Treatment of Central Nervous System Degenerative Disorders
Neurodegenerative disorders are characterized by progressive and irreversible loss of neurons from specific regions of the brain. Prototypical neurodegenerative disorders include Parkinson disease (PD) and Huntington disease (HD), where loss of neurons from structures of the basal ganglia results in abnormalities in the control of movement; Alzheimer disease (AD), where the loss of hippocampal and cortical neurons leads to impairment of memory and cognitive ability; and amyotrophic lateral sclerosis (ALS), where muscular weakness results from the degeneration of spinal, bulbar, and cortical motor neurons. Currently available therapies for neurodegenerative disorders alleviate the disease symptoms but do not alter the underlying neurodegenerative process.
SELECTIVE VULNERABILITY AND NEUROPROTECTIVE STRATEGIES
SELECTIVE VULNERABILITY. A striking feature of neurodegenerative disorders is the exquisite specificity of the disease processes for particular types of neurons. For example, in PD there is extensive destruction of the dopaminergic neurons of the substantia nigra, whereas neurons in the cortex and many other areas of the brain are unaffected. In contrast, neural injury in AD is most severe in the hippocampus and neocortex, and even within the cortex, the loss of neurons is not uniform but varies dramatically in different functional regions. In HD the mutant gene responsible for the disorder is expressed throughout the brain and in many other organs, yet the pathological changes are most prominent in the neostriatum. In ALS, there is loss of spinal motor neurons and the cortical neurons that provide their descending input. The diversity of these patterns of neural degeneration suggests that the process of neural injury results from the interaction of genetic and environmental influences.
GENETICS AND ENVIRONMENT. Each of the major neurodegenerative disorders may be familial in nature. HD is exclusively familial; it is transmitted by autosomal dominant inheritance, and the molecular mechanism of the genetic defect has been defined. Nevertheless, environmental factors importantly influence the age of onset and rate of progression of HD symptoms. PD, AD, and ALS are mostly sporadic without clear pattern of inheritance. But for each there are well-recognized genetic forms. For example, there are both dominant (α-synuclein, LRRK2) and recessive (parkin, DJ-1, PINK1) gene mutations that may give rise to PD. In AD, mutations in the genes coding for the amyloid precursor protein (APP) and proteins known as the presenilins (involved in APP processing) lead to inherited forms of the disease. Mutations in the gene coding for copper-zinc superoxide dismutase (SOD1) account for about 2% of the cases of adult-onset ALS. There are also genetic risk factors that influence the probability of disease onset and modify the phenotype. For example, the apolipoprotein E (apoE) genotype constitutes an important risk factor for AD. Three distinct isoforms of this protein exist. Although all isoforms carry out their primary role in lipid metabolism equally well, individuals who are homozygous for the apoE4 allele (“4/4”) have a much higher lifetime risk of AD than do those homozygous for the apoE2 allele (“2/2”).
Environmental factors including infectious agents, environmental toxins, and acquired brain injury have been proposed in the etiology of neurodegenerative disorders. Traumatic brain injury has been suggested as a trigger for neurodegenerative disorders. At least one toxin, N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), can induce a condition closely resembling PD. More recently, evidence has linked pesticide exposure with PD. Exposure of soldiers to neurotoxic chemicals has been implicated in ALS (as part of “Gulf War syndrome”).
COMMON CELLULAR MECHANISMS OF NEURODEGENERATION. Despite their varied phenotypes, the neurodegenerative disorders share some common features. For example, misfolded and aggregated proteins are found in every major neurodegenerative disorder: alpha-synuclein, in PD; amyloid-β (Aβ) and tau in AD; huntingtin in HD; and SOD and TDP-43 in ALS. The accumulation of misfolded proteins may result from either genetic mutations producing abnormal structure, or from impaired cellular clearance.
The term excitotoxicity describes the neural injury that results from the presence of excess glutamate in the brain. Glutamate is used as a neurotransmitter to mediate most excitatory synaptic transmission in the mammalian brain (see Table 14–1). The presence of excessive amounts of glutamate can lead to excitotoxic cell death (see Figure 14–8). The destructive effects of glutamate are mediated by glutamate receptors, particularly those of the N-methyl-D-aspartate (NMDA) type. Excitotoxic injury contributes to the neuronal death that occurs in acute processes such as stroke and head trauma. The role of excitotoxicity is less certain in the chronic neurodegenerative disorders; nevertheless, regional and cellular differences in susceptibility to excitotoxic injury, conveyed, e.g., by differences in types of glutamate receptors, may contribute to selective vulnerability. This has led to the development of glutamate antagonists as neuroprotective therapies, with 2 such agents (memantine and riluzole, described later) currently in clinical use.
