Cytotoxic Agents
I. Alkylating Agents and Platinum Coordination Complexes
In 1942, Louis Goodman and Alfred Gilman, the originators of this text, began clinical studies of intravenous nitrogen mustards in patients with lymphoma, launching the modern era of cancer chemotherapy. Six major types of alkylating agents are used in the chemotherapy of neoplastic diseases:
• Nitrogen mustards
• Ethyleneimines
• Alkyl sulfonates
• Nitrosoureas
• The triazenes
• DNA-methylating drugs, including procarbazine, temozolomide, and dacarbazine
In addition, because of similarities in their mechanisms of action and resistance, platinum complexes are discussed with classical alkylating agents, even though they do not alkylate DNA but instead form covalent metal adducts with DNA.
The chemotherapeutic alkylating agents have in common the property of forming highly reactive carbonium ion intermediates. These reactive intermediates covalently link to sites of high electron density, such as phosphates, amines, sulfhydryl, and hydroxyl groups. Their chemotherapeutic and cytotoxic effects are directly related to the alkylation of reactive amines, oxygens, or phosphates on DNA. The general mechanisms actions of alkylating agents on DNA are illustrated in Figure 61–1 with mechlorethamine (nitrogen mustard). The extreme cytotoxicity of bifunctional alkylators correlates very closely with interstrand cross-linkage of DNA.
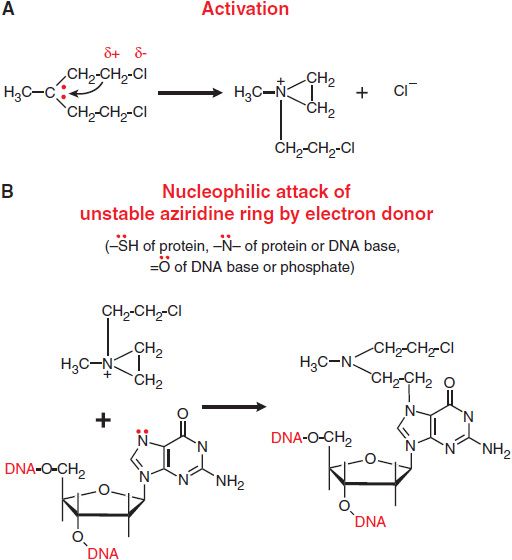
Figure 61–1 Mechanism of action of alkylating agents. A. Activation reaction. B. Alkylation of N7 of guanine.
The ultimate cause of cell death related to DNA damage is not known. Specific cellular responses include cell-cycle arrest and attempts to repair DNA. The specific repair enzyme complex utilized will depend on 2 factors: the chemistry of the adduct formed and the repair capacity of the cell involved. The process of recognizing and repairing DNA generally requires an intact nucleotide excision repair (NER) complex, but may differ with each drug and with each tumor. Alternatively, recognition of extensively damaged DNA by p53 can trigger apoptosis. Mutations of p53 lead to alkylating agent resistance.
Structure-Activity Relationships. Although these alkylating agents share the capacity to alkylate biologically important molecules, modification of the basic chloroethylamino structure changes reactivity, lipophilicity, active transport across biological membranes, sites of macromolecular attack, and mechanisms of DNA repair, all of which determine drug activity in vivo. With several of the most valuable agents (e.g., cyclophosphamide, ifosfamide), the active alkylating moieties are generated in vivo through hepatic metabolism (Figure 61–2). Consult Chapter 61 of the 12th edition of the parent text for details on metabolic activation and structure-activity relationships among these compounds.
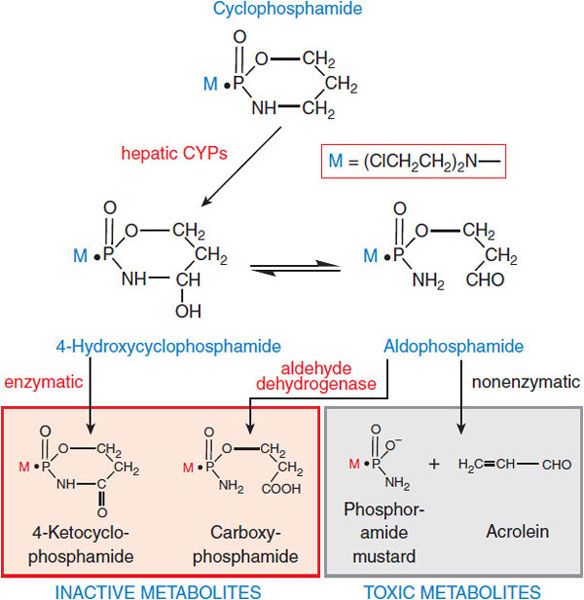
Figure 61–2 Metabolic activation of cyclophosphamide. Cyclophosphamide is activated (hydroxylated) by CYP2B, with subsequent transport of the activated intermediate to sites of action. The selectivity of cyclophosphamide against certain malignant tissues may result in part from the capacity of normal tissues to degrade the activated intermediates via aldehyde dehydrogenase, glutathione transferase, and other pathways. Ifosfamide is structurally similar to cyclophosphamide: whereas cyclophosphamide has 2 chloroethyl groups on the exocyclic nitrogen atom, 1 of the 2-chloroethyl groups of ifosfamide is on the cyclic phosphoramide nitrogen of the oxazaphosphorine ring. Ifosfamide is activated by hepatic CYP3A4. The activation of ifosfamide proceeds more slowly, with greater production of de-chlorinated metabolites and chloroacetaldehyde. These differences in metabolism likely account for the higher doses of ifosfamide required for equitoxic effects, the greater neurotoxicity of ifosfamide, and perhaps differences in the antitumor spectra of cyclophosphamide and ifosfamide.
The newest approved alkylating agent, bendamustine, has the typical chloroethyl reactive groups attached to a benzimidazole backbone. The unique properties and activity of this drug may derive from this purine-like structure; the agent produces slowly repaired DNA cross-links, lacks cross-resistance with other classical alkylators, and has significant activity in chronic lymphocytic leukemia (CLL) and large-cell lymphomas refractory to standard alkylators. One class of alkylating agents transfers methyl rather than ethyl groups to DNA. The triazene derivative 5-(3,3-dimethyl-1-triazeno)-imidazole-4-carboxamide, usually called dacarbazine or DTIC, is prototypical of methylating agents. Dacarbazine requires initial activation by hepatic CYPs through an N-demethylation reaction. In the target cell, spontaneous cleavage of the metabolite, methyl-triazeno-imidazole-carboxamide (MTIC), yields an alkylating moiety, a methyl diazonium ion. A related triazene, temozolomide, undergoes spontaneous, nonenzymatic activation to MTIC and has significant activity against gliomas.
The nitrosoureas, which include compounds such as 1,3-bis-(2-chloroethyl)-1-nitrosourea (carmustine [BCNU]), 1-(2-chloroethyl)-3-cyclohexyl-1-nitrosourea (lomustine [CCNU]), and its methyl derivative (semustine [methyl-CCNU]), as well as the antibiotic streptozocin (streptozotocin), exert their cytotoxicity through the spontaneous breakdown to an alkylating intermediate, the 2-chloroethyl diazonium ion. As with the nitrogen mustards, interstrand cross-linking appears to be the primary lesion responsible for the cytotoxicity of nitrosoureas. The reactions of the nitrosoureas with macromolecules are shown in Figure 61–3.
