Topical and transdermal drug delivery
Adrian C. Williams
Chapter contents
Permeant properties affecting permeation
Mathematics of skin permeation
Experimental methods for studying transdermal drug delivery
Transdermal and topical preparations
Enhancement of transdermal and topical drug delivery
Key points
• Transdermal drug delivery the skin to deliver the drug to the systemic circulation.
 between 1 and 4, and an effective daily dose of < 10 mg/day.
between 1 and 4, and an effective daily dose of < 10 mg/day.• Patches, of varying complexities, are the most common transdermal drug delivery systems.
Introduction
Topical and transdermal formulations have a long history of use. Over 2000 years ago, Greek physicians used formulations containing salt, vinegar, honey and resins to treat skin lesions and ulcers. Chinese, Egyptian and Roman medical histories describe numerous remedies applied topically as pastes and poultices.
Topical and transdermal products remain key formulations for delivering drugs not only to the skin, but also through it for systemic action. An estimated 40 million topical items are dispensed annually in England and Wales, including 16 million emollient products and 13 million topical corticosteroid preparations. In addition, many other products are dispensed for topical anaesthesia and antisepsis or for transdermal delivery, such as fentanyl patches. Additionally, ‘over-the-counter’ products are widely sought by patients and range from emollients to non-steroidal anti-inflammatory creams and gels, to treatments for warts, verrucae and fungal infections, such as athletes’ foot. Thus, pharmacists often supply topical and transdermal formulations which contain a broad variety of active ingredients; indeed it has been reported that up to 20% of all repeat prescriptions are for application to the skin. The efficacy of these products is critically dependent on biological factors, such as the integrity of the skin, on the physicochemical properties of the active ingredient and on the formulation designed to release and deliver the active into or across the skin.
Terminology
The literature occasionally contains different terminology relating to transdermal and topical drug delivery. For this Chapter, it may be helpful to clarify the following at this point:
Topical drug delivery.
The application of a formulation to the skin to treat a local disorder, i.e. the intention is to retain the active pharmaceutical ingredient (API) within the skin, for example, a locally acting hydrocortisone cream.
Transdermal drug delivery.
The application of a formulation to the skin to deliver a drug to the systemic circulation, for example, estradiol patches.
Locally acting.
The active pharmaceutical ingredient acts directly on the skin.
Regionally acting.
The active pharmaceutical ingredient acts in the area close to where the formulation is applied. This is often also described as locally acting, but here the drug does not act directly on the skin, for example topically applied ibuprofen gels to treat musculoskeletal conditions.
Permeant.
The chemical species that is moving through or into the tissue. This will be the drug, but may also be other ingredients within the formulation.
Permeation.
Movement of drug through the membrane.
Penetration.
Entry into the tissue. Penetration does not necessarily require the molecules to pass out of the tissue.
Diffusion.
Movement of molecules through a domain, from high concentration to low concentration, by random molecular movement.
Diffusivity.
This is a property of the permeant in the membrane and is a measure of how easily it will traverse through the tissue. It is expressed in units of area/time (usually cm2/h or cm2/s).
Diffusion coefficient (D).
This is the diffusion coefficient of the permeant, and is sometimes a term used interchangeably with diffusivity. As with diffusivity, its units are area/time (usually cm2/h or cm2/s).
Permeability coefficient (kp).
Describes the speed of permeant transport, given in units of distance/time (usually cm/h).
Partition coefficient (P).
This is a measure of the distribution of molecules between two phases. For transdermal delivery studies, a partition coefficient (usually expressed as a log10, hence ‘log P’) between octanol and water is often used as a guide to how well a molecule will distribute between water and stratum corneum lipids. In some texts, the symbol K is used for the partition coefficient; here, and to avoid confusion with the permeability coefficient (kp), the symbol P (also widely used in the literature) has been employed.
Partitioning.
The process of molecules distributing between two domains. In transdermal drug delivery, partitioning is generally used to describe molecular redistribution from one domain to another, such as from an aqueous domain to a lipid domain.
Flux (J).
The rate of a permeant crossing the skin (or entering the systemic circulation). It is given in units of mass/area/time (usually µg/cm2/h).