Aging is associated with a progressive impairment in the capacity of neurons for oxidative metabolism with consequent production of reactive compounds such as hydrogen peroxide and oxygen radicals. These reactive species can lead to DNA damage, peroxidation of membrane lipids, and neuronal death. This has led to pursuit of drugs that can enhance cellular metabolism (such as the mitochondrial cofactor coenzyme Q10) and antioxidant strategies as treatments to prevent or retard degenerative diseases.
PARKINSON DISEASE (PD)
CLINICAL OVERVIEW. Parkinsonism is a clinical syndrome with 4 cardinal features:
• Bradykinesia (slowness and poverty of movement)
• Muscular rigidity
• Resting tremor (which usually abates during voluntary movement)
• An impairment of postural balance leading to disturbances of gait and to falling
The most common form of parkinsonism is idiopathic PD, first described by James Parkinson in 1817 as paralysis agitans, or the “shaking palsy.” The pathological hallmark of PD is the loss of the pigmented, dopaminergic neurons of the substantia nigra pars compacta, with the appearance of intracellular inclusions known as Lewy bodies. A loss of 70-80% of these dopamine-containing neurons accompanies symptomatic PD.
Without treatment, PD progresses over 5-10 years to a rigid, akinetic state in which patients are incapable of caring for themselves. Death frequently results from complications of immobility, including aspiration pneumonia or pulmonary embolism. The availability of effective pharmacological treatment has radically altered the prognosis of PD; in most cases, good functional mobility can be maintained for many years. Life expectancy of adequately treated patients is increased substantially, but overall mortality remains higher than that of the general population. In addition, while DA neuron loss is the most prominent feature of the disease, the disorder affects a wide range of other brain structures, including the brainstem, hippocampus, and cerebral cortex. This pathology is likely responsible for the “non-motor” features of PD, which include sleep disorders, depression, and memory impairment.
Several disorders other than idiopathic PD also may produce parkinsonism, including some relatively rare neurodegenerative disorders, stroke, and intoxication with DA-receptor antagonists. Drugs that may cause parkinsonism include antipsychotics such as haloperidol and chlorpromazine (see Chapter 16) and antiemetics such as prochlorperazine and metoclopramide (see Chapter 46). The distinction between idiopathic PD and other causes of parkinsonism is important because parkinsonism arising from other causes usually is refractory to all forms of treatment.
PATHOPHYSIOLOGY. The dopaminergic deficit in PD arises from a loss of the neurons in the substantia nigra pars compacta that provide innervation to the striatum (caudate and putamen). The current understanding of the pathophysiology of PD is based on the finding that the striatal DA content is reduced in excess of 80%, with a parallel loss of neurons from the substantia nigra, suggesting that replacement of DA could restore function. We now have a model of the function of the basal ganglia that, while incomplete, is still useful.
DOPAMINE SYNTHESIS, METABOLISM, AND RECEPTORS. DA, a catecholamine, is synthesized in the terminals of dopaminergic neurons from tyrosine and stored, released, reaccumulated, and metabolized by processes described in Chapter 13 and summarized in Figure 22–1. The actions of DA in the brain are mediated by DA receptor, of which there are 2 broad classes. D1 and D2, with 5 distinct subtypes, D1-D5 (see Figure 13–6). All the DA receptors are G protein–coupled receptors (GPCRs). Receptors of the D1 group (D1 and D5 subtypes) couple to Gs and thence to activation of the cyclic AMP pathway. The D2 group (D2, D3, and D4 receptors) couple to Gi to reduce the adenylyl cyclase activity and voltage-gated Ca2+ currents while activating K+ currents (see Chapters 3 and 13 for details). Each of the 5 DA receptors has a distinct anatomical pattern of expression in the brain (see Figure 13–6). D1 and D2 proteins are abundant in the striatum and are the most important receptor sites with regard to the causes and treatment of PD. The D4 and D5 proteins are largely extrastriatal, whereas D3 expression is low in the caudate and putamen but more abundant in the nucleus accumbens and olfactory tubercle.