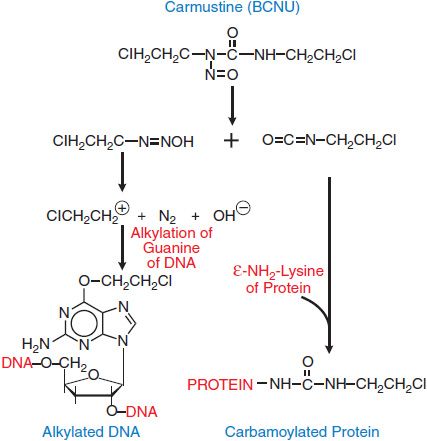
Figure 61–3 Generation of alkylating and carbamylating intermediates from carmustine (BCNU). The 2-chloroethyl diazonium ion, a strong electrophile, can alkylate guanine, cytidine, and adenine bases. Displacement of the halogen atom that can then lead to interstrand or intrastrand cross-linking of DNA. The formation of cross-links after the initial alkylation reaction proceeds slowly and can be reversed by the DNA repair enzyme O6-alkyl, methyl guanine methyltransferase (MGMT), which displaces the chloroethyl adduct from its binding to guanine in a suicide reaction. The same enzyme, when expressed in human gliomas, produces resistance to nitrosoureas and to other methylating agents, including DTIC, temozolomide, and procarbazine.
Stable ethyleneimine derivatives have antitumor activity; triethylenemelamine (TEM) and triethylenethiophosphoramide (thiotepa) have been used clinically. In standard doses, thiotepa produces little toxicity other than myelosuppression; it also is used for high-dose chemotherapy regimens, in which it causes both mucosal and CNS toxicity. Altretamine (hexamethylmelamine [HMM]) is chemically similar to TEM. The methylmelamines are N-demethylated by hepatic microsomes with the release of formaldehyde; there is a direct relationship between the degree of the demethylation and their antitumor activity in model systems.
Esters of alkane sulfonic acids alkylate DNA through the release of methyl radicals. Busulfan is of value in high-dose chemotherapy.
GENERAL PHARMACOLOGICAL ACTIONS
Cytotoxic Actions. The capacity of alkylating agents to interfere with DNA integrity and function and to induce cell death in rapidly proliferating tissues provides the basis for their therapeutic and toxic properties. Acute effects manifest primarily against rapidly proliferating tissues; however, certain alkylating agents may have damaging effects on tissues with normally low mitotic indices (e.g., liver, kidney, and mature lymphocytes); effects in these tissues usually are delayed. Lethality of DNA alkylation depends on the recognition of the adduct, the creation of DNA strand breaks by repair enzymes, and an intact apoptotic response. In non-dividing cells, DNA damage activates a checkpoint that depends on the presence of a normal p53 gene. Cells thus blocked in the G1/S interface either repair DNA alkylation or undergo apoptosis. Malignant cells with mutant or absent p53 fail to suspend cell-cycle progression, do not undergo apoptosis, and exhibit resistance to these drugs.
Although DNA is the ultimate target of all alkylating agents, there is a crucial distinction between the bifunctional agents, in which cytotoxic effects predominate, and the monofunctional methylating agents (procarbazine, temozolomide), which have greater capacity for mutagenesis and carcinogenesis. This suggests that the cross-linking of DNA strands represents a much greater threat to cellular survival than do other effects, such as single-base alkylation and the resulting depurination and single-chain scission. Conversely, simple methylation may be bypassed by DNA polymerases, leading to mispairing reactions that permanently modify DNA sequence. These new sequences are transmitted to subsequent generations and may result in mutagenesis or carcinogenesis. Some methylating agents, such as procarbazine, are highly carcinogenic.
Adduct recognition systems and DNA repair systems play important roles in removing adducts, and thereby determine the selectivity of action against particular cell types and the acquisition of resistance to alkylating agents. Alkylation of a single strand of DNA (mono-adducts) is repaired by the nucleotide excision repair pathway; the less frequent cross-links require participation of nonhomologous end joining, an error-prone pathway, or the error-free homologous recombination pathway. After drug infusion in humans, mono-adducts appear rapidly and peak within 2 h of drug exposure, while cross-links peak at 8 h. The t1/2 for repair of adducts varies among normal tissues and tumors; in peripheral blood mononuclear cells, both mono-adducts and cross-links disappear with a t1/2 of 12-16 h.
The repair process depends on the presence and accurate functioning of multiple proteins. Their absence or mutation, as in Fanconi anemia or ataxia telangiectasia, leads to extreme sensitivity to DNA cross-linking agents such as mitomycin, cisplatin, or classical alkylators. Other repair enzymes are specific for removing methyl and ethyl adducts from the O-6 of guanine (MGMT) and for repair of alkylation of the N-3 of adenine and N-7 of guanine (3-methyladenine-DNA glycosylase). High expression of MGMT protects cells from cytotoxic effects of nitrosoureas and methylating agents and confers drug resistance, while methylation and silencing of the gene in brain tumors are associated with clinical response to BCNU and temozolomide. Bendamustine differs from classical chloroethyl alkylators in activating base excision repair, rather than the more complex double-strand break repair or MGMT. It impairs physiological arrest of adduct-containing cells at mitotic checkpoints and leads to mitotic catastrophe rather than apoptosis, and does not require an intact p53 to cause cytotoxicity.
Recognition of DNA adducts is an essential step in promoting attempts at repair and ultimately leading to apoptosis. The Fanconi pathway, consisting of 12 proteins, recognizes adducts and signals the need for repair of a broad array of DNA-damaging drugs and irradiation. Absence or inactivation of components of this pathway leads to increased sensitivity to DNA damage. Conversely, for the methylating drugs, nitrosoureas, cisplatin and carboplatin, and thiopurine analogs, the mismatch repair (MMR) pathway is essential for cytotoxicity, causing strand breaks at sites of adduct formation, creating mispairing of thymine residues, and triggering apoptosis.
Mechanisms of Resistance to Alkylating Agents. Resistance to an alkylating agent develops rapidly when it is used as a single agent. Specific biochemical changes implicated in the development of resistance include:
• Decreased permeation of actively transported drugs (mechlorethamine and melphalan).
• Increased intracellular concentrations of nucleophilic substances, principally thiols such as glutathione, which can conjugate with and detoxify electrophilic intermediates.
• Increased activity of DNA repair pathways, which may differ for the various alkylating agents.
• Increased rates of metabolic degradation of the activated forms of cyclophosphamide and ifosfamide to their inactive keto and carboxy metabolites by aldehyde dehydrogenase (see Figure 61–2), and detoxification of most alkylating intermediates by glutathione transferases.
• Loss of ability to recognize adducts formed by nitrosoureas and methylating agents, as the result of defective mismatch repair (MMR) proteins capability, confers resistance, as does defective checkpoint function, for virtually all alkylating drugs.
• Impaired apoptotic pathways, with overexpression of bcl-2 as an example, confer resistance.
TOXICITIES OF ALKYLATING AGENTS
BONE MARROW. Alkylating agents differ in their patterns of antitumor activity and in the sites and severity of their side effects. Most cause dose-limiting toxicity to bone marrow elements and, to a lesser extent, intestinal mucosa. Most alkylating agents (i.e., melphalan, chlorambucil, cyclophosphamide, and ifosfamide) cause acute myelosuppression, with a nadir of the peripheral blood granulocyte count at 6-10 days and recovery in 14-21 days. Cyclophosphamide has lesser effects on peripheral blood platelet counts than do the other agents. Busulfan suppresses all blood elements, particularly stem cells, and may produce a prolonged and cumulative myelosuppression lasting months or even years. For this reason, it is used as a preparative regimen in allogenic bone marrow transplantation. Carmustine and other chloroethylnitrosoureas cause delayed and prolonged suppression of both platelets and granulocytes, reaching a nadir 4-6 weeks after drug administration and reversing slowly thereafter. Both cellular and humoral immunity are suppressed by alkylating agents, which have been used to treat various autoimmune diseases. Immunosuppression is reversible at usual doses, but opportunistic infections may occur with extended treatment.