Lag time (L).
This is obtained from a permeation profile by extrapolating the steady state flux line to the time axis. Older texts have used the symbol τ whereas others have used tL for the lag time, but most modern texts use the abbreviation L.
Vehicle.
The base formulation in which the drug is applied to the skin.
Thermodynamic activity.
Used here as a measure of the ‘escaping tendency’ of a molecule from its formulation. A thermodynamic activity of 1 equates to a saturated solution, or suspension, since the molecules in a saturated solution have the greatest ‘escape tendency’.
Skin structure and function
Human skin is a highly complex multilayered organ designed to ‘keep the outside out and the insides in’. It is the largest organ of the body, comprising around 10% of the body mass and covers an area of approximately 1.8 m2 in a typical adult. As a self-repairing barrier, skin permits terrestrial life by preventing the ingress of microorganisms and chemicals whilst regulating heat and water loss from the body. Indeed, the body continually loses water and transepidermal water loss (TEWL) is in the region of 1 mg/cm2/h, but its value varies with body site and external conditions (temperature, humidity).
For drug delivery and therapy, the intact skin presents a formidable barrier and a difficult challenge to formulation scientists. The properties of the skin limit the range of active ingredients that can be delivered through the barrier to achieve therapeutic levels. However, skin can be relatively easily damaged through mechanical, chemical or microbiological assault and by radiation, such as sun damage. In these cases, drug delivery may be enhanced and could in fact lead to adverse reactions.
Structure of the skin
In terms of drug delivery, human skin can be considered as a series of layers which potentially provide a series of barriers to a molecule traversing the tissue (Fig. 39.1).
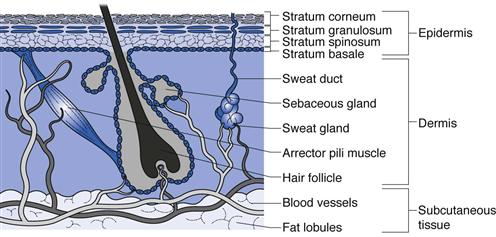
Fig. 39.1 A diagrammatical cross-section through human skin showing the different skin layers and appendages.
The subcutaneous layer
The inner subcutaneous fatty layer is typically several millimetres thick, except for some areas such as the eyelids where it is mostly absent. This subcutaneous layer of adipose tissue provides mechanical protection against physical shock, insulates the body, provides a store of high-energy molecules and carries the principal blood vessels and nerves to the skin. The subcutaneous layer is seldom an important barrier to transdermal and topical drug delivery.
The dermis
Overlying the fatty layer is the dermis, a layer typically 3–5 mm thick that is the major component of human skin. The dermis is composed of a network of mainly collagen and elastin in a mucopolysaccharide gel; essentially this combination provides an aqueous environment similar to a hydrogel. The dermis has several structures embedded within it, termed appendages, in particular nerve endings, pilosebaceous units (hair follicles and sebaceous glands) and eccrine and apocrine sweat glands (see below).
The dermis is metabolically active and requires extensive vasculature for this, as well as for regulating body temperature, for wound repair, to deliver oxygen and nutrients to the tissue and to remove waste products. The blood supply reaches to approximately 0.2 mm below the skin surface, near the dermis-epidermis boundary, and so most molecules passing through the outer layer of the skin are rapidly diluted and are carried systemically by the blood. This rich blood flow keeps the dermal concentration of most transdermally delivered drugs low, which in turn provides a concentration gradient from the outside of the body into the skin and it is this concentration gradient (more accurately, it is the chemical potential gradient) that allows drug delivery through the skin.
The epidermis
The epidermis overlies the dermis and is itself a multiple layer containing various cell types, including keratinocytes, melanocytes and Langerhans cells. Keratinocytes in the basal layer (stratum basale) undergo division and then differentiate as they migrate outwards, forming the stratum spinosum, then the stratum granulosum and finally the stratum corneum. Differentiation is complex and essentially changes the metabolically active basal cells that contain typical organelles, such as mitochondria and ribosomes, into stratum corneum that comprises anucleate flattened corneocytes packed into multiple lipid bilayers.