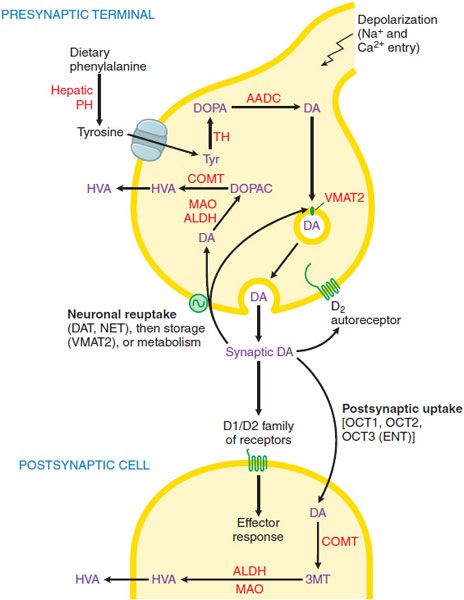
Figure 22–1 Dopaminergic nerve terminal. Dopamine (DA) is synthesized from tyrosine in the nerve terminal by the sequential actions of tyrosine hydrolase (TH) and aromatic amino acid decarboxylase (AADC). DA is sequestered by VMAT2 in storage granules and released by exocytosis. Synaptic DA activates presynaptic autoreceptors and postsynaptic D1 and D2 receptors. Synaptic DA may be taken up into the neuron via the DA and NE transporters (DAT, NET), or removed by postsynaptic uptake via OCT3 transporters. Cytosolic DA is subject to degradation by monoamine oxidase (MAO) and aldehyde dehydrogenase (ALDH) in the neuron, and by catechol-O-methyl transferase (COMT) and MAO/ALDH in non-neuronal cells; the final metabolic product is homovanillic acid (HVA). See structures in Figure 22–4. PH, phenylalanine hydroxylase.
NEURAL MECHANISM OF PARKINSONISM: A MODEL OF BASAL GANGLIA FUNCTION. Considerable effort has been devoted to understanding how the loss of dopaminergic input to the neurons of the neostriatum gives rise to the clinical features of PD. The basal ganglia can be viewed as a modulatory side loop that regulates the flow of information from the cerebral cortex to the motor neurons of the spinal cord (Figure 22–2).
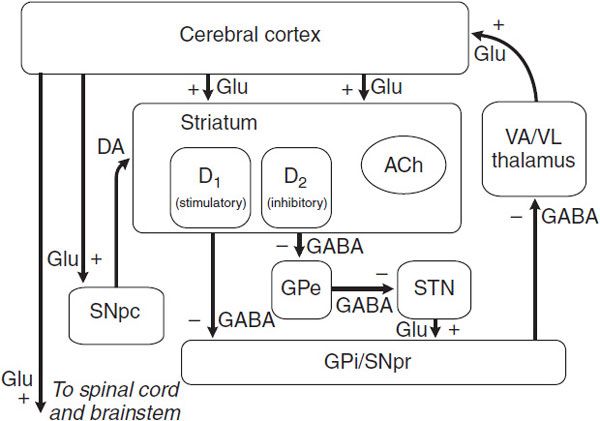
Figure 22–2 Schematic wiring diagram of the basal ganglia. The striatum is the principal input structure of the basal ganglia and receives excitatory glutamatergic input from many areas of cerebral cortex. The striatum contains projection neurons expressing predominantly D1 or D2 dopamine receptors, as well as interneurons that use ACh as a neurotransmitter. Outflow from the striatum proceeds along 2 routes. The direct pathway, from the striatum to the substantia nigra pars reticulata (SNpr) and globus pallidus interna (GPi), uses the inhibitory transmitter GABA. The indirect pathway, from the striatum through the globus pallidus externa (GPe) and the subthalamic nucleus (STN) to the SNpr and GPi, consists of 2 inhibitory GABA-ergic links and 1 excitatory glutamatergic projection (Glu). The substantia nigra pars compacta (SNpc) provides dopaminergic innervation to the striatal neurons, giving rise to both the direct and indirect pathways, and regulates the relative activity of these 2 paths. The SNpr and GPi are the output structures of the basal ganglia and provide feedback to the cerebral cortex through the ventroanterior and ventrolateral nuclei of the thalamus (VA/VL).
The neostriatum is the principal input structure of the basal ganglia and receives excitatory glutamatergic input from many areas of the cortex. Most neurons within the striatum are projection neurons that innervate other basal ganglia structures. A small but important subgroup of striatal neurons consists of interneurons that connect neurons within the striatum but do not project beyond its borders. Acetylcholine (ACh) and neuropeptides are used as transmitters by these striatal interneurons.
The outflow of the striatum proceeds along 2 distinct routes, termed the direct and indirect pathways. The direct pathway is formed by neurons in the striatum that project directly to the output stages of the basal ganglia, the substantia nigra pars reticulata (SNpr) and the globus pallidus interna (GPi); these, in turn, relay to the ventroanterior and ventrolateral thalamus, which provides excitatory input to the cortex. The neurotransmitter of both links of the direct pathway is γ-aminobutyric acid (GABA), which is inhibitory, so that the net effect of stimulation of the direct pathway at the level of the striatum is to increase the excitatory outflow from the thalamus to the cortex.