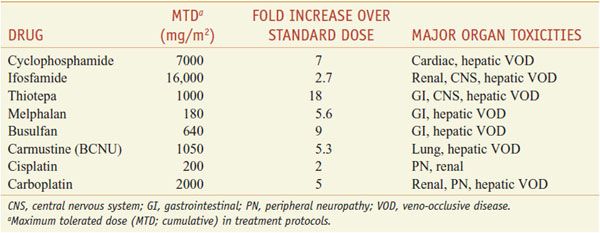
MUCOSA. Alkylating agents are highly toxic to dividing mucosal cells and to hair follicles, leading to oral mucosal ulceration, intestinal denudation, and alopecia. Mucosal effects are particularly damaging in high-dose chemotherapy protocols associated with bone marrow reconstitution, as they predispose to bacterial sepsis arising from the GI tract. In these protocols, cyclophosphamide, melphalan, and thiotepa have the advantage of causing less mucosal damage than the other agents. In high-dose protocols, however, other toxicities become limiting (Table 61–1).
Table 61–1
Dose-Limiting Extramedullary Toxicities of Single Alkylating Agents
NERVOUS SYSTEM. Nausea and vomiting commonly follow agent administration of nitrogen mustard or BCNU. Ifosfamide is the most neurotoxic agent of this class and may produce altered mental status, coma, generalized seizures, and cerebellar ataxia. These side effects result from the release of chloroacetaldehyde from the phosphate-linked chloroethyl side chain of ifosfamide. High-dose busulfan can cause seizures; in addition, it accelerates the clearance of phenytoin, an antiseizure medication.
OTHER ORGANS. All alkylating agents, including temozolomide, have caused pulmonary fibrosis, usually several months after treatment. In high-dose regimens, particularly those employing busulfan or BCNU, vascular endothelial damage may precipitate veno-occlusive disease (VOD) of the liver, an often fatal side effect that is successfully reversed by the investigational drug defibrotide. The nitrosoureas and ifosfamide, after multiple cycles of therapy, may lead to renal failure. Cyclophosphamide and ifosfamide release a nephrotoxic and urotoxic metabolite, acrolein, which causes a severe hemorrhagic cystitis in high-dose regimens. This adverse effect can be prevented by coadministration of 2-mercaptoethanesulfonate (mesna [MESNEX]), which conjugates acrolein in urine. Ifosfamide in high doses for transplant causes a chronic, and often irreversible, renal toxicity. Nephrotoxicity correlates with the total dose of drug received and increases in frequency in children <5 years of age. The syndrome may be due to chloroacetaldehyde and/or acrolein excreted in the urine.
The more unstable alkylating agents (e.g., mechlorethamine and the nitrosoureas) have strong vesicant properties, damage veins with repeated use and, if extravasated, produce ulceration. All alkylating agents have toxic effects on the male and female reproductive systems, causing an often permanent amenorrhea, particularly in perimenopausal women, and an irreversible azoospermia in men.
Leukemogenesis. Alkylating agents are highly leukemogenic. Acute nonlymphocytic leukemia, often associated with partial or total deletions of chromosome 5 or 7, peaks in incidence ~4 years after therapy and may affect up to 5% of patients treated on regimens containing alkylating drugs. Leukemia often is preceded by a period of neutropenia or anemia and by bone marrow morphology consistent with myelodysplasia. Melphalan, the nitrosoureas, and the methylating agent procarbazine have the greatest propensity to cause leukemia, while it is less common after cyclophosphamide.
CLINICAL PHARMACOLOGY
MECHLORETHAMINE
Mechlorethamine HCl (MUSTARGEN) was the first clinically used nitrogen mustard and is the most reactive of the drugs in this class. It is used topically for treatment of cutaneous T-cell lymphoma (CTCL) as a solution that is rapidly mixed and applied to affected areas. It has been largely replaced by cyclophosphamide, melphalan, and other, more stable alkylating agents.
Severe local reactions of exposed tissues necessitate rapid intravenous injection of mechlorethamine for most uses. Mechlorethamine, rapidly and within minutes, undergoes chemical degradation (affected markedly by pH) as it combines with either water or cellular nucleophiles. The major acute toxic manifestations of mechlorethamine are nausea and vomiting, lacrimation, and myelosuppression. Leukopenia and thrombocytopenia limit the amount of drug that can be given in a single course.
CYCLOPHOSPHAMIDE
ADME. Cyclophosphamide is well absorbed orally and is activated to the 4-hydroxy intermediate (see Figure 61–2). Its rate of metabolic activation exhibits significant interpatient variability and increases with successive doses in high-dose regimens but appears to be saturable at infusion rates of >4 g/90 min and concentrations of the parent compound >150 μM. 4-Hydroxycyclophosphamide may be oxidized further by aldehyde oxidase, either in liver or in tumor tissue, to inactive metabolites. The hydroxyl metabolite of ifosfamide similarly is inactivated by aldehyde dehydrogenase. 4-Hydroxycyclophosphamide and its tautomer, aldophosphamide, travel in the circulation to tumor cells where aldophosphamide cleaves spontaneously, generating stoichiometric amounts of phosphoramide mustard and acrolein. Phosphoramide mustard is responsible for antitumor effects, while acrolein causes hemorrhagic cystitis often seen during therapy with cyclophosphamide. Patients should receive vigorous intravenous hydration during high-dose treatment. Brisk hematuria in a patient receiving daily oral therapy should lead to immediate drug discontinuation. Refractory bladder hemorrhage can become life-threatening and may require cystectomy for control of bleeding. Inappropriate secretion of antidiuretic hormone has been observed, usually at doses >50 mg/kg (see Chapter 25). It is important to be aware of the possibility of water intoxication, because these patients usually are vigorously hydrated.
Pretreatment with CYP inducers such as phenobarbital enhances the rate of activation of the azoxyphosphorenes but does not alter total exposure to active metabolites over time and does not affect toxicity or efficacy. Cyclophosphamide can be used in full doses in patients with renal dysfunction, because it is eliminated by hepatic metabolism. Patients with significant hepatic dysfunction (bilirubin <3 mg/dL) should receive reduced doses. Maximal CP is achieved 1 hour after oral administration; the t1/2 of parent drug in plasma is ~7 h.
Therapeutic Uses. Cyclophosphamide (LYOPHILIZED CYTOXAN) is administered orally or intravenously. Recommended doses vary widely, and standard protocols for determining the schedule and dose of cyclophosphamide in combination with other chemotherapeutic agents should be consulted. As a single agent, a daily oral dose of 100 mg/m2 for 14 days has been recommended for patients with lymphomas and CLL. Higher doses of 500 mg/m2 intravenously every 2-4 weeks are used in combination with other drugs in the treatment of breast cancer and lymphomas. The neutrophil nadir of 500-1000 cells/mm3 generally serves as a lower limit for dosage adjustments in prolonged therapy. In regimens associated with bone marrow or peripheral stem cell rescue, cyclophosphamide may be given in total doses of 5-7 g/m2 over a 3-5-day period. GI ulceration, cystitis (counteracted by mesna and diuresis), and, less commonly, pulmonary, renal, hepatic, and cardiac toxicities (hemorrhagic myocardial necrosis) may occur after high-dose therapy with total doses >200 mg/kg.
The clinical spectrum of activity for cyclophosphamide is very broad. It is an essential component of many effective drug combinations for non-Hodgkin lymphomas, other lymphoid malignancies, breast and ovarian cancers, and solid tumors in children. Complete remissions and presumed cures have been reported when cyclophosphamide was given as a single agent for Burkitt lymphoma. It frequently is used in combination with doxorubicin and a taxane as adjuvant therapy after surgery for breast cancer. Because of its potent immunosuppressive properties, cyclophosphamide has been used to treat autoimmune disorders, including Wegener granulomatosis, rheumatoid arthritis, and the nephrotic syndrome. Caution is advised when the drug is considered for non-neoplastic conditions, not only because of its acute toxic effects but also because of its potential for inducing sterility, teratogenic effects, and leukemia.
IFOSFAMIDE
Ifosfamide (IFEX, others) is an analog of cyclophosphamide. Severe urinary tract and CNS toxicity initially limited the use of ifosfamide, but adequate hydration and coadministration of mesna have reduced its bladder toxicity.