The stratum corneum
This outer skin layer is predominantly responsible for the barrier properties of human skin and limits drug delivery into and across the skin. The stratum corneum typically comprises only 10 to 15 cell layers and is around 10 µm thick when dry (although it can swell to several times this when wet). The stratum corneum is thinnest on the lips and eyelids and thickest on the load-bearing areas of the body such as the soles of the feet and palms of the hands. The lipid bilayers in which the keratin filled cells are embedded are uniquely different to other lipid bilayers in the body since they are comprised largely of ceramides, fatty acids, triglycerides and cholesterol/cholesterol sulphate, whilst phospholipids are largely absent. Longer chain ceramides act as ‘rivets’ connecting bilayers together and corneo-desmosomes interconnect corneocytes. The resulting structure can be likened to a brick wall (Fig. 39.1) where the keratin-filled cells act as the bricks in a mortar of multiply bilayered lipids.
In normal skin, it takes approximately 14 days for a daughter cell in the stratum basale to migrate and differentiate into a stratum corneum cell, and these cells are then retained in the stratum corneum for a further 2 weeks before they are shed.
The appendages
In terms of drug delivery, the appendages can be viewed as shunt routes or ‘short cuts’ through which molecules can pass across the stratum corneum barrier.
Specialized apocrine glands are found at specific body sites, such as the axillae and nipples, whereas eccrine glands are found over most of the body surface at a density of 100–200 glands per cm2. When stimulated by heat or emotional stress, eccrine glands secrete sweat, which is a dilute salt solution of around pH 5.
The largest appendages are the hair follicles and associated sebaceous glands which secrete sebum, composed of fatty acids, waxes and triglycerides. These lubricate the skin surface and help to maintain the skin surface pH at around 5. Skin typically has 50 to 100 hair follicles per cm2 but the load bearing areas of the soles and palms are largely devoid of these appendages.
Whilst these shunt routes offer a potential route through intact skin, the fractional area that they occupy is relatively small; for example, on the forearm, hair follicles occupy approximately 0.1% of the surface area although on the forehead this may be as much as 13%. The ducts are seldom empty, being occupied by sweat or sebum flowing out of the body which again inhibits drug delivery. However, formulators are able to target these structures, for example by delivering nano-sized drug delivery systems, such as liposomes, to the follicles in order to treat acne.
The shunt routes are important for electrical enhancement of transdermal drug delivery (iontophoresis) and also play a role in the early time course of passive drug delivery through the skin, where diffusion through the intact stratum corneum barrier has yet to reach steady state; rub a cut clove of garlic on your leg and it can be tasted within minutes, which is too fast for molecules to diffuse across the intact stratum corneum.
Transport through the skin
From the above discussion on the structure of skin it is clear that delivery of drug molecules from a topically applied formulation into the systemic circulation is complex, with numerous processes occurring and several routes of transport in operation, as illustrated in Figure 39.2.
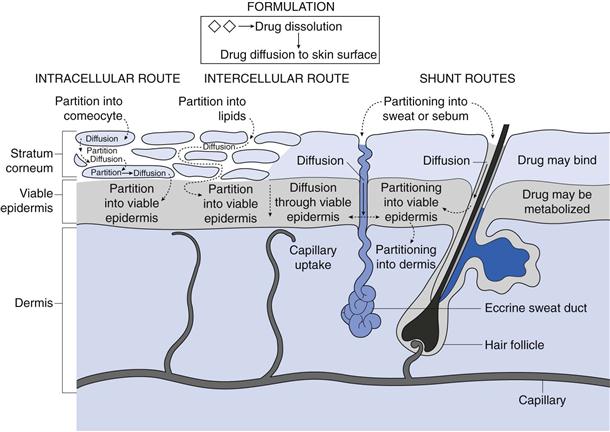
Fig. 39.2 Some of the processes occurring during transdermal drug delivery from a suspension formulation.
