitroprusside is rapidly metabolized by the liver to thiocyanate, which is then renally excreted. Thiocyanate and/or cyanide toxicity is uncommon but may occur in the setting of hepatic or renal failure, or following prolonged periods of high-dose drug infusion (see Chapter 27 for details). Typical symptoms include unexplained abdominal pain, mental status changes, convulsions, and lactic acidosis. Methemoglobinemia is another unusual complication and is due to the oxidation of hemoglobin by NO•.
Intravenous Nitroglycerin. Intravenous nitroglycerin is a vasoactive NO donor that is used in the intensive care unit setting. Unlike nitroprusside, nitroglycerin is relatively selective for venous capacitance vessels, particularly at low infusion rates. In CHF, intravenous nitroglycerin is most commonly used in the treatment of LV dysfunction due to an acute myocardial ischemia. Parenteral nitroglycerin also is used in the treatment of nonischemic cardiomyopathy when expeditious LV filling pressure reduction is desired. At higher infusion rates, this drug also may decrease systemic arterial resistance. Nitroglycerin therapy may be limited by headache and nitrate tolerance; tolerance may be partially offset by increasing the dosage.
Hydralazine. Hydralazine is a direct vasodilator whose mechanism of action is poorly understood. Hydralazine is an effective antihypertensive drug (see Chapter 27
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