Therapeutic Uses. Ifosfamide is approved for treatment of relapsed germ cell testicular cancer and is frequently used for first-time treatment of pediatric and adult sarcomas. It is a common component of high-dose chemotherapy regimens with bone marrow or stem cell rescue; in these regimens, in total doses of 12-14 g/m2, it may cause severe neurological toxicity, including hallucinations, coma, and death, with symptoms appearing 12 h to 7 days after beginning the ifosfamide infusion. This toxicity may result from a metabolite, chloroacetaldehyde. Ifosfamide also causes nausea, vomiting, anorexia, leukopenia, nephrotoxicity, and VOD of the liver. In nonmyeloablative regimens, ifosfamide is infused intravenously over at least 30 min at a dose of ≤1.2 g/m2/day for 5 days. Intravenous mesna is given as bolus injections in a dose equal to 20% of the ifosfamide dose concomitantly and an additional 20% again 4 and 8 h later, for a total mesna dose of 60% of the ifosfamide dose. Alternatively, mesna may be given concomitantly in a single dose equal to the ifosfamide dose. Patients also should receive at least 2 L of oral or intravenous fluid daily. Treatment cycles are repeated every 3-4 weeks.
Pharmacokinetics. Ifosfamide has a plasma elimination t1/2 ~1.5 h after doses of 3.8-5 g/m2 and a somewhat shorter t1/2 at lower doses; its pharmacokinetics are highly variable due to variable rates of hepatic metabolism (see legend to Figure 61–2).
Toxicity. Ifosfamide has virtually the same toxicity profile as cyclophosphamide, although it causes greater platelet suppression, neurotoxicity, nephrotoxicity, and in the absence of mesna, urothelial damage.
MELPHALAN
This alkylating agent primarily is used to treat multiple myeloma and, less commonly, in high-dose chemotherapy with marrow transplantation. The general pharmacological and cytotoxic actions of melphalan are similar to those of other bifunctional alkylators. The drug is not a vesicant.
ADME. Oral melphalan is absorbed in an inconsistent manner and, for most indications, is given as an intravenous infusion. The drug has a plasma t1/2 ~45-90 min; 10-15% of an administered dose is excreted unchanged in the urine. Patients with decreased renal function may develop unexpectedly severe myelosuppression.
Therapeutic Uses. Melphalan (ALKERAN) for multiple myeloma is given in doses of 4-10 mg orally for 4-7 days every 28 days, with dexamethasone or thalidomide. Treatment is repeated at 4-week intervals based on response and tolerance. Dosage adjustments should be based on blood cell counts. Melphalan also may be used in myeloablative regimens followed by bone marrow or peripheral blood stem cell reconstitution, at a dose of 180-200 mg/m2. The toxicity of melphalan is mostly hematological and is similar to that of other alkylating agents. Nausea and vomiting are less frequent. The drug causes less alopecia and, rarely, renal or hepatic dysfunction.
CHLORAMBUCIL
The cytotoxic effects of chlorambucil on the bone marrow, lymphoid organs, and epithelial tissues are similar to those observed with other nitrogen mustards. As an orally administered agent, chlorambucil is well tolerated in small daily doses and provides flexible titration of blood counts. Nausea and vomiting may result from single oral doses of ≥20 mg.
ADME. Oral absorption of chlorambucil is adequate and reliable. The drug has a t1/2 in plasma of ~1.5 h and is hydrolyzed to inactive products.
Therapeutic Uses. Chlorambucil is almost exclusively used in treating CLL, for which it has largely been replaced by fludarabine and cyclophosphamide. In treating CLL, the initial daily dose of chlorambucil (LEUKERAN) is 0.1-0.2 mg/kg, given once daily and continued for 3-6 weeks. With a fall in the peripheral total leukocyte count or clinical improvement, the dosage is titrated to maintain neutrophils and platelets at acceptable levels. Maintenance therapy (usually 2 mg daily) often is required to maintain clinical response. Chlorambucil treatment may continue for months or years, achieving its effects gradually and often without significant toxicity to a compromised bone marrow. Marked hypoplasia of the bone marrow may be induced with excessive doses, but the myelosuppressive effects are moderate, gradual, and rapidly reversible. GI discomfort, azoospermia, amenorrhea, pulmonary fibrosis, seizures, dermatitis, and hepatotoxicity rarely may be encountered. A marked increase in the incidence of acute myelocytic leukemia (AML) and other tumors was noted in the treatment of polycythemia vera and in patients with breast cancer receiving chlorambucil as adjuvant chemotherapy.
BENDAMUSTINE
This drug is approved for treatment of CLL and non-Hodgkin lymphoma. Bendamustine is given as a 30-min intravenous infusion in dosages of 100 mg/m2/day on days 1 and 2 of a 28-day cycle. Lower doses may be indicated in heavily pretreated patients. Bendamustine is rapidly degraded through sulfhydryl interaction and adduct formation with macromolecules; <5% of the parent drug is excreted in the urine intact. N-demethylation and oxidation produces metabolites that have antitumor activity, but less than that of the parent molecule. The parent drug has a plasma t1/2 ~30 min. The clinical toxicity pattern of bendamustine is typical of alkylators, with a rapidly reversible myelosuppression and mucositis, both generally tolerable.
MISCELLANEOUS ALKYLATING DRUGS
Although nitrogen mustards containing chloroethyl groups constitute the most widely used class of alkylating agents, alternative alkylators with greater chemical stability and well-defined activity in specific types of cancer have value in clinical practice.
ETHYLENEIMINES AND METHYLMELAMINES
ALTRETAMINE
Altretamine (hexamethylmelamine [HEXALEN]) is structurally similar to TEM (tretamine). Its precise mechanism of cytotoxicity is unknown. It is a palliative treatment for persistent or recurrent ovarian cancer following cisplatin-based combination therapy. The usual dosage of altretamine as a single agent in ovarian cancer is 260 mg/m2/day in 4 divided doses, for 14 or 21 consecutive days out of a 28-day cycle, for up to 12 cycles.
ADME. Altretamine is well absorbed from the GI tract; its elimination t1/2 is 4-10 h. The drug undergoes rapid demethylation in the liver; the principal metabolites are pentamethylmelamine and tetramethyl melamine.
Clinical Toxicities. The main toxicities of altretamine are myelosuppression and neurotoxicity. Altretamine causes both peripheral and central neurotoxicity (ataxia, depression, confusion, drowsiness, hallucinations, dizziness, and vertigo). Neurological symptoms abate upon discontinuation of therapy. Peripheral blood counts and a neurological examination should be performed prior to the initiation of each course of therapy. Therapy should be interrupted for at least 14 days and subsequently restarted at a lower dosage of 200 mg/m2/day if the white cell count falls to <2000 cells/mm3 or the platelet count falls to <75,000 cells/mm3 or if neurotoxic or intolerable GI symptoms occur. If neurological symptoms fail to stabilize on the reduced-dose schedule, altretamine should be discontinued. Nausea and vomiting also are common side effects and may be dose limiting. Renal dysfunction may necessitate discontinuing the drug. Other rare adverse effects include rashes, alopecia, and hepatic toxicity. Severe, life-threatening orthostatic hypotension may develop in patients who receive MAO-inhibitors, amitriptyline, imipramine, or phenelzine concurrently with altretamine.
THIOTEPA
Thiotepa (THIOPLEX, others) consists of 3 ethyleneimine groups stabilized by attachment to the nucleophilic thiophosphoryl base. Its current use is primarily for high-dose chemotherapy regimens. Both thiotepa and its desulfurated primary metabolite, triethylenephosphoramide (TEPA), to which it is rapidly converted by hepatic CYPs, form DNA cross-links.