Initially, drug molecules must be presented to the skin surface. Consequently, if the formulation contains solid drug, then dissolution and diffusion through the formulation is the initial step in delivery. If the formulation contains dissolved drug, then as the molecules nearest to the skin surface enter the tissue these must be replaced by other molecules diffusing within the formulation towards the skin surface. Once at the outer layer of the stratum corneum, the drug molecule has three potential routes to cross the skin. Firstly it can pass via the shunt routes as described above. In this case molecules will partition into sweat or sebum before diffusing against the outflow from the glands.
More usually, the molecule encounters the intact stratum corneum ‘brick wall’ where transport can either be via an intracellular (also termed transcellular) route or transport can be intercellular.
Considering the intracellular route, the drug molecule initially partitions into a keratin-filled corneocyte, which is essentially an aqueous environment, they then diffuse through the corneocyte before partitioning into the intercellular lipid domains. For transcellular transport to continue, the molecule must then diffuse through the lipoidal region before repeatedly partitioning into and diffusing through the aqueous keratin in corneocytes and then intercellular lipids.
In contrast, the intercellular route requires the molecule to partition into the lipid bilayers between the corneocytes and then diffusion is via a tortuous route within the continuous lipid domain, i.e. following the mortar in the ‘brick wall’.
Having travelled through the stratum corneum, molecules diffuse through the lower epidermal layers before being cleared by the capillaries at the epidermal-dermal junction. During transport, there is potential for the permeant to bind to skin components such as keratin, in which case it may not reach the systemic circulation but could be sloughed off. In addition, skin is metabolically active and contains esterases, peptidases and hydrolases that can reduce the bioavailability of topically applied drugs such that, for example, only around 70% of topically applied glyceryl trinitrate (nitroglycerin) may be bioavailable.
It is important to recognize that, whilst three different routes exist for drugs to cross the skin (intercellular, transcellular, and shunt routes), for any permeant ALL three routes operate but the proportion of molecules crossing by the different routes will vary depending on the physicochemical properties of the permeant.
Permeant properties affecting permeation
Considering the processes described above, it is evident that the physicochemical properties of the permeant will control its transport into and through the skin. For both the transcellular and intercellular routes, the drug molecule has to cross the multiple lipid bilayers between the corneocytes and hence partitioning into, and diffusion through these lipid environments is essential. However, to reach the systemic circulation the molecule must also pass through the more aqueous environment of the viable epidermal cells and enter the blood. Thus, molecules which are lipophilic are usually seen as better candidates for transdermal delivery than hydrophilic compounds, but high lipophilicity is problematic for clearance.
The molecular weight of the permeant also impacts dramatically on its transport through the skin. The skin is designed to act as a barrier to external chemicals and so prevents the entry of large molecules, such as larger peptides and proteins. Not only is molecular weight an important factor in diffusion, but molecular structure (in particular hydrogen-bonding potential) can control the extent of binding to skin constituents and hence affect bioavailability. Naturally, the drug must have some solubility in the formulation and whilst transport through the stratum corneum is usually the rate limiting step in transdermal delivery, poor drug release from a formulation can occasionally limit drug transport. Finally, the effective dose of the drug must be relatively low to allow the application of appropriately sized patches/formulations.
The above processes restrict the range of drugs that can be delivered transdermally to therapeutically useful levels and some generic ‘rules of thumb’ can be used to predict whether transdermal delivery is viable for an active pharmaceutical ingredient. These include:
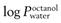 ) should be in the range 1–4, though more lipophilic molecules may be effectively delivered, such as fentanyl
) should be in the range 1–4, though more lipophilic molecules may be effectively delivered, such as fentanylIndeed, the active pharmaceutical ingredients currently used in transdermal formulations have many of the above properties; estradiol MW is 272 and it is lipophilic, with a 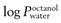 of 2.7; fentanyl MW is 336 with a log
of 2.7; fentanyl MW is 336 with a log 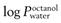 of 4.4; nicotine MW is 162 with a
of 4.4; nicotine MW is 162 with a 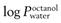 of 1.2.
of 1.2.
Mathematics of skin permeation
With such a highly complex multiple layered organ as skin, and numerous factors affecting transdermal drug delivery, it appears daunting to apply mathematical principles to describe such a complex process. However, simple mathematical principles can be used to understand the basic principles of permeation through membranes, including skin, and these assist in designing dosage forms for transdermal and topical drug delivery.