The indirect pathway is composed of striatal neurons that project to the globus pallidus externa (GPe). This structure, in turn, innervates the subthalamic nucleus (STN), which provides outflow to the SNpr and GPi output stage. The first 2 links—the projections from striatum to GPe and GPe to STN—use the inhibitory transmitter GABA; however, the final link—the projection from STN to SNpr and GPi—is an excitatory glutamatergic pathway. Thus, the net effect of stimulating the indirect pathway at the level of the striatum is to reduce the excitatory outflow from the thalamus to the cerebral cortex. The key feature of this model of basal ganglia function, which accounts for the symptoms observed in PD as a result of loss of dopaminergic neurons, is the differential effect of DA on the direct and indirect pathways (Figure 22–3).
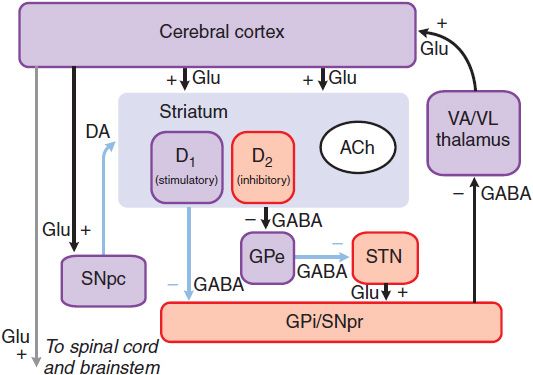
Figure 22–3 The basal ganglia in Parkinson disease. The primary defect is destruction of the dopaminergic neurons of the SNpc. The striatal neurons that form the direct pathway from the striatum to the SNpr and GPi express primarily the excitatory D1 DA receptor, whereas the striatal neurons that project to the GPe and form the indirect pathway express the inhibitory D2 dopamine receptor. Thus, loss of the dopaminergic input to the striatum has a differential effect on the 2 outflow pathways; the direct pathway to the SNpr and GPi is less active (structures in purple), whereas the activity in the indirect pathway is increased (structures in red). The net effect is that neurons in the SNpr and GPi become more active. This leads to increased inhibition of the VA/VL thalamus and reduced excitatory input to the cortex. Light blue lines indicate primary pathways with reduced activity. (See legend to Figure 22–2 for definitions of anatomical abbreviations.)
The dopaminergic neurons of the substantia nigra pars compacta (SNpc) innervate all parts of the striatum; however, the target striatal neurons express distinct types of DA receptors. The striatal neurons giving rise to the direct pathway express primarily the excitatory D1 dopamine receptor protein, whereas the striatal neurons forming the indirect pathway express primarily the inhibitory D2 type. Thus, DA released in the striatum tends to increase the activity of the direct pathway and reduce the activity of the indirect pathway, whereas the depletion that occurs in PD has the opposite effect. The net effect of the reduced dopaminergic input in PD is to increase markedly the inhibitory outflow from the SNpr and GPi to the thalamus and reduce excitation of the motor cortex. There are several limitations of this model of basal ganglia function. The anatomical connections are considerably more complex and many of the pathways involved use several neurotransmitters. Nevertheless, the model is useful and has important implications for the rational design and use of pharmacological agents in PD.
TREATMENT OF PARKINSON DISEASE
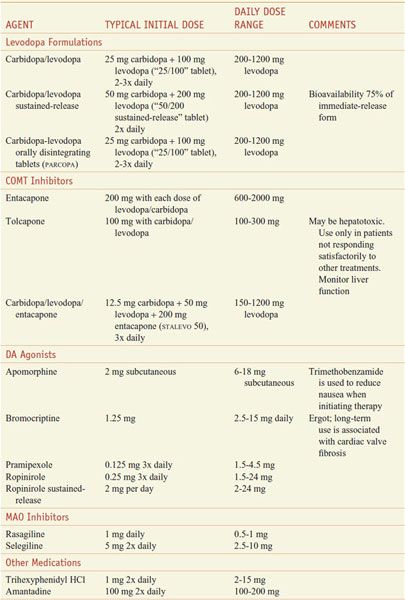
Table 22–1 summarizes commonly used medications for the treatment of PD.
Table 22–1
Commonly Used Medications for the Treatment of Parkinson Disease
LEVODOPA. Levodopa (L-dopa, LARODOPA, L-3,4-dihydroxyphenylalanine), the metabolic precursor of DA, is the single most effective agent in the treatment of PD.
The effects of levodopa result from its decarboxylation to DA. When administered orally, levodopa is absorbed rapidly from the small bowel by the transport system for aromatic amino acids. Concentrations of the drug in plasma usually peak between 0.5 and 2 h after an oral dose. The t1/2
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