ADME. TEPA becomes the predominant form of the drug present in plasma within hours of thiotepa administration. The parent compound has a plasma t1/2 of 1.2-2 h, TEPA has a longer t1/2, 3-24 h. Thiotepa pharmacokinetics essentially are the same in children as in adults at conventional doses (<80 mg/m2), and drug and metabolite t1/2 are unchanged in children receiving high-dose therapy of 300 mg/m2/day for 3 days. Less than 10% of the administered drug appears in urine as the parent drug or the primary metabolite.
Clinical Toxicities. Toxicities include myelosuppression and, to a lesser extent, mucositis. Myelosuppression tends to develop somewhat later than with cyclophosphamide, with leukopenic nadirs at 2 weeks and platelet nadirs at 3 weeks. In high-doses, thiotepa may cause neurotoxic symptoms, including coma and seizures.
ALKYL SULFONATES
BUSULFAN
Busulfan (MYLERAN, BUSULFEX) exerts few pharmacological actions other than myelosuppression at conventional doses and, prior to the advent of imatinib mesylate (GLEEVEC), was a standard agent for patients in the chronic phase of myelocytic leukemia and caused a severe and prolonged pancytopenia in some patients. Busulfan now is primarily used in high-dose regimens, in which pulmonary fibrosis, GI mucosal damage, and hepatic veno-occlusive disease (VOD) are important toxicities.
ADME. Busulfan is well absorbed after oral administration in dosages of 2-6 mg/day and has a plasma t1/2 of 2-3 h. The drug is conjugated to GSH by GSTA1A and further metabolized by CYP-dependent pathways; its major urinary metabolite is methane sulfonic acid. In high doses, children <18 years of age clear the drug 2-4 times faster than adults and tolerate higher doses. Busulfan clearance varies considerably among patients. VOD is associated with high area under the curve (AUC) (AUC > 1500 1M × min) peak drug levels and slow clearance, leading to recommendations for dose adjustment based on drug level monitoring. A target steady-state concentration of 600-900 ng/mL in plasma in adults or AUC <1000 1M × min in children achieves an appropriate balance between toxicity and therapeutic benefit.
Therapeutic Uses. In treating chronic myelogenous leukemia (CML), the initial oral dose of busulfan varies with the total leukocyte count and the severity of the disease; daily doses of 2-8 mg for adults (~60 μg/kg or 1.8 mg/m2 for children) are used to initiate therapy and are adjusted appropriately to subsequent hematological and clinical responses, with the aim of reducing the total leukocyte count to ≤10,000 cells/mm3. A decrease in the leukocyte count usually is not seen during the first 10-15 days of treatment, and the leukocyte count may actually increase during this period. Because the leukocyte count may fall for >1 month after discontinuing the drug, it is recommended that busulfan be withdrawn when the total leukocyte count has declined to ~15,000 cells/mm3. A normal leukocyte count usually is achieved within 12-20 weeks. During remission, daily treatment resumes when the total leukocyte count reaches ~50,000 cells/mm3. Daily maintenance doses are 1-3 mg.
In high-dose therapy, doses of 1 mg/kg are given every 6 h for 4 days, with adjustment based on pharmacokinetics. Anticonvulsants must be used concomitantly to protect against acute CNS toxicities, including tonic-clonic seizures that may occur several hours after each dose. Although phenytoin is a frequent choice, phenytoin induces the GSTs that metabolize busulfan, reducing its AUC by ~20%. In patients requiring concomitant anti-seizure medication, non-enzyme-inducing benzodiazepines such as lorazepam and clonazepam are recommended as an alternative to phenytoin. If phenytoin is used concurrently, plasma busulfan levels should be monitored and the busulfan dose adjusted accordingly.
Clinical Toxicity. The toxic effects of busulfan are related to its myelosuppressive properties; prolonged thrombocytopenia may occur. Occasionally, patients experience nausea, vomiting, and diarrhea. Long-term use leads to impotence, sterility, amenorrhea, and fetal malformation. Rarely, patients develop asthenia and hypotension. High-dose busulfan causes VOD of the liver in ≤10% of patients, as well as seizures, hemorrhagic cystitis, permanent alopecia, and cataracts. The coincidence of VOD and hepatotoxicity is increased by its coadministration with drugs that inhibit CYPs, including imidazoles and metronidazole, possibly through inhibition of the clearance of busulfan and/or its toxic metabolites.
NITROSOUREAS
The nitrosoureas have an important role in the treatment of brain tumors and find occasional use in treating lymphomas and in high-dose regimens with bone marrow reconstitution. They function as bifunctional alkylating agents but differ from conventional nitrogen mustards in both pharmacological and toxicological properties.
Carmustine (BCNU) and lomustine (CCNU) are highly lipophilic and thus readily cross the blood-brain barrier, an important property in the treatment of brain tumors. Unfortunately, with the exception of streptozocin, nitrosoureas cause profound and delayed myelosuppression with recovery 4-6 weeks after a single dose. Long-term treatment with the nitrosoureas, especially semustine (methyl-CCNU), has resulted in renal failure. As with other alkylating agents, the nitrosoureas are highly carcinogenic and mutagenic. They generate both alkylating and carbamylating moieties (see Figure 61–3).
CARMUSTINE (BCNU)
Carmustine’s major action is its alkylation of DNA at the O6-guanine position, an adduct repaired by MGMT. Methylation of the MGMT promoter inhibits its expression in ~30% of primary gliomas and is associated with sensitivity to nitrosoureas. In high doses with bone marrow rescue, carmustine produces hepatic VOD, pulmonary fibrosis, renal failure, and secondary leukemia.
ADME. Carmustine is unstable in aqueous solution and in body fluids. After intravenous infusion, it disappears from the plasma with a highly variable t1/2 of ≥15-90 min. Approximately 30-80% of the drug appears in the urine within 24 h as degradation products. The alkylating metabolites enter rapidly into the cerebrospinal fluid (CSF), and their concentrations in the CSF reach 15-30% of the concurrent plasma values.
Therapeutic Uses. Carmustine (BICNU) is administered intravenously at doses of 150-200 mg/m2, given by infusion over 1-2 h and repeated every 6 weeks. Because of its ability to cross the blood-brain barrier, carmustine has been used in the treatment of malignant gliomas. An implantable carmustine wafer (GLIADEL) is available for use as an adjunct to surgery and radiation in newly diagnosed high-grade malignant glioma patients and as an adjunct to surgery for recurrent glioblastoma multiforme.
STREPTOZOCIN
Streptozocin (or streptozotocin) has a methylnitrosourea (MNU) moiety attached to the 2-carbon of glucose. It has a high affinity for cells of the islets of Langerhans and causes diabetes in experimental animals.
ADME. Streptozocin is rapidly degraded following intravenous administration. The t1/2 of the drug is ~15 min. Only 10-20% of a dose is recovered intact in the urine.
Therapeutic Uses. Streptozocin (ZANOSAR) is used in the treatment of human pancreatic islet cell carcinoma and malignant carcinoid tumors. It is administered intravenously, 500 mg/m2 once daily for 5 days; this course is repeated every 6 weeks. Alternatively, 1000 mg/m2 can be given weekly for 2 weeks, and the weekly dose then can be increased to a maximum of 1500 mg/m2 as tolerated. Nausea is frequent. Mild, reversible renal or hepatic toxicity occurs in approximately two-thirds of cases; in <10% of patients, renal toxicity may be cumulative with each dose and may lead to irreversible renal failure. Streptozocin should not be given with other nephrotoxic drugs. Hematological toxicities (anemia, leukopenia, thrombocytopenia) occur in 20% of patients.
TRIAZENES
DACARBAZINE (DTIC)
Dacarbazine functions as a methylating agent after metabolic activation to the monomethyl triazeno metabolite, MTIC. It kills cells in all phases of the cell cycle. Resistance has been ascribed to the removal of methyl groups from the O6– guanine bases in DNA by MGMT.