Fick’s laws of diffusion
Considering simple passive diffusion where molecules move by random motion from one region to another in the direction of decreasing concentration, then transport can be described by Fick’s First Law of Diffusion (Chapter 2):
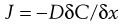 (39.1)
(39.1)
where J is the flux (the mass flow rate at which the material passes through unit area of the surface), C is the concentration of diffusing substance, x is the space co-ordinate measured normal to the section and D is the diffusion coefficient of the permeant. The negative sign demonstrates that the flux of molecules is in the direction of decreasing concentration. When a topically applied drug enters the skin, it is usually assumed that the diffusion gradient is from the outer surface into the tissue, i.e. is unidirectional.
Fick’s Second Law of Diffusion gives:
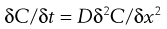 (39.2)
(39.2)
where t is time. Essentially this equation shows that the rate of change of concentration with time at a point within a diffusional field is proportional to the rate of change in the concentration gradient at that point.
The above laws assume that diffusion is through an isotropic material (i.e. one that has the same structural and diffusional properties in all directions); skin clearly is not isotropic with multiple layers, different permeation routes, etc. and indeed it is remarkable that Fickian diffusion can be used to generate valuable approximations from transdermal drug delivery data.
Experimental estimation of skin penetration
Experimentally, it is usually difficult to study transdermal drug delivery in vivo, so most researchers use in vitro protocols to mimic as closely as possible the in vivo situation. Most commonly, a membrane (for example human epidermis) is used to separate two compartments in a diffusion cell. The drug in a vehicle (for example water, buffer or in a formulation) is then applied to the uppermost skin surface (stratum corneum). This is usually termed the ‘donor’ solution. The other compartment contains a ‘receptor’ (or receiver) solution that is a good solvent for the drug, but which will not affect the skin barrier. This receptor solution thus provides essentially sink conditions (near zero concentration) of the permeant and allows a concentration gradient to exist between the donor and receptor phase, which in turn provides the driving force for diffusion across the membrane. If the cumulative mass of permeant that crosses the membrane is plotted as a function of time, then a typical permeation profile can be drawn, as illustrated in Figure 39.3.
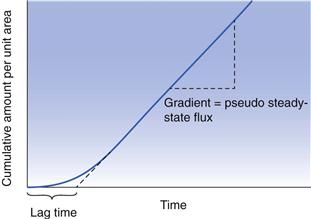
As can be seen, after sufficient time the plot approaches a straight line and from the slope we can obtain the steady state flux, dm/dt and Equation 39.2 can then be simplified to:
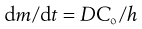 (39.3)
(39.3)
where dm/dt is the flux, usually termed J which is the cumulative mass of permeant that passes per unit area of the membrane in time t, Co is the concentration of permeant in the first layer of the membrane (at the skin surface, in contact with the donor solution) and h is the membrane thickness.
It is difficult to measure Co, the concentration of permeant in the first layer of the skin, but the concentration of the drug in the vehicle (donor solution), termed Cv, which bathes the skin membrane is usually known or can be determined easily. Differences in drug concentration between the donor solution and the first skin layer arise due to partitioning of the molecule between the membrane and donor solution so:
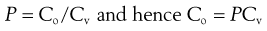 (39.4)
(39.4)
where P is the partition coefficient of the permeant between the membrane and the vehicle. Simply substituting Equation 39.4 into Equation 39.3 gives:
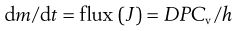 (39.5)
(39.5)
Equation 39.5 thus reiterates that the flux of a permeant through skin will be high for molecules with a high diffusion coefficient (e.g. generally having a relatively small molecular weight), will increase with increasing partitioning between the membrane and the donor solution (e.g. for lipophilic molecules) and will increase with increasing effective concentration in the donor solution (which increases the chemical potential gradient), whereas the flux through thicker membranes is reduced.
Figure 39.3 also shows that the lag time can be evaluated experimentally. The lag time (L
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