ADME. Dacarbazine is administered intravenously. After an initial rapid phase (t1/2 of ~20 min), dacarbazine is cleared from plasma with a terminal t1/2 of ~5 h. The t1/2 is prolonged in the presence of hepatic or renal disease. Almost 50% of the compound is excreted intact in the urine by tubular secretion.
Therapeutic Uses. The primary clinical indication for dacarbazine (DTIC-DOME) is in the chemotherapy of Hodgkin disease. In combination with other drugs for Hodgkin disease, dacarbazine is given in dosages of 150 mg/m2/day for 5 days, repeated every 4 weeks; it can be used alone as a single dose of 375 mg/m2, repeated every 15 days. It is modestly effective against malignant melanoma and adult sarcomas. Dacarbazine (DTIC-DOME) for malignant melanoma is given in dosages of 2-4.5 mg/kg/day for a 10-day period, repeated every 28 days; alternatively, 250 mg/m2 can be given daily for 5 days and repeated every 3 weeks. Extravasation of the drug may cause tissue damage and severe pain.
Toxicity. DTIC induces nausea and vomiting in >90% of patients; vomiting usually develops 1-3 h after treatment and may last up to 12 h. Myelosuppression, with both leukopenia and thrombocytopenia, is mild and readily reversible within 1-2 weeks. A flu-like syndrome may occur. Hepatotoxicity, alopecia, facial flushing, neurotoxicity, and dermatological reactions are less common adverse effects.
TEMOZOLOMIDE
Temozolomide (TEMODAR) is the standard agent in combination with radiation therapy for patients with malignant glioma and for astrocytoma. Temozolomide, like dacarbazine, forms the methylating metabolite MTIC and kills cells in all phases of the cell cycle.
ADME. Temozolomide is administered orally or intravenously in dosages of ~200 mg/day; it has a bioavailability approaching 100%. Plasma levels of the parent drug decline with a t1/2 of 1-2 h. The primary active metabolite MTIC reaches a maximum plasma concentration (150 ng/mL) 90 min after a dose and declines with a t1/2 of 2 h. Little intact drug is recovered in the urine, the primary urinary metabolite being the inactive imidazole carboxamide.
Toxicity. The toxicities of temozolomide mirror those of DTIC. Hematological monitoring is necessary to guide dosing adjustments.
METHYLHYDRAZINES
PROCARBAZINE
Procarbazine is employed for second-line therapy in malignant brain tumors.
Cytotoxic Action. The antineoplastic activity of procarbazine results from its conversion by CYP-mediated hepatic oxidative metabolism to highly reactive alkylating species that methylate DNA. Activated procarbazine can produce chromosomal damage, including chromatid breaks and translocations, consistent with its mutagenic and carcinogenic actions. Resistance to procarbazine develops rapidly when it is used as a single agent; one mechanism of resistance results from increased expression of MGMT, which repairs methylation of guanine.
Pharmacokinetics. The pharmacokinetic behavior of procarbazine has not yet been thoroughly defined. The drug is extensively metabolized by CYP isoenzymes to azo, methylazoxy, and benzylazoxy intermediates, which are found in the plasma and yield the alkylating metabolites in tumor cells. In brain cancer patients, the concurrent use of anti-seizure drugs that induce hepatic CYPs does not significantly alter the pharmacokinetics of the parent drug.
Therapeutic Uses. The recommended dosage of procarbazine (MATULANE) for adults is 100 mg/m2/day for 10-14 days in combination regimens such as MOPP (nitrogen mustard, Oncovin, procarbazine, and prednisone) for Hodgkin disease. The drug rarely is used in current practice.
Toxicity. The most common toxic effects include leukopenia and thrombocytopenia, which begin during the second week of therapy and reverse within 2 weeks off treatment. GI symptoms such as mild nausea and vomiting occur in most patients; diarrhea and rash are noted in 5-10% of cases. Behavioral disturbances also have been reported. Because procarbazine augments sedative effects, the concomitant use of CNS depressants should be avoided. The drug is a weak MAO inhibitor; it blocks the metabolism of catecholamines, sympathomimetics, and dietary tyramine and may provoke hypertension in patients concurrently exposed to these. Procarbazine has disulfiram-like actions, and therefore the ingestion of alcohol should be avoided. The drug is highly carcinogenic, mutagenic, and teratogenic and is associated with a 5-10% risk of acute leukemia in patients treated with MOPP. The highest risk is for patients who also receive radiation therapy. Procarbazine is a potent immunosuppressive agent. It causes infertility, particularly in males.
PLATINUM COORDINATION COMPLEXES
Platinum coordination complexes have broad antineoplastic activity and have become the foundation for treatment of ovarian, head and neck, bladder, esophagus, lung, and colon cancers. Although cisplatin and other platinum complexes do not form carbonium ion intermediates like other alkylating agents or formally alkylate DNA, they covalently bind to nucleophilic sites on DNA and share many pharmacological attributes with alkylators.
MECHANISM OF ACTION. Cisplatin, carboplatin, and oxaliplatin enter cells by an active Cu2+ transporter, CTR1, and in doing so rapidly degrade the transporter. The compounds are actively extruded from cells by ATP7A and ATP7B copper transporters and by multidrug resistance protein 1 (MRP1); variable expression of these transporters may contribute to clinical resistance. Inside the cell, the chloride, cyclohexane, or oxalate ligands of the analogs are displaced by water molecules, yielding a positively charged molecule that reacts with nucleophilic sites on DNA and proteins.
Aquation of cisplatin is favored at the low concentrations of Cl– inside the cell and in the urine. High concentrations of Cl– stabilize the drug, explaining the effectiveness of Cl– diuresis in preventing nephrotoxicity. The activated platinum complexes can react with electron-rich regions, such as sulfhydryls, and with various sites on DNA, forming both intrastrand and interstrand cross-links. The DNA-platinum adducts inhibit replication and transcription, lead to single- and double-stranded breaks and miscoding, and if recognized by p53 and other checkpoint proteins, cause induction of apoptosis. Adduct formation is an important predictor of clinical response. The analogs differ in the conformation of their adducts and the effects of adduct on DNA structure and function. Oxaliplatin and carboplatin are slower to form adducts. The oxaliplatin adducts are bulkier and less readily repaired, create a different pattern of distortion of the DNA helix, and differ from cisplatin adducts in the pattern of hydrogen bonding to adjacent segments of DNA.
Unlike the other platinum analogs, oxaliplatin exhibits a cytotoxicity that does not depend on an active MMR system, which may explain its greater activity in colorectal cancer. It also seems less dependent on the presence of high mobility group (HMG) proteins that are required by the other platinum derivatives. Testicular cancers have a high concentration of HMG proteins and are quite sensitive to cisplatin. Basal-type breast cancers, such as those with BRCA1 and BRCA2 mutations, lack Her 2 amplification and hormone-receptor expression and appear to be uniquely susceptible to cisplatin through their upregulation of apoptotic pathways governed by p63 and p73. The cell cycle specificity of cisplatin differs among cell types; the effects of cross-linking are most pronounced during the S phase. The platinum analogs are mutagenic, teratogenic, and carcinogenic. Cisplatin- or carboplatin-based chemotherapy for ovarian cancer is associated with a 4-fold increased risk of developing secondary leukemia.
Resistance to Platinum Analogs. Resistance to the platinum analogs likely is multifactorial; the compounds differ in their degree of cross-resistance. Carboplatin shares cross-resistance with cisplatin in most experimental tumors, while oxaliplatin does not. A number of factors influence sensitivity to platinum analogs in experimental cells, including intracellular drug accumulation and intracellular levels of glutathione and other sulfhydryls such as metallothionein that bind to and inactivate the drug and rates of repair of DNA adducts. Repair of platinum-DNA adducts requires participation of the NER pathway. Inhibition or loss of NER increases sensitivity to cisplatin in ovarian cancer patients, while overexpression of NER components is associated with poor response to cisplatin or oxaliplatin-based therapy in lung, colon, and gastric cancer.
Resistance to cisplatin, but not oxaliplatin, appears to be partly mediated through loss of function in the MMR proteins. In the absence of effective repair of DNA-platinum adducts, sensitive cells cannot replicate or transcribe affected portions of the DNA strand. Some DNA polymerases can bypass adducts, possibly contributing to resistance. Overexpression of copper efflux transporters, ATP7A and ATP7B, correlates with poor survival after cisplatin-based therapy for ovarian cancer.
CISPLATIN
ADME. After intravenous administration, cisplatin has an initial plasma elimination t1/2 of 25-50 min; concentrations of total (bound and unbound) drug fall thereafter, with a t1/2 of ≥24 h. More than 90% of the platinum in the blood is covalently bound to plasma proteins. High concentrations of cisplatin are found in the kidney, liver, intestine, and testes; cisplatin penetrates poorly into the CNS. Only a small portion of the drug is excreted by the kidney during the first 6 h; by 24 h, up to 25% is excreted, and by 5 days, up to 43% of the administered dose is recovered in the urine, mostly covalently bound to protein and peptides. Biliary or intestinal excretion of cisplatin is minimal.
Therapeutic Uses. Cisplatin (PLATINOL) is given only intravenously. The usual dosage is 20 mg/m2/day for 5 days, 20-30 mg weekly for 3-4 weeks, or 100 mg/m2 given once every 4 weeks. To prevent renal toxicity, it is important to establish a chloride diuresis by the infusion of 1-2 L of normal saline prior to treatment. The appropriate amount of cisplatin then is diluted in a solution containing dextrose, saline, and mannitol and administered intravenously over 4-6 h. Because aluminum inactivates cisplatin, the drug should not come in contact with needles or other infusion equipment that contain aluminum during its preparation or administration.
Cisplatin, in combination with bleomycin, etoposide, ifosfamide, or vinblastine, cures 90% of patients with testicular cancer. Used with paclitaxel, cisplatin or carboplatin induces complete response in the majority of patients with carcinoma of the ovary. Cisplatin produces responses in cancers of the bladder, head and neck, cervix, and endometrium; all forms of carcinoma of the lung; anal and rectal carcinomas; and neoplasms of childhood. The drug also sensitizes cells to radiation therapy and enhances control of locally advanced lung, esophageal, and head and neck tumors when given with irradiation.
TOXICITY. Cisplatin-induced nephrotoxicity has been largely abrogated by adequate pretreatment hydration and chloride diuresis. Amifostine (ETHYOL), a thiophosphate cytoprotective agent, reduces renal toxicity associated with repeated administration of cisplatin. Ototoxicity caused by cisplatin is unaffected by diuresis and is manifested by tinnitus and high-frequency hearing loss. Marked nausea and vomiting occur in almost all patients and usually can be controlled with 5HT3 antagonists, NK1-receptor antagonists, and high-dose corticosteroids (see Table 46–6).
At higher doses or after multiple cycles of treatment, cisplatin causes a progressive peripheral motor and sensory neuropathy that may worsen after discontinuation of the drug and may be aggravated by subsequent or simultaneous treatment with taxanes or other neurotoxic drugs. Cisplatin causes mild to moderate myelosuppression, with transient leukopenia and thrombocytopenia. Anemia may become prominent after multiple cycles of treatment. Electrolyte disturbances, including hypomagnesemia, hypocalcemia, hypokalemia, and hypophosphatemia, are common. Hypocalcemia and hypomagnesemia secondary to tubular damage and renal electrolyte wasting may produce tetany if untreated. Routine measurement of Mg2+ concentrations in plasma is recommended. Hyperuricemia, hemolytic anemia, and cardiac abnormalities are rare side effects. Anaphylactic-like reactions, characterized by facial edema, bronchoconstriction, tachycardia, and hypotension, may occur within minutes after administration and should be treated by intravenous injection of epinephrine and with corticosteroids or anti-histamines. Cisplatin has been associated with the development of AML, usually ≥4 years after treatment.
CARBOPLATIN
The mechanisms of action and resistance and the spectrum of clinical activity of carboplatin (PARAPLATIN; CBDCA, JIM-8) are similar to cisplatin. However, the 2 drugs differ significantly in their chemical, pharmacokinetic, and toxicological properties.
ADME. Because carboplatin is much less reactive than cisplatin, the majority of drug in plasma remains in its parent form, unbound to proteins. Most drug is eliminated via renal excretion, with a t1/2 ~2 h. A small fraction of platinum binds irreversibly to plasma proteins and disappears slowly, with a t1/2 ≥5 days.
Therapeutic Uses. Carboplatin and cisplatin are equally effective in the treatment of suboptimally debulked ovarian cancer, non–small cell lung cancer, and extensive-stage small cell lung cancer; however, carboplatin may be less effective than cisplatin in germ cell, head and neck, and esophageal cancers. Carboplatin is an effective alternative for responsive tumors in patients unable to tolerate cisplatin because of impaired renal function, refractory nausea, significant hearing impairment, or neuropathy, but doses must be adjusted for renal function. In addition, it may be used in high-dose therapy with bone marrow or peripheral stem cell rescue. Carboplatin is administered as an intravenous infusion over at least 15 min and is given once every 21-28 days; the dose of carboplatin should be adjusted in proportion to the reduction in creatinine clearance (CrCl) for patients with a CrCl <60 mL/min.
Toxicity. Carboplatin is relatively well tolerated clinically, causing less nausea, neurotoxicity, ototoxicity, and nephrotoxicity than cisplatin. The dose-limiting toxicity of carboplatin is myelosuppression, primarily thrombocytopenia. It may cause a hypersensitivity reaction; in patients with a mild reaction, premedication, graded doses of drug, and more prolonged infusion lead to desensitization.
OXALIPLATIN
ADME. Oxaliplatin has a short t1/2 in plasma, probably as a result of its rapid uptake by tissues and its reactivity; the initial t1/2 ~17 min. No dose adjustment is required for hepatic dysfunction or for patients with a CrCl ≥20 mL/min.
Therapeutic Uses. Oxaliplatin exhibits a range of antitumor activity (colorectal and gastric cancer) that differs from other platinum agents. Oxaliplatin’s effectiveness in colorectal cancer is perhaps due to its MMR- and HMG-independent effects. It also suppresses expression of thymidylate synthase (TS), the target enzyme of 5-fluorouracil (5-FU) action, which may promote synergy of these 2 drugs. In combination with 5-FU, it is approved for treatment of patients with colorectal cancer.
Toxicity. The dose-limiting toxicities of oxaliplatin are peripheral neuropathies. An acute form, often triggered by exposure to cold liquids, manifests as paresthesias and/or dysesthesias in the upper and lower extremities, mouth, and throat. A second type relates to cumulative dose and has features similar to cisplatin neuropathy; 75% of patients receiving a cumulative dose of 1560 mg/m2 experience some progressive sensory neurotoxicity, with dysesthesias, ataxia, and numbness of the extremities. Hematological toxicity is mild to moderate, except for rare immune-mediated cytopenias; nausea is well controlled with 5HT3-receptor antagonists. Oxaliplatin causes leukemia and pulmonary fibrosis months to years after administration. Oxaliplatin may cause an acute allergic response with urticaria, hypotension, and bronchoconstriction.
II. Antimetabolites
FOLIC ACID ANALOGS
Folic acid is an essential dietary factor that is converted by enzymatic reduction to a series of tetrahydrofolate (FH4) cofactors that provide methyl groups for the synthesis of precursors of DNA (thymidylate and purines) and RNA (purines). Interference with FH4 metabolism reduces the cellular capacity for one-carbon transfer and the necessary methylation reactions in the synthesis of purine ribonucleotides and thymidine monophosphate (TMP), thereby inhibiting DNA replication.
Antifolate chemotherapy produced the first striking, although temporary, remissions in leukemia and the first cure of a solid tumor, choriocarcinoma. Recognition that methotrexate (MTX), an inhibitor of dihydrofolate reductase (DHFR), also directly inhibits the folate-dependent enzymes of de novo purine and thymidylate synthesis led to development of antifolate analogs that specifically target these other folate-dependent enzymes (Figure 61–4). New congeners have greater capacity for transport into tumor cells (pralatrexate) and exert their primary inhibitory effect on TS (raltitrexed, TOMUDEX), early steps in purine biosynthesis (lometrexol), or both (the multitargeted antifolate, pemetrexed, ALIMTA).
Mechanism of Action. The primary target of MTX is the enzyme DHFR (see Figure 61–4). To function as a cofactor in 1-carbon transfer reactions, folate must be reduced by DHFR to FH4. Inhibitors such as MTX, with a high affinity for DHFR (Ki 0.01-0.2 nM), cause partial depletion of the FH4 cofactors (5-10 methylene tetrahydrofolic acid and N-10 formyl tetrahydrofolic acid) required for the synthesis of thymidylate and purines. In addition, MTX, like cellular folates, undergoes conversion to a series of polyglutamates (MTX-PGs) in both normal and tumor cells. These MTX-PGs constitute an intracellular storage form of folates and folate analogs and dramatically increase inhibitory potency of the analog for additional sites, including TS and 2 early enzymes in the purine biosynthetic pathway. The dihydrofolic acid polyglutamates that accumulate in cells behind the blocked DHFR reaction also act as inhibitors of TS and other enzymes (see Figure 61–4).
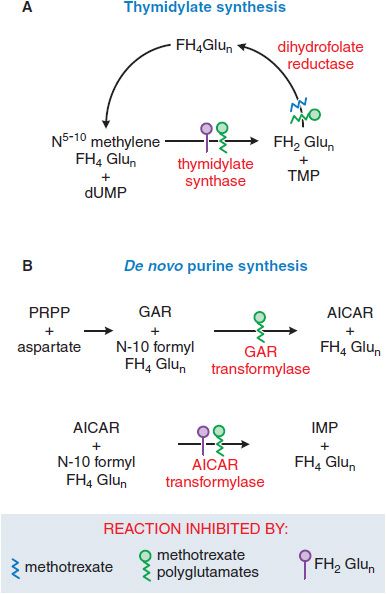
Figure 61–4 Actions of methotrexate and its polyglutamates. AICAR, aminoimidazole carboxamide; dUMP, deoxyuridine monophosphate; FH2Glun/FH4Glun, dihydro-/tetrahydro-folate polyglutamates; GAR, glycinamide ribonucleotide; IMP, inosine monophosphate; PRPP, 5-phosphoribosyl-1-pyrophosphate; TMP, thymidine monophosphate.
Selective Toxicity; Rescue. As with most antimetabolites, MTX is only partially selective for tumor cells and kills rapidly dividing normal cells, such as those of the intestinal epithelium and bone marrow. Folate antagonists kill cells during the S phase of the cell cycle and are most effective when cells are proliferating rapidly. The toxic effects of MTX may be terminated by administering leucovorin, a fully reduced folate coenzyme that repletes the intracellular pool of FH4 cofactors.
Cellular Entry and Retention. Because folic acid and many of its analogs are polar, they cross the blood-brain barrier poorly and require specific transport mechanisms to enter mammalian cells. Three inward folate transport systems are found on mammalian cells: (1) a folate receptor, which has high affinity for folic acid but much reduced ability to transport MTX and other analogs; (2) the reduced folate transporter, the major transit protein for MTX, raltitrexed, pemetrexed, and most analogs; and (3) a transporter that is active at low pH. The reduced folate transporter is highly expressed in the hyperdiploid subtype of acute lymphoblastic leukemia (ALL), which has extreme sensitivity to MTX. Once in the cell, additional glutamyl residues are added to the molecule by the enzyme folylpolyglutamate synthetase. Because these higher polyglutamates are strongly charged and cross cellular membranes poorly, polyglutamation serves as a mechanism of entrapment and may account for the prolonged retention of MTX in chorionic epithelium (where it is a potent abortifacient); in tumors derived from this tissue, such as choriocarcinoma cells; and in normal tissues subject to cumulative drug toxicity, such as liver. Polyglutamyl folates and analogs have substantially greater affinity than the monoglutamate form for folate-dependent enzymes that are required for purine and thymidylate synthesis and have at least equal affinity for DHFR.
Newer Congeners. New folate antagonists that are better substrates for the reduced folate carrier have been identified. In efforts to bypass the obligatory membrane transport system and to facilitate penetration of the blood-brain barrier, lipid-soluble folate antagonists also have been synthesized. Trimetrexate (NEUTREXIN), a lipid-soluble analog that lacks a terminal glutamate, has modest antitumor activity, primarily in combination with leucovorin rescue. However, it is beneficial in the treatment of Pneumocystis jiroveci (Pneumocystis carinii) pneumonia where leucovorin provides differential rescue of the host but not the parasite. The most important new folate analog, MTA or pemetrexed (ALIMTA), is a pyrrole–pyrimidine structure. It is avidly transported into cells via the reduced folate carrier and is converted to polyglutamates that inhibit TS and glycine amide ribonucleotide transformylase, as well as DHFR. It has activity against ovarian cancer, mesothelioma, and non–small cell adenocarcinomas of the lung. Pemetrexed and its polyglutamates have a somewhat different spectrum of biochemical actions. Like MTX, pemetrexed inhibits DHFR, but as a polyglutamate, it even more potently inhibits glycinamide ribonucleotide formyltransferase (GART) and TS. Unlike MTX, it produces little change in the pool of reduced folates, indicating that the distal sites of inhibition (TS and GART) predominate. Its pattern of deoxynucleotide depletion also differs; it causes a greater fall in thymidine triphosphate (TTP) than in other triphosphates. Like MTX, it induces p53 and cell-cycle arrest, but this effect does not depend on induction of p21. A newer congener, pralatrexate, is more effectively taken up and polyglutamated than MTX and is approved for treatment of cutaneous T-cell lymphoma.
MECHANISMS OF RESISTANCE TO ANTIFOLATES. Resistance to MTX can involve alterations in each known step in MTX action, including:
• Impaired transport of MTX into cells
• Production of altered forms of DHFR that have decreased affinity for the inhibitor
• Increased concentrations of intracellular DHFR through gene amplification or altered gene regulation
• Decreased ability to synthesize MTX polyglutamates
• Increased expression of a drug efflux transporter of the MRP class (see Chapter 5)
DHFR levels in leukemic cells increase within 24 h after treatment of patients with MTX, probably as a result of induction of DHFR synthesis. Unbound DHFR protein may bind to its own message and reduce its own translation, while the DHFR-MTX complex is ineffective in blocking the DHFR translation. With longer periods of drug exposure, tumor cell populations that contain markedly increased levels of DHFR emerge. These cells contain multiple gene copies of DHFR either in mitotically unstable double-minute chromosomes (extrachromosomal elements) or in stably integrated, homogeneously staining chromosomal regions or amplicons. Similar gene amplification target proteins have been implicated in the resistance to many antitumor agents, including 5-FU and pentostatin (2′-deoxycoformycin).
High doses of MTX may permit entry of the drug into transport-defective cells and may permit the intracellular accumulation of MTX in concentrations that inactivate high levels of DHFR. The understanding of resistance to pemetrexed is incomplete. In various cell lines, resistance to this agent seems to arise from loss of influx transport, TS amplification, changes in purine biosynthetic pathways, or loss of polyglutamation.
ADME. MTX is readily absorbed from the GI tract at doses of M25 mg/m2; larger doses are absorbed incompletely and are routinely administered intravenously. After intravenous administration, the drug disappears from plasma in a triphasic fashion. The rapid distribution phase is followed by a second phase, which reflects renal clearance (t1/2 of 2-3 h). A third phase has a t1/2
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


